Label: SOTYKTU- deucravacitinib tablet, film coated

- NDC Code(s): 0003-0895-11, 0003-0895-91
- Packager: E.R. Squibb & Sons, L.L.C.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated September 9, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SOTYKTU safely and effectively. See full prescribing information for SOTYKTU.
SOTYKTU™ (deucravacitinib) tablets, for oral use
Initial U.S. Approval: 2022INDICATIONS AND USAGE
SOTYKTU is a tyrosine kinase 2 (TYK2) inhibitor indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. (1)
Limitations of Use:
Not recommended for use in combination with other potent immunosuppressants.DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 6 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- •
- Hypersensitivity: Hypersensitivity reactions such as angioedema have been reported. Discontinue if a clinically significant hypersensitivity reaction occurs. (5.1)
- •
- Infections: SOTYKTU may increase the risk of infection. Avoid use in patients with active or serious infection. If a serious infection develops, discontinue SOTYKTU until the infection resolves. (5.2)
- •
- Tuberculosis: Evaluate for TB prior to initiating treatment with SOTYKTU. (5.3)
- •
- Malignancy: Malignancies including lymphomas were observed in clinical trials with SOTYKTU (5.4)
- •
- Rhabdomyolysis and elevated CPK. (5.5)
- •
- Laboratory Abnormalities: Periodically evaluate serum triglycerides. Evaluate liver enzymes at baseline and thereafter in patients with known or suspected liver disease. (5.6)
- •
- Immunizations: Avoid use with live vaccines. (5.7)
- •
- Potential Risks Related to JAK Inhibition: It is not known whether TYK2 inhibition may be associated with the observed or potential adverse reactions of JAK inhibition. Higher rates of all-cause mortality, including sudden cardiovascular death, major adverse cardiovascular events, overall thrombosis, deep venous thrombosis, pulmonary embolism, and malignancies (excluding non-melanoma skin cancer) were observed in patients treated with a JAK inhibitor compared to those treated with TNF blockers in rheumatoid arthritis (RA) patients. SOTYKTU is not approved for use in RA. (5.8)
ADVERSE REACTIONS
Most common adverse reactions (≥ 1%) are upper respiratory infections, blood creatine phosphokinase increased, herpes simplex, mouth ulcers, folliculitis, and acne. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
SOTYKTU is not recommended in patients with severe hepatic impairment (Child-Pugh C). (2.3)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Evaluations and Immunizations Prior to Treatment Initiation
2.2 Recommended Dosage
2.3 Recommended Dosage in Patients with Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
5.2 Infections
5.3 Tuberculosis
5.4 Malignancy including Lymphomas
5.5 Rhabdomyolysis and Elevated CPK
5.6 Laboratory Abnormalities
5.7 Immunizations
5.8 Potential Risks Related to JAK Inhibition
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Plaque Psoriasis
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Evaluations and Immunizations Prior to Treatment Initiation
Evaluate patients for active and latent tuberculosis (TB) infection prior to initiating treatment with SOTYKTU. If positive, start treatment for TB prior to SOTYKTU use [see Warnings and Precautions (5.3)].
Update immunizations according to current immunization guidelines [see Warnings and Precautions (5.7)].
2.2 Recommended Dosage
The recommended dosage of SOTYKTU is 6 mg taken orally once daily, with or without food.
Do not crush, cut, or chew the tablets.
2.3 Recommended Dosage in Patients with Hepatic Impairment
SOTYKTU is not recommended in patients with severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
No dosage adjustment is needed for patients with mild to moderate hepatic impairment.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
SOTYKTU is contraindicated in patients with a history of hypersensitivity reaction to deucravacitinib or to any of the excipients in SOTYKTU [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Hypersensitivity reactions such as angioedema have been reported in subjects receiving SOTYKTU. If a clinically significant hypersensitivity reaction occurs, institute appropriate therapy and discontinue SOTYKTU [see Adverse Reactions (6.1)].
5.2 Infections
SOTYKTU may increase the risk of infections.
Serious infections have been reported in subjects with psoriasis who received SOTYKTU. The most common serious infections reported with SOTYKTU included pneumonia and COVID-19 [see Adverse Reactions (6.1)].
Avoid use of SOTYKTU in patients with an active or serious infection.
Consider the risks and benefits of treatment prior to initiating SOTYKTU in patients:
- •
- with chronic or recurrent infection
- •
- who have been exposed to tuberculosis
- •
- with a history of a serious or an opportunistic infection
- •
- with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with SOTYKTU. A patient who develops a new infection during treatment with SOTYKTU should undergo prompt and complete diagnostic testing; appropriate antimicrobial therapy should be initiated; and the patient should be closely monitored. Interrupt SOTYKTU if a patient develops a serious infection. Do not resume SOTYKTU until the infection resolves or is adequately treated.
Viral Reactivation
Herpes virus reactivation (e.g., herpes zoster, herpes simplex), was reported in clinical trials with SOTYKTU. In the 16‑week placebo-controlled period, herpes simplex infections were reported in 17 subjects (6.8 per 100 patient‑years) treated with SOTYKTU, and 1 subject (0.8 per 100 patient-years) treated with placebo.
Multidermatomal herpes zoster was reported in an immunocompetent subject who received SOTYKTU.
During PSO-1, PSO-2, and the open-label extension trial in which subjects who completed the controlled trials could enroll, the majority of subjects who reported events of herpes zoster while receiving SOTYKTU were under 50 years of age.
The impact of SOTYKTU on chronic viral hepatitis reactivation is unknown. Subjects with positive screening tests for hepatitis B or C, or chronic hepatitis B, or untreated hepatitis C were excluded from clinical trials. Consider viral hepatitis screening and monitoring for reactivation in accordance with clinical guidelines before starting therapy and during therapy with SOTYKTU. If signs of reactivation occur, consult a hepatitis specialist. SOTYKTU is not recommended for use in patients with active hepatitis B or hepatitis C.
5.3 Tuberculosis
In clinical trials, of 4 subjects with latent tuberculosis (TB) who were treated with SOTYKTU and received appropriate TB prophylaxis, no subjects developed active TB (during the mean follow-up of 34 weeks). One subject, who did not have latent TB, developed active TB after receiving 54 weeks of SOTYKTU.
Evaluate patients for latent and active TB infection prior to initiating treatment with SOTYKTU. Do not administer SOTYKTU to patients with active TB. Initiate treatment of latent TB prior to administering SOTYKTU.
Consider anti-TB therapy prior to initiation of SOTYKTU in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor patients receiving SOTYKTU for signs and symptoms of active TB during treatment.
5.4 Malignancy including Lymphomas
Malignancies, including lymphomas, were observed in clinical trials with SOTYKTU [see Adverse Reactions (6.1)].
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with SOTYKTU, particularly in patients with a known malignancy (other than a successfully treated non-melanoma skin cancer) and patients who develop a malignancy when on treatment with SOTYKTU.
5.5 Rhabdomyolysis and Elevated CPK
Cases of rhabdomyolysis were reported in subjects treated with SOTYKTU resulting in interruption or discontinuation of SOTYKTU dosing.
Treatment with SOTYKTU was associated with an increased incidence of asymptomatic creatine phosphokinase (CPK) elevation and rhabdomyolysis compared to treatment with placebo. Discontinue SOTYKTU if markedly elevated CPK levels occur or myopathy is diagnosed or suspected. Instruct patients to promptly report any unexplained muscle pain, tenderness or weakness, particularly if accompanied by malaise or fever [see Adverse Reactions (6.1)].
5.6 Laboratory Abnormalities
Triglyceride Elevations - Treatment with SOTYKTU was associated with increases in triglyceride levels. The effect of this elevated parameter on cardiovascular morbidity and mortality has not been determined. Periodically evaluate serum triglycerides according to the clinical guidelines for hyperlipidemia while patients are receiving treatment with SOTYKTU. Manage patients according to clinical guidelines for the management of hyperlipidemia [see Adverse Reactions (6.1)].
Liver Enzyme Elevations - Treatment with SOTYKTU was associated with an increase in the incidence of liver enzyme elevation compared to treatment with placebo. Liver serum transaminase elevations ≥ 3 times the ULN were reported in subjects treated with SOTYKTU. Evaluate liver enzymes at baseline and thereafter in patients with known or suspected liver disease according to routine patient management. If treatment-related increases in liver enzymes occur and drug-induced liver injury is suspected, interrupt SOTYKTU until a diagnosis of liver injury is excluded [see Adverse Reactions (6.1)].
5.7 Immunizations
Prior to initiating therapy with SOTYKTU, consider completion of all age-appropriate immunizations according to current immunization guidelines including prophylactic herpes zoster vaccination. Avoid use of live vaccines in patients treated with SOTYKTU. The response to live or non-live vaccines has not been evaluated.
5.8 Potential Risks Related to JAK Inhibition
It is not known whether TYK2 inhibition may be associated with the observed or potential adverse reactions of Janus Kinase (JAK) inhibition. In a large, randomized, postmarketing safety trial of a JAK inhibitor in rheumatoid arthritis (RA), patients 50 years of age and older with at least one cardiovascular risk factor, higher rates of all-cause mortality, including sudden cardiovascular death, major adverse cardiovascular events, overall thrombosis, deep venous thrombosis, pulmonary embolism, and malignancies (excluding non-melanoma skin cancer) were observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. SOTYKTU is not approved for use in RA.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of labeling:
- •
- Infections [see Warnings and Precautions (5.2)]
- •
- Malignancy including lymphomas [see Warnings and Precautions (5.4)]
- •
- Laboratory Abnormalities [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of SOTYKTU was evaluated in two placebo- and active-controlled trials (PSO-1 and PSO-2) and an open-label extension trial in which subjects who completed PSO-1 or PSO-2 could enroll [see Clinical Studies (14)]. In these clinical trials, a total of 1,519 subjects with moderate-to-severe plaque psoriasis who were candidates for systemic therapy or phototherapy received SOTYKTU 6 mg orally once daily. Of these, 1,141 subjects were exposed to SOTYKTU for at least one year.
In trials PSO-1 and PSO-2, 1,681 subjects were randomized to receive SOTYKTU 6 mg (840 subjects), placebo (419 subjects), or apremilast 30 mg twice daily (422 subjects). All subjects randomized to placebo switched to SOTYKTU at Week 16. All other subjects remained in their original treatment group until Week 24, at which point subjects could have continued on the same treatment or be switched to SOTYKTU or placebo. The mean age of subjects was 47 years. The majority of subjects were White (87%) and male (67%).
In the 16-week placebo-controlled period of the pooled clinical trials (PSO-1 and PSO-2), discontinuation of therapy due to adverse reactions in subjects who received SOTYKTU was 2.4%, compared to 3.8% for placebo.
Table 1 summarizes the adverse reactions that occurred in at least 1% of subjects in the SOTYKTU group and at a higher rate than the placebo group during the 16-week controlled period.
Table 1: Adverse Reactions that Occurred in ≥ 1% of Subjects with Plaque Psoriasis in the SOTYKTU Group and More Frequently than in the Placebo Group in Trials PSO-1 and PSO-2 through Week 16 a Includes upper respiratory tract infection (viral, bacterial, and unspecified), nasopharyngitis, pharyngitis (including viral, streptococcal, and unspecified), sinusitis (includes acute, viral, bacterial), rhinitis, rhinotracheitis, tracheitis, laryngitis, and tonsillitis (including bacterial, streptococcal)
b Includes oral herpes, genital herpes, herpes simplex, and herpes virus infection
c Includes mouth ulceration, aphthous ulcer, tongue ulceration, and stomatitis
d Includes acne, acne cystic, and dermatitis acneiformAdverse Reaction
SOTYKTU
6 mg once dailyPlacebo
N=840
n (%)N=419
n (%)Upper respiratory infectionsa
161 (19.2)
62 (14.8)
Blood creatine phosphokinase increased
23 (2.7)
5 (1.2)
Herpes simplexb
17 (2.0)
1 (0.2)
Mouth ulcersc
16 (1.9)
0 (0.0)
Folliculitis
14 (1.7)
0 (0.0)
Acned
12 (1.4)
1 (0.2)
Adverse reactions that occurred in < 1% of subjects in the SOTYKTU group were herpes zoster.
Specific Adverse Reactions
Exposure adjusted incidence rates are reported for all the adverse reactions presented below.
Infections
In the 16-week placebo-controlled period, infections occurred in 29% of the SOTYKTU group (116 events per 100 person-years) compared to 22% of the placebo group (83.7 events per 100 person-years). The majority of infections were non-serious and mild to moderate in severity and did not lead to discontinuation of SOTYKTU.
In the 16-week placebo-controlled period, serious infections were reported in 5 subjects (2.0 per 100 patient-years) treated with SOTYKTU, and 2 subjects (1.6 per 100 patient-years) treated with placebo.
The most common serious infections reported during the 52-week treatment period were pneumonia and COVID-19.
Malignancies
During the 0-to-52-week treatment period of the two clinical trials, PSO-1 and PSO-2 (total exposure of 986 patient-years with SOTYKTU), malignancies (excluding non-melanoma skin cancer) were reported in 3 subjects treated with SOTYKTU (0.3 per 100 patient-years), including single cases each of breast cancer, hepatocellular carcinoma, and lymphoma after 24, 32, and 25 weeks of treatment, respectively.
During PSO-1, PSO-2, and the open-label extension trial in which subjects who completed the controlled trials could enroll, a total of 3 subjects (0.1 per 100 patient-years), developed lymphoma while receiving SOTYKTU after 25, 77, and 98 weeks of treatment.
Laboratory Abnormalities
Creatine Phosphokinase (CPK)
In the 16-week placebo-controlled period, increased CPK (including Grade 4) was reported in 23 subjects (9.3 per 100 patient-years) treated with SOTYKTU, and 5 subjects (4.1 per 100 patient-years) treated with placebo.
Liver Enzyme Elevations
Events of increases in liver enzymes ≥3 times the ULN were observed in subjects treated with SOTYKTU [see Warnings and Precautions (5.5)]. In the 16-week placebo-controlled period:
- •
- ALT elevations ≥3 times the ULN was reported in 9 subjects (3.6 per 100 patient- years) treated with SOTYKTU, and 2 subjects (1.6 per 100 patient-years) treated with placebo.
- •
- AST elevations ≥3 times the ULN was reported in 13 subjects (5.2 per 100 patient- years) treated with SOTYKTU, and 2 subjects (1.6 per 100 patient-years) treated with placebo.
Decreased Glomerular Filtration Rate (GFR)
In the 16-week placebo-controlled period in subjects who had moderate renal impairment (eGFR 30-59 mL/min) at baseline, decreased GFR was reported in 4 subjects (1.6 per 100 patient-years) treated with SOTYKTU, and 1 subject (0.8 per 100 patient-years) treated with placebo. Two of the deucravacitinib-treated subjects had worsening of baseline proteinuria.
Lipid Elevations
Mean triglycerides increased by 10.3 mg/dL during the 16-week treatment period in subjects treated with SOTYKTU and by 9.1 mg/dL during the 52-week treatment period.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from case reports on SOTYKTU use during pregnancy are insufficient to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
In animal reproduction studies, no effects on embryo-fetal development were observed with oral administration of deucravacitinib to rats and rabbits during organogenesis at doses that were at least 91 times the maximum recommended human dose (MRHD) of 6 mg once daily (see Data).
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Report pregnancies to the Bristol-Myers Squibb Company’s Adverse Event reporting line at 1-800-721-5072.
Data
Animal data
Deucravacitinib was administered orally during the period of organogenesis at doses of 5, 15, or 75 mg/kg/day in rats and 1, 3, or 10 mg/kg/day in rabbits. Deucravacitinib was not associated with embryo-fetal lethality or fetal malformations in either species. These doses resulted in maternal exposures (AUC) that were 266 times (rat) or 91 times (rabbit) the exposure at the MRHD.
In a pre- and post-natal development study in rats, deucravacitinib was administered orally from gestation day 6 through lactation day 20, at doses of 5, 15, or 50 mg/kg/day. At 50 mg/kg/day, F1 offspring had reduced body weight gains during the pre-weaning period. After weaning, body weights of affected F1 offspring gradually normalized to control levels. No maternal effects were observed at 50 mg/kg/day (110 times the MRHD based on AUC comparison). No deucravacitinib-related effects on postnatal developmental, neurobehavioral, or reproductive performance of offspring were noted at doses up to 15 mg/kg/day (19 times the MRHD based on AUC comparison).
8.2 Lactation
Risk Summary
There are no data on the presence of deucravacitinib in human milk, the effects on the breastfed infant, or the effects on milk production. Deucravacitinib is present in rat milk. When a drug is present in animal milk, it is likely that the drug will be present in human milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SOTYKTU and any potential adverse effects on the breastfed infant from SOTYKTU or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of SOTYKTU in pediatric patients have not been established.
8.5 Geriatric Use
Of the 1,519 subjects with plaque psoriasis treated with SOTYKTU, 152 (10%) subjects were 65 years or older and 21 (1.4%) subjects were 75 years or older.
During the Week 0-16 period, for those subjects (80 subjects ≥ 65 years old, including 12 subjects ≥ 75 years old) who received SOTYKTU without switching treatment arms, there was a higher rate of overall serious adverse reactions, including serious infections, and discontinuations due to adverse reactions compared with younger adults.
No overall differences in effectiveness of SOTYKTU have been observed between patients 65 years of age and older and younger adult patients.
8.6 Renal Impairment
No dose adjustment of SOTYKTU is recommended in patients with mild, moderate, or severe renal impairment or in patients with end stage renal disease (ESRD) on dialysis [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment of SOTYKTU is recommended in patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment. SOTYKTU is not recommended for use in patients with severe hepatic impairment (Child-Pugh C) [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no experience regarding human overdosage with SOTYKTU. In case of overdose, consider contacting the Poison Help line (1-800-222-1222) for additional overdosage management recommendations.
The extent of deucravacitinib elimination by hemodialysis was small (5.4% of dose per dialysis treatment), and thus hemodialysis for treatment of overdose with SOTYKTU is limited.
-
11 DESCRIPTION
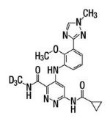
Deucravacitinib is a tyrosine kinase 2 (TYK2) inhibitor and is described chemically as:
6-(cyclopropanecarbonylamido)-4-[2-methoxy-3-(1-methyl-1,2,4-triazol-3-yl)anilino]-N-(trideuteriomethyl)pyridazine-3-carboxamide.
The molecular formula is C20H19D3N8O3 and the molecular weight of the free base is 425.47. Deucravacitinib has the structural formula:
Deucravacitinib is a white to yellow powder. The solubility of deucravacitinib is pH dependent. Solubility decreases with increasing pH.
SOTYKTU (deucravacitinib) tablets are supplied in 6 mg strength for oral administration. Each tablet contains deucravacitinib as the active ingredient and the following inactive ingredients: anhydrous lactose, croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, microcrystalline cellulose, and silicon dioxide. In addition, the film coating Opadry® II Pink contains the following inactive ingredients: iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Deucravacitinib is an inhibitor of tyrosine kinase 2 (TYK2). TYK2 is a member of the Janus kinase (JAK) family. Deucravacitinib binds to the regulatory domain of TYK2, stabilizing an inhibitory interaction between the regulatory and the catalytic domains of the enzyme. This results in allosteric inhibition of receptor-mediated activation of TYK2 and its downstream activation of Signal Transducers and Activators of Transcription (STATs) as shown in cell-based assays. JAK kinases, including TYK2, function as pairs of homo- or heterodimers in the JAK-STAT pathways. TYK2 pairs with JAK1 to mediate multiple cytokine pathways and also pairs with JAK2 to transmit signals as shown in cell-based assays. The precise mechanism linking inhibition of TYK2 enzyme to therapeutic effectiveness in the treatment of adults with moderate-to-severe plaque psoriasis is not currently known.
12.2 Pharmacodynamics
In patients with psoriasis, deucravacitinib reduced psoriasis-associated gene expression in psoriatic skin in a dose dependent manner, including reductions in IL-23-pathway and type I IFN pathway regulated genes. Deucravacitinib reduced IL-17A, IL-19 and beta-defensin by 47 to 50%, 72%, and 81 to 84% respectively following 16 weeks of once daily treatment. The relationship between these pharmacodynamic markers and the mechanism(s) by which deucravacitinib exerts its clinical effects is unknown.
12.3 Pharmacokinetics
Following oral administration, deucravacitinib plasma Cmax and AUC increased proportionally over a dose range from 3 mg to 36 mg (0.5 to 6 times the approved recommended dosage) in healthy subjects. The accumulation of deucravacitinib was <1.4-fold following once daily dosing in healthy subjects. The PK of deucravacitinib and its active metabolite, BMT‑153261, were comparable between healthy subjects and subjects with psoriasis. The steady state Cmax and AUC24 of deucravacitinib following administration of 6 mg once daily were 45 ng/mL and 473 ng·hr/mL, respectively. The steady state Cmax and AUC24 of the active deucravacitinib metabolite, BMT-153261, following administration of 6 mg once daily were 5 ng/mL and 95 ng·hr/mL, respectively.
Absorption
The absolute oral bioavailability of deucravacitinib was 99% and the median Tmax ranged from 2 to 3 hours in healthy subjects.
Food Effect
No clinically significant differences in the pharmacokinetics of deucravacitinib were observed following administration of a high-fat, high-calorie meal (951 kcal in total, with approximate distribution of 52% fat, 33% carbohydrate and 15% protein). Cmax and AUC of deucravacitinib when administered with food were decreased by approximately 24% and 11%, respectively, and Tmax was prolonged by 1 hour. Cmax and AUC of BMT-153261 when administered with food were decreased by approximately 23% and 10%, respectively, and Tmax was prolonged by 2 hours.
Distribution
The volume of distribution of deucravacitinib at steady state is 140 L. Protein binding of deucravacitinib was 82 to 90% and the blood-to-plasma concentration ratio was 1.26.
Elimination
The terminal half-life of deucravacitinib was 10 hours. The renal clearance of deucravacitinib ranged from 27 to 54 mL/minute.
Metabolism
Deucravacitinib is metabolized by cytochrome P-450 (CYP) 1A2 to form major metabolite BMT-153261. Deucravacitinib is also metabolized by CYP2B6, CYP2D6, carboxylesterase (CES) 2, and uridine glucuronyl transferase (UGT) 1A9.
The active deucravacitinib metabolite, BMT-153261, has comparable potency to the parent drug, but the circulating exposure of BMT-153261 accounts for approximately 20% of the systemic exposure of the total drug-related components.
Specific Populations
Patients with Renal Impairment
Deucravacitinib Cmax was 14% lower and 6% higher in patients with mild (eGFR ≥60 to <90 mL/min/1.73m2) and moderate (eGFR ≥30 to <60 mL/min/1.73m2) renal impairment, compared to subjects with normal renal function (eGFR ≥ 90 mL/min/1.73m2); no change in Cmax was observed in patients with severe (eGFR <30 mL/min/1.73m2) renal impairment, and ESRD (eGFR <15 mL/min/1.73m2) on dialysis. Deucravacitinib AUCinf was unchanged in patients with mild renal impairment but higher by 39%, 28% and 34% in patients with moderate, severe and ESRD on dialysis, respectively, compared to subjects with normal renal function.
BMT-153261 Cmax was 11% lower, 8% lower, 28% higher and 9% higher in patients with mild, moderate, severe renal impairment and ESRD on dialysis, respectively, compared to subjects with normal renal function. BMT-153261 AUCinf was 2% lower, 24% higher, 81% higher and 27% higher in patients with mild, moderate, severe renal impairment and ESRD on dialysis, respectively, compared to subjects with normal renal function.
Dialysis did not substantially clear deucravacitinib from systemic circulation (5.4% of dose cleared per dialysis).
Patients with Hepatic Impairment
Deucravacitinib Cmax was higher by 4%, 10% and 1% in patients with mild (Child-Pugh Class A), moderate (Child-Pugh Class B), and severe (Child-Pugh Class C) hepatic impairment, respectively, compared to subjects with normal hepatic function. Deucravacitinib AUCinf was higher by 10%, 40% and 43% in patients with mild, moderate, and severe hepatic impairment, respectively, compared to subjects with normal hepatic function.
BMT-153261 Cmax was lower by 25%, 59% and 79% in patients with mild, moderate, and severe hepatic impairment, respectively, compared to subjects with normal hepatic function. BMT-153261 AUCinf was lower by 3%, 20% and 50% in patients with mild, moderate, and severe hepatic impairment, respectively, compared to subjects with normal hepatic function [see Use in Specific Populations (8.7)].
Drug Interaction Studies
Clinical Trials
No clinically significant differences in the pharmacokinetics of deucravacitinib were observed when co-administered with the following drugs: Cyclosporine (dual Pgp/BCRP inhibitor), fluvoxamine (CYP1A2 inhibitor), ritonavir (CYP1A2 inducer), diflunisal (UGT 1A9 inhibitor), pyrimethamine (OCT1 inhibitor), famotidine (H2 receptor antagonist), or rabeprazole (proton pump inhibitor).
No clinically significant differences in the pharmacokinetics of the following drugs were observed when co-administered with deucravacitinib: Rosuvastatin, methotrexate, mycophenolate mofetil (MMF) and oral contraceptives (norethindrone acetate and ethinyl estradiol).
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Deucravacitinib is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4. Deucravacitinib is not an inducer of CYP1A2, CYP2B6, or CYP3A4.
Carboxylesterase (CES) Enzymes: Deucravacitinib is not an inhibitor of CES2.
Uridine diphosphate (UDP)-glucuronosyl transferase (UGT) Enzymes: Deucravacitinib is not an inhibitor of UGT1A1, UGT1A4, UGT1A6, UGT1A9, or UGT2B7.
Transporter Systems: Deucravacitinib is a substrate of Pgp, BCRP, and OCT1, but not OATP, NTCP, OAT1, OAT3, OCT2, MATE1, or MATE2K. Deucravacitinib is an inhibitor of BCRP and OATP1B3, but not an inhibitor of Pgp, OATP1B1, NTCP, BSEP, MRP2, OAT1, OAT3, OCT1, OCT2, MATE1, or MATE2-K.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of deucravacitinib was assessed in 2-year rat and 6-month rasH2 transgenic mouse studies. No evidence of tumorigenicity was observed in male or female rats that received deucravacitinib at oral doses up to 15 mg/kg/day (51 times the MRHD based on AUC comparison). No evidence of tumorigenicity was observed in male or female Tg.rasH2 mice that received deucravacitinib at oral doses up to 60 mg/kg/day.
Deucravacitinib was not mutagenic in a bacterial mutagenicity assay (Ames test) or clastogenic in an in vitro chromosomal aberration assay (cultured Chinese hamster ovary cells) or in an in vivo rat peripheral blood micronucleus assay.
In male rats, deucravacitinib had no effects on reproductive parameters (mating, fertility, and sperm morphology) or early embryonic development of their offspring at oral doses up to 50 mg/kg/day (224 times the MRHD based on AUC comparison).
In female rats, deucravacitinib had no effects on mating, fertility, or early embryonic parameters at oral doses up to 50 mg/kg/day (171 times the MRHD based on AUC comparison).
-
14 CLINICAL STUDIES
14.1 Plaque Psoriasis
The efficacy and safety of SOTYKTU 6 mg once daily were assessed in two multicenter, randomized, double-blind, placebo- and active-controlled clinical trials, PSO-1 (NCT03624127) and PSO-2 (NCT03611751) which enrolled subjects 18 years of age and older with moderate-to-severe plaque psoriasis who were eligible for systemic therapy or phototherapy. Subjects had a body surface area (BSA) involvement of ≥ 10%, a Psoriasis Area and Severity Index (PASI) score ≥ 12, and a static Physician’s Global Assessment (sPGA) ≥ 3 (moderate or severe).
In PSO-1 and PSO-2, efficacy was assessed in 1,684 subjects randomized to either SOTYKTU (6 mg orally once daily), placebo, or apremilast (30 mg orally twice daily).
Endpoints
Both trials assessed the responses at Week 16 compared to placebo for the two co-primary endpoints:
- •
- proportion of subjects who achieved a sPGA score of 0 (clear) or 1 (almost clear) with at least a 2-grade improvement from baseline;
- •
- the proportion of subjects who achieved at least a 75% improvement in PASI scores from baseline (PASI 75).
Other comparisons between SOTYKTU and placebo that were secondary endpoints at Week 16:
- •
- the proportion of subjects who achieved PASI 90, PASI 100, sPGA 0, scalp severity PGA (ss-PGA) score of 0 (clear) or 1 (almost clear) with at least 2-grade improvement, and Psoriasis Symptoms and Signs Diary (PSSD) Symptom Score of 0 (symptom-free).
Comparisons between SOTYKTU and apremilast were made for the following secondary endpoints at these time points:
- •
- at Week 16 and Week 24 (PSO-1 and PSO-2), the proportion of subjects who achieved PASI 75, PASI 90, and sPGA 0/1 with at least a 2-grade improvement from baseline
- •
- at Week 16 (PSO-1 and PSO-2), the proportion of subjects who achieved sPGA 0 and ss-PGA 0/1 with at least a 2-grade improvement from baseline (scalp).
Results
In both trials, the mean age was 47 years, the mean weight was 91 kg, 67% of subjects were male, 13% were Hispanic or Latino, 87% were White, 2% were Black, and 10% were Asian. At baseline, subjects had a median affected BSA of 20% and a median PASI score of 19. The proportion of subjects with sPGA score of 3 (moderate) and 4 (severe) at baseline were 80% and 20%, respectively. Approximately 18% of subjects had a history of psoriatic arthritis.
Across both trials, 40% of subjects had received prior phototherapy, 42% were naive to any systemic therapy (including biologic and/or non-biologic treatment), 41% received prior non-biologic systemic treatment, and 35% had received prior biologic therapy.
Table 2 presents the efficacy results of SOTYKTU compared to apremilast and placebo in PSO-1. Table 3 presents the efficacy results in PSO-2.
Table 2: Efficacy Results in Adults with Moderate to Severe Plaque Psoriasis (NRIa) in PSO-1 CI = Confidence interval; PASI = Psoriasis Area and Severity Index; sPGA = Static Physician Global Assessment; ss-PGA = Scalp Specific Physician’s Global Assessment
a NRI = Non-Responder Imputation
b Adjusted difference in proportions is the weighted average of the treatment differences across region, body weight and prior biologic use with the Cochran-Mantel-Haenszel weights.
c Co-primary endpoints comparing SOTYKTU to placebo
d Includes only subjects with baseline ss-PGA score of ≥ 3Endpoint
SOTYKTU
(N=330)
n (%)
Placebo
(N=166)
n (%)
Apremilast
(N=168)
n (%)
Difference, % (95% CI)b
Difference from Placebo
Difference from Apremilast
sPGA response of 0/1 (clear or almost clear)
Week 16c
178 (54)
12 (7)
54 (32)
47 (40, 53)
22 (13, 30)
Week 24
194 (59)
-
52 (31)
-
27 (19, 36)
sPGA response of 0
Week 16
58 (18)
1 (1)
8 (5)
17 (13, 21)
13 (8, 18)
PASI 75 response
Week 16c
193 (58)
21 (13)
59 (35)
46 (39, 53)
23 (14, 32)
Week 24
228(69)
-
64 (38)
-
31 (22, 40)
PASI 90 response
Week 16
118 (36)
7 (4)
33 (20)
32 (26, 38)
16 (8, 24)
Week 24
140 (42)
-
37 (22)
-
20 (12, 28)
PASI 100 response
Week 16
47 (14)
1 (1)
-
14 (10, 18)
-
ss-PGA response of 0/1 (scalp)d
(N=209)
(N=121)
(N=110)
Week 16
147 (70)
21 (17)
43 (39)
53 (44, 62)
30 (19, 41)
Table 3: Efficacy Results in Adults with Moderate to Severe Plaque Psoriasis (NRIa) in PSO-2 CI = Confidence interval; PASI = Psoriasis Area and Severity Index; sPGA = Static Physician Global Assessment; ss-PGA = Scalp Specific Physician’s Global Assessment
a NRI = Non-Responder Imputation
b Adjusted difference in proportions is the weighted average of the treatment differences across region, body weight and prior biologic use with the Cochran-Mantel-Haenszel weights.
c Co-primary endpoints comparing SOTYKTU to placebo
d Includes only subjects with baseline ss-PGA score of ≥ 3Endpoint
SOTYKTU
(N=511)
n (%)
Placebo
(N=255)
n (%)
Apremilast
(N=254)
n (%)
Difference, % (95% CI)b
Difference from Placebo
Difference from Apremilast
sPGA response of 0/1 (clear or almost clear)
Week 16c
253 (50)
22 (9)
86 (34)
41 (35, 46)
16 (9, 23)
Week 24
251 (49)
-
75 (30)
-
20 (13, 27)
sPGA response of 0
Week 16
80 (16)
3 (1)
16 (6)
14 (11, 18)
9 (5, 14)
PASI 75 response
Week 16c
271 (53)
24 (9)
101 (40)
44 (38, 49)
13 (6, 21)
Week 24
296 (58)
-
96 (38)
-
20 (13, 27)
PASI 90 response
Week 16
138 (27)
7 (3)
46 (18)
24 (20, 29)
9 (3, 15)
Week 24
164 (32)
-
50 (20)
-
13 (6, 19)
PASI 100 response
Week 16
52 (10%)
3 (1)
-
9 (6, 12)
-
ss-PGA response of 0/1 (scalp)d
(N=305)
(N=173)
(N=166)
Week 16
182 (60)
30 (17)
61 (37)
42 (34, 50)
23 (14, 33)
Examination of age, gender, race, body weight, baseline disease severity, and prior systemic therapy did not identify differences in response to SOTYKTU at Week 16 among these subgroups.
Maintenance and Durability of Response
In PSO-1, among subjects who received SOTYKTU and had sPGA 0/1 response at Week 24, the sPGA 0/1 response at Week 52 was 78% (151/194). Among subjects who received SOTYKTU and had PASI 75 response at Week 24, the PASI 75 response at Week 52 was 82% (187/228). Among subjects who received SOTYKTU and had PASI 90 response at Week 24, the PASI 90 response at Week 52 was 74% (103/140).
In PSO-2, to evaluate maintenance and durability of response, subjects who were originally randomized to SOTYKTU and were PASI 75 responders at Week 24, were re-randomized to either continue treatment on SOTYKTU or be withdrawn from therapy (i.e., receive placebo).
For subjects who were re-randomized and also had a sPGA score of 0 or 1 at Week 24, 70% (83/118) of subjects who continued on SOTYKTU maintained this response (sPGA 0 or 1) at Week 52 compared to 24% (28/119) of subjects who were re-randomized to placebo. In addition, at Week 52, 80% (119/148) of subjects who continued on SOTYKTU maintained PASI 75 compared to 31% (47/150) of subjects who were withdrawn from SOTYKTU.
For sPGA 0 or 1 responders at Week 24 who were re-randomized to treatment withdrawal (i.e., placebo), the median time to loss of sPGA score of 0 or 1 was approximately 8 weeks. For PASI 75 responders at Week 24 who were re-randomized to treatment withdrawal (i.e., placebo), the median time to loss of PASI 75 was approximately 12 weeks.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
SOTYKTU™ (deucravacitinib) tablets are available as listed in the table below:
Tablet Strength
Tablet Color/Shape
Tablet Markings
Package Size
NDC Code
6 mg
Pink, round, biconvex, film-coated tablet
Laser printed with “BMS 895” and “6 mg” on one side
Bottles of 30 with child-resistant closure
0003-0895-11
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide) before starting SOTYKTU therapy and each time the prescription is renewed, as there may be new information they need to know. Advise patients of the potential benefits and risks of SOTYKTU.
Hypersensitivity Reactions
Advise patients to discontinue SOTYKTU and seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions [see Warnings and Precautions (5.1)].
Infections
Inform patients that SOTYKTU may lower the ability of their immune system to fight infections and to contact their healthcare provider immediately if they develop any signs or symptoms of infection [see Warnings and Precautions (5.2)].
Inform patients that herpes infections, including serious, may occur with use of SOTYKTU [see Warnings and Precautions (5.2)].
Malignancies including Lymphomas
Inform patients that SOTYKTU may increase their risk of developing malignancies including lymphomas. Instruct patients to inform their healthcare provider if they have ever had any type of cancer [see Warnings and Precautions (5.4)].
Rhabdomyolysis
Inform patients that SOTYKTU may increase their risk of developing rhabdomyolysis. Instruct patients to immediately inform their healthcare provider if they develop unexplained muscle pain, tenderness or weakness, particularly if accompanied by malaise or fever [see Warnings and Precautions (5.5)].
Laboratory Abnormalities
Inform patients that SOTYKTU may affect certain lab tests, and that blood tests may be required before and during SOTYKTU treatment [see Warnings and Precautions (5.6)].
Immunizations
Advise patients that vaccination with live vaccines is not recommended during SOTYKTU treatment. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Instruct patients to inform the healthcare practitioner that they are taking SOTYKTU prior to a potential vaccination [see Warnings and Precautions (5.7)].
Pregnancy
Advise patients to report their pregnancy to Bristol-Myers Squibb Company at 1-800-721-5072 [see Use in Specific Populations (8.1)].
-
MEDICATION GUIDE
SOTYKTU™ (soh-tik-too)
(deucravacitinib)
tablets
What is the most important information I should know about SOTYKTU?
SOTYKTU may cause serious side effects, including:
- •
- Serious allergic reactions. Stop taking SOTYKTU and get emergency medical help right away if you develop any of the following symptoms of a serious allergic reaction:
- o
- feel faint
- o
- swelling of your face, eyelids, lips, mouth, tongue, or throat
- o
- trouble breathing or throat tightness
- o
- chest tightness
- o
- skin rash, hives
- •
-
Infections. SOTYKTU is a medicine that affects your immune system. SOTYKTU can lower the ability of your immune system to fight infections and can increase your risk of infections. Some people have had serious infections while taking SOTYKTU, such as infections of the lungs, including pneumonia and tuberculosis (TB), and COVID-19.
- o
- Your healthcare provider should check you for infections and TB before starting treatment with SOTYKTU.
- o
- Your healthcare provider may treat you for TB before you begin treatment with SOTYKTU if you have a history of TB or have active TB.
- o
- Your healthcare provider should watch you closely for signs and symptoms of TB during treatment with SOTYKTU.
- o
- If you get a serious infection, your healthcare provider may tell you to stop taking SOTYKTU until your infection is controlled.
- SOTYKTU should not be used in people with an active, serious infection, including localized infections. You should not start taking SOTYKTU if you have any kind of infection unless your healthcare provider tells you it is okay.
- You may be at a higher risk of developing shingles (herpes zoster).
- Before starting SOTYKTU, tell your healthcare provider if you:
-
- o
- are being treated for an infection
- o
- have had an infection that does not go away or keeps coming back
- o
- have TB or have been in close contact with someone with TB
- o
- have or have had hepatitis B or C
- o
- think you have an infection or have symptoms of an infection such as:
- ▪
- fever, sweats, or chills
- ▪
- muscle aches
- ▪
- weight loss
- ▪
- cough
- ▪
- shortness of breath
- ▪
- blood in your phlegm (mucus)
- ▪
- warm, red, or painful skin or sores on your body different from your psoriasis
- ▪
- diarrhea or stomach pain
- ▪
- burning when you urinate or urinating more often than normal
- ▪
- feeling very tired
- After you start taking SOTYKTU, call your healthcare provider right away if you have an infection or have symptoms of an infection.
- SOTYKTU can make you more likely to get infections or make any infections you have worse.
- •
-
Cancer. Certain kinds of cancer including lymphoma have been reported in people taking SOTYKTU.
- o
- Tell your healthcare provider if you have ever had any type of cancer.
- •
- Muscle problems (rhabdomyolysis). SOTYKTU can cause muscle problems that can be severe. Treatment with SOTYKTU may increase the level of an enzyme in your blood called creatine phosphokinase (CPK) and can be a sign of muscle damage. Increased CPK is common in people taking SOTYKTU. Your healthcare provider may tell you to stop taking SOTYKTU if the amount of CPK in your blood gets too high or if you have signs and symptoms of severe muscle problems. Tell your healthcare provider right away if you have any of these signs or symptoms of severe muscle problems:
- o
- unexplained muscle pain, tenderness, or weakness
- o
- feeling very tired
- o
- fever
- o
- dark-colored urine
See “What are the possible side effects of SOTYKTU?” for more information about side effects.
What is SOTYKTU?
SOTYKTU is a prescription medicine used to treat adults with moderate to severe plaque psoriasis who may benefit from taking injections or pills (systemic therapy) or treatment using ultraviolet or UV light (phototherapy).
It is not known if SOTYKTU is safe and effective in children under 18 years of age.
Do not take SOTYKTU if you are allergic to deucravacitinib or any of the ingredients in SOTYKTU. See the end of this Medication Guide for a complete list of ingredients in SOTYKTU.
Before taking SOTYKTU, tell your healthcare provider about all of your medical conditions, including if you:
- •
- See “What is the most important information I should know about SOTYKTU?”
- •
- have liver problems or kidney problems
- •
- have high levels of fat in your blood (triglycerides)
- •
- have recently received or are scheduled to receive an immunization (vaccine). You should avoid receiving live vaccines during treatment with SOTYKTU.
- •
- are pregnant or plan to become pregnant. It is not known if SOTYKTU can harm your unborn baby.
- o
- Report pregnancies to the Bristol-Myers Squibb Company’s Adverse Event reporting line at 1-800-721-5072.
- •
- are breastfeeding or plan to breastfeed. It is not known if SOTYKTU passes into your breast milk.
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take SOTYKTU?
- •
- Take SOTYKTU exactly as your healthcare provider tells you to take it.
- •
- Take SOTYKTU 1 time every day.
- •
- Take SOTYKTU with or without food.
- •
- Do not crush, cut, or chew the SOTYKTU tablets.
What are the possible side effects of SOTYKTU?
SOTYKTU may cause serious side effects, including:
- •
- See “What is the most important information I should know about SOTYKTU?”
- •
-
Changes in certain laboratory test results. Changes in laboratory tests have happened in some people taking SOTYKTU. Your healthcare provider may do blood tests before you start taking SOTYKTU and during treatment with SOTYKTU to check for the following:
- o
- Increased triglycerides. Triglycerides are a type of fat found in your blood. Too much fat in your blood can cause problems with your heart.
- o
- Increased liver enzymes. Liver enzymes are found in your blood and help to tell if your liver is functioning normally. If your liver enzymes increase too much, your healthcare provider may need to do additional tests on your liver and may tell you to stop taking SOTYKTU if they think that SOTYKTU is harming your liver.
- •
- Potential risks from Janus kinase (JAK) inhibition. SOTYKTU is a tyrosine kinase 2 (TYK2) inhibitor. TYK2 is in the JAK family. It is not known whether taking SOTYKTU has the same risks as taking JAK inhibitors. Increased risk of death (all causes) has happened in people who were 50 years of age and older with at least 1 heart disease (cardiovascular) risk factor who were taking a JAK inhibitor used to treat rheumatoid arthritis (RA) compared to people taking another medicine in a class of medicines called TNF blockers. SOTYKTU is not for use in people with RA.
The most common side effects of SOTYKTU include:
- •
- common cold, sore throat, and sinus infection (upper respiratory infections)
- •
- cold sores (herpes simplex)
- •
- sores on inner lips, gums, tongue, or roof of the mouth (canker sores)
- •
- inflamed hair pores (folliculitis)
- •
- acne
These are not all of the possible side effects of SOTYKTU.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store SOTYKTU?
Store SOTYKTU at room temperature between 68°F to 77°F (20°C to 25°C).
Keep SOTYKTU and all medicines out of the reach of children.
General information about the safe and effective use of SOTYKTU.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use SOTYKTU for a condition for which it was not prescribed. Do not give SOTYKTU to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about SOTYKTU that is written for health professionals.
What are the ingredients in SOTYKTU?
Active ingredient: deucravacitinib.
Inactive ingredients: anhydrous lactose, croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, microcrystalline cellulose and silicon dioxide. In addition, the film coating Opadry® II Pink contains the following inactive ingredients: polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, iron oxide red and yellow.
Distributed by:
Bristol-Myers Squibb Company
Princeton, New Jersey 08543 USA
For more product information about SOTYKTU, go to website (www.sotyktu.com) or call SOTYKTU 360 SUPPORT at 1.888.SOTYKTU (768.9588).
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Issued: 9/2022
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SOTYKTU
deucravacitinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0003-0895 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEUCRAVACITINIB (UNII: N0A21N6RAU) (DEUCRAVACITINIB - UNII:N0A21N6RAU) DEUCRAVACITINIB 6 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MM2/S) (UNII: 6N003M473W) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color PINK Score no score Shape ROUND (biconvex) Size 8mm Flavor Imprint Code BMS895;6mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0003-0895-11 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 09/09/2022 2 NDC:0003-0895-91 30 in 1 BLISTER PACK; Type 0: Not a Combination Product 09/09/2022 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214958 09/09/2022 Labeler - E.R. Squibb & Sons, L.L.C. (011550092)