Label: BRUKINSA- zanubrutinib capsule
BRUKINSA- zanubrutinib tablet, film coated


- NDC Code(s): 72579-011-01, 72579-011-02, 72579-122-01
- Packager: BeOne Medicines USA, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated September 24, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BRUKINSA safely and effectively. See full prescribing information for BRUKINSA.
BRUKINSA® (zanubrutinib) capsules, for oral use
BRUKINSA® (zanubrutinib) tablets, for oral use
Initial U.S. Approval: 2019RECENT MAJOR CHANGES
INDICATIONS AND USAGE
BRUKINSA is a kinase inhibitor indicated for the treatment of adult patients with:
- Mantle cell lymphoma (MCL) who have received at least one prior therapy. (1.1)
This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. - Waldenström's macroglobulinemia (WM). (1.2)
- Relapsed or refractory marginal zone lymphoma (MZL) who have received at least one anti–CD20-based regimen. (1.3)
This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. - Chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). (1.4)
- Relapsed or refractory follicular lymphoma (FL), in combination with obinutuzumab, after two or more lines of systemic therapy. (1.5)
This indication is approved under accelerated approval based on response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
DOSAGE AND ADMINISTRATION
- Recommended dosage: 160 mg orally twice daily or 320 mg orally once daily with or without food; swallow whole with water.
- Tablets can be split in half as prescribed by the healthcare provider. (2.1)
- Reduce BRUKINSA dose in patients with severe hepatic impairment. (2.2, 8.7)
- Advise patients not to open, break, or chew capsules. (2.1)
- Advise patients not to chew or crush tablets. (2.1)
- Manage toxicity using treatment interruption, dose reduction, or discontinuation. (2.4)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Hemorrhage: Monitor for bleeding and manage appropriately. (5.1)
- Infections: Monitor patients for signs and symptoms of infection, including opportunistic infections, and treat as needed. (5.2)
- Cytopenias: Monitor complete blood counts during treatment. (5.3)
- Second Primary Malignancies: Other malignancies have developed including skin cancers and non-skin carcinomas. Monitor and advise patients to use sun protection. (5.4)
- Cardiac Arrhythmias: Monitor for signs and symptoms of arrhythmias and manage appropriately. (5.5)
- Hepatotoxicity, Including Drug-Induced Liver Injury: Monitor hepatic function throughout treatment. (5.6)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise women of the potential risk to a fetus and to use effective contraception. (5.7)
ADVERSE REACTIONS
The most common adverse reactions (≥30%), including laboratory abnormalities, are neutrophil count decreased, platelet count decreased, upper respiratory tract infection, hemorrhage, and musculoskeletal pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact BeOne Medicines at 1-877-828-5596 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2025
- Mantle cell lymphoma (MCL) who have received at least one prior therapy. (1.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Mantle Cell Lymphoma
1.2 Waldenström's Macroglobulinemia
1.3 Marginal Zone Lymphoma
1.4 Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma
1.5 Follicular Lymphoma
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modification for Use in Hepatic Impairment
2.3 Dosage Modifications for Drug Interactions
2.4 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
5.2 Infections
5.3 Cytopenias
5.4 Second Primary Malignancies
5.5 Cardiac Arrhythmias
5.6 Hepatotoxicity, Including Drug-Induced Liver Injury
5.7 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on BRUKINSA
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Mantle Cell Lymphoma
14.2 Waldenström's Macroglobulinemia
14.3 Marginal Zone Lymphoma
14.4 Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma
14.5 Follicular Lymphoma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Mantle Cell Lymphoma
BRUKINSA is indicated for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
1.2 Waldenström's Macroglobulinemia
BRUKINSA is indicated for the treatment of adult patients with Waldenström's macroglobulinemia (WM) [see Clinical Studies (14.2)].
1.3 Marginal Zone Lymphoma
BRUKINSA is indicated for the treatment of adult patients with relapsed or refractory marginal zone lymphoma (MZL) who have received at least one anti–CD20-based regimen.
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies (14.3)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
1.4 Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma
BRUKINSA is indicated for the treatment of adult patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) [see Clinical Studies (14.4)].
1.5 Follicular Lymphoma
BRUKINSA is indicated for the treatment of adult patients with relapsed or refractory follicular lymphoma (FL), in combination with obinutuzumab, after two or more lines of systemic therapy.
This indication is approved under accelerated approval based on response rate and durability of response [see Clinical Studies (14.5)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of BRUKINSA for monotherapy or in combination with obinutuzumab is 160 mg taken orally twice daily or 320 mg taken orally once daily until disease progression or unacceptable toxicity.
Capsule Administration Instructions
- Administer BRUKINSA capsules with or without food [see Clinical Pharmacology (12.3)]. Advise patients to swallow capsules whole with water and not to open, break, or chew capsules.
Tablet Administration Instructions
- Administer BRUKINSA tablets with or without food [see Clinical Pharmacology (12.3)]. Advise patients to swallow tablets whole with water and not to chew or crush the tablets. The tablets can be split in half as prescribed by the healthcare provider.
2.2 Dosage Modification for Use in Hepatic Impairment
The recommended dosage of BRUKINSA for patients with severe hepatic impairment (Child-Pugh class C) is 80 mg orally twice daily; no dosage modification is recommended for patients with mild or moderate hepatic impairment (Child-Pugh class A or B) [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
2.3 Dosage Modifications for Drug Interactions
Recommended dosage modifications of BRUKINSA for drug interactions are provided in Table 1 [see Drug Interactions (7.1)].
Table 1: Dosage Modifications for Use with CYP3A Inhibitors or Inducers Coadministered Drug Recommended BRUKINSA Dosage
(Starting Dose: 160 mg twice daily or 320 mg once daily)- *
- Since clarithromycin 250 mg twice daily acts as a moderate CYP3A inhibitor, it is recommended that patients be administered clarithromycin 250 mg twice daily with 80 mg BRUKINSA twice daily [see Clinical Pharmacology (12.3)].
- †
- Modify or interrupt zanubrutinib dose as recommended for adverse reactions [see Dosage and Administration (2.4)].
Clarithromycin 250 mg twice daily* 80 mg twice daily† Clarithromycin 500 mg twice daily 80 mg once daily† Posaconazole suspension 100 mg once daily 80 mg twice daily† Posaconazole suspension dosage higher than 100 mg once daily Posaconazole delayed-release tablets 300 mg once daily
Posaconazole intravenous 300 mg once daily80 mg once daily† Other strong CYP3A inhibitor 80 mg once daily† Moderate CYP3A inhibitor 80 mg twice daily† Strong CYP3A inducer Avoid concomitant use. Moderate CYP3A inducer Avoid concomitant use.
If these inducers cannot be avoided, increase BRUKINSA dose to 320 mg twice daily.After discontinuation of a CYP3A inhibitor or moderate CYP3A inducer, resume previous dose of BRUKINSA [see Dosage and Administration (2.1, 2.2) and Drug Interactions (7.1)].
2.4 Dosage Modifications for Adverse Reactions
Recommended dosage modifications of BRUKINSA for Grade 3 or higher adverse reactions are provided in Table 2.
Table 2: Recommended Dosage Modifications for Adverse Reaction Adverse Reaction Adverse Reaction Occurrence Dosage Modification
(Starting Dose: 160 mg twice daily or 320 mg once daily)- *
- Evaluate the benefit-risk before resuming treatment at the same dosage for Grade 4 nonhematological toxicity.
Hematological toxicities [see Warnings and Precautions (5.3)] Grade 3 or Grade 4 febrile neutropenia
Platelet count decreased to 25,000-50,000/mm3 with significant bleeding
Neutrophil count decreased to <500/mm3 (lasting more than 10 consecutive days)
Platelet count decreased to <25,000/mm3 (lasting more than 10 consecutive days)First Interrupt BRUKINSA
Once toxicity has resolved to Grade 1 or lower or baseline: Resume at 160 mg twice daily or 320 mg once daily.Second Interrupt BRUKINSA
Once toxicity has resolved to Grade 1 or lower or baseline: Resume at 80 mg twice daily or 160 mg once daily.Third Interrupt BRUKINSA
Once toxicity has resolved to Grade 1 or lower or baseline: Resume at 80 mg once daily.Fourth Discontinue BRUKINSA Nonhematological toxicities [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)] Severe or life-threatening nonhematological toxicities* First Interrupt BRUKINSA
Once toxicity has resolved to Grade 1 or lower or baseline: Resume at 160 mg twice daily or 320 mg once daily.*Second Interrupt BRUKINSA
Once toxicity has resolved to Grade 1 or lower or baseline: Resume at 80 mg twice daily or 160 mg once daily.Third Interrupt BRUKINSA
Once toxicity has resolved to Grade 1 or lower or baseline: Resume at 80 mg once daily.Fourth Discontinue BRUKINSA Asymptomatic lymphocytosis in CLL and MCL should not be regarded as an adverse reaction, and these patients should continue taking BRUKINSA.
Refer to the obinutuzumab prescribing information for management of obinutuzumab toxicities.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
Fatal and serious hemorrhage has occurred in patients with hematological malignancies treated with BRUKINSA. Grade 3 or higher hemorrhage including intracranial and gastrointestinal hemorrhage, hematuria, and hemothorax was reported in 3.8% of patients treated with BRUKINSA in clinical trials, with fatalities occurring in 0.2% of patients. Bleeding of any grade, excluding purpura and petechiae, occurred in 32% of patients.
Bleeding has occurred in patients with and without concomitant antiplatelet or anticoagulation therapy. Coadministration of BRUKINSA with antiplatelet or anticoagulant medications may further increase the risk of hemorrhage.
Monitor for signs and symptoms of bleeding. Discontinue BRUKINSA if intracranial hemorrhage of any grade occurs. Consider the benefit-risk of withholding BRUKINSA for 3-7 days before and after surgery depending upon the type of surgery and the risk of bleeding.
5.2 Infections
Fatal and serious infections (including bacterial, viral, or fungal infections) and opportunistic infections have occurred in patients with hematological malignancies treated with BRUKINSA. Grade 3 or higher infections occurred in 26% of patients, most commonly pneumonia (7.9%), with fatal infections occurring in 3.2% of patients. Infections due to hepatitis B virus (HBV) reactivation have occurred.
Consider prophylaxis for herpes simplex virus, pneumocystis jirovecii pneumonia, and other infections according to standard of care in patients who are at increased risk for infections. Monitor and evaluate patients for fever or other signs and symptoms of infection and treat appropriately.
5.3 Cytopenias
Grade 3 or 4 cytopenias, including neutropenia (21%), thrombocytopenia (8%), and anemia (8%) based on laboratory measurements, developed in patients treated with BRUKINSA [see Adverse Reactions (6.1)]. Grade 4 neutropenia occurred in 10% of patients, and Grade 4 thrombocytopenia occurred in 2.5% of patients.
Monitor complete blood counts regularly during treatment and interrupt treatment, reduce the dose, or discontinue treatment as warranted [see Dosage and Administration (2.4)]. Treat using growth factor or transfusions, as needed.
5.4 Second Primary Malignancies
Second primary malignancies, including non-skin carcinoma, have occurred in 14% of patients treated with BRUKINSA. The most frequent second primary malignancy was non-melanoma skin cancers (8%), followed by other solid tumors in 7% of the patients (including melanoma in 1% of patients) and hematologic malignancies (0.7%). Advise patients to use sun protection and monitor patients for the development of second primary malignancies.
5.5 Cardiac Arrhythmias
Serious cardiac arrhythmias have occurred in patients treated with BRUKINSA. Atrial fibrillation and atrial flutter were reported in 4.4% of patients treated with BRUKINSA, including Grade 3 or higher cases in 1.9% of patients. Patients with cardiac risk factors, hypertension, and acute infections may be at increased risk. Grade 3 or higher ventricular arrhythmias were reported in 0.3% of patients.
Monitor for signs and symptoms of cardiac arrhythmias (e.g., palpitations, dizziness, syncope, dyspnea, chest discomfort), manage appropriately [see Dosage and Administration (2.4)], and consider the risks and benefits of continued BRUKINSA treatment.
5.6 Hepatotoxicity, Including Drug-Induced Liver Injury
Hepatotoxicity, including severe, life-threatening, and potentially fatal cases of drug-induced liver injury (DILI), has occurred in patients treated with Bruton tyrosine kinase inhibitors, including BRUKINSA.
Evaluate bilirubin and transaminases at baseline and throughout treatment with BRUKINSA. For patients who develop abnormal liver tests after BRUKINSA, monitor more frequently for liver test abnormalities and clinical signs and symptoms of hepatic toxicity. If DILI is suspected, withhold BRUKINSA. Upon confirmation of DILI, discontinue BRUKINSA.
5.7 Embryo-Fetal Toxicity
Based on findings in animals, BRUKINSA can cause fetal harm when administered to a pregnant woman. Administration of zanubrutinib to pregnant rats during the period of organogenesis caused embryo-fetal toxicity, including malformations at exposures that were 5 times higher than those reported in patients at the recommended dose of 160 mg twice daily. Advise women to avoid becoming pregnant while taking BRUKINSA and for 1 week after the last dose. Advise men to avoid fathering a child during treatment and for 1 week after the last dose. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in more detail in other sections of the labeling:
- Hemorrhage [see Warnings and Precautions (5.1)]
- Infections [see Warnings and Precautions (5.2)]
- Cytopenias [see Warnings and Precautions (5.3)]
- Second Primary Malignancies [see Warnings and Precautions (5.4)]
- Cardiac Arrhythmias [see Warnings and Precautions (5.5)]
- Hepatotoxicity, including DILI [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the WARNINGS AND PRECAUTIONS reflect exposure to BRUKINSA in nine monotherapy and 2 combination clinical trials, administered at 160 mg twice daily in 1608 patients and at 320 mg once daily in 121 patients. Among these 1729 patients, the median duration of exposure was 27.6 months, 78% of patients were exposed for at least 12 months, and 60% of patients were exposed for at least 24 months.
In this pooled safety population, the most common adverse reactions (≥30%), including laboratory abnormalities, were neutrophil count decreased (51%), platelet count decreased (41%), upper respiratory tract infection (38%), hemorrhage (32%), and musculoskeletal pain (31%).
Mantle Cell Lymphoma (MCL)
The safety of BRUKINSA was evaluated in 118 patients with MCL who received at least one prior therapy in two single-arm clinical trials, BGB-3111-206 [NCT03206970] and BGB-3111-AU-003 [NCT02343120] [see Clinical Studies (14.1)]. The median age of patients who received BRUKINSA in studies BGB-3111-206 and BGB-3111-AU-003 was 62 years (range: 34 to 86), 75% were male, 75% were Asian, 21% were White, and 94% had an ECOG performance status of 0 to 1. Patients had a median of 2 prior lines of therapy (range: 1 to 4). The BGB-3111-206 trial required a platelet count ≥75 × 109/L and an absolute neutrophil count ≥1 × 109/L independent of growth factor support, hepatic enzymes ≤2.5 × upper limit of normal, total bilirubin ≤1.5 × ULN. The BGB-3111-AU-003 trial required a platelet count ≥50 × 109/L and an absolute neutrophil count ≥1 × 109/L independent of growth factor support, hepatic enzymes ≤3 × upper limit of normal, total bilirubin ≤1.5 × ULN. Both trials required a creatinine clearance (CLcr) ≥30 mL/min. Both trials excluded patients with prior allogeneic hematopoietic stem cell transplant, exposure to a BTK inhibitor, known infection with HIV, and serologic evidence of active hepatitis B or hepatitis C infection, and patients requiring strong CYP3A inhibitors or strong CYP3A inducers. Patients received BRUKINSA 160 mg twice daily or 320 mg once daily. Among patients receiving BRUKINSA, 79% were exposed for 6 months or longer, and 68% were exposed for greater than one year.
Fatal adverse reactions within 30 days of the last dose of BRUKINSA occurred in 8 (7%) of 118 patients with MCL. Fatal cases included pneumonia in 2 patients and cerebral hemorrhage in one patient.
Serious adverse reactions were reported in 36 patients (31%). The most frequent serious adverse reactions that occurred were pneumonia (11%) and hemorrhage (5%).
Of the 118 patients with MCL treated with BRUKINSA, 8 (7%) patients discontinued treatment due to adverse reactions in the trials. The most frequent adverse reaction leading to treatment discontinuation was pneumonia (3.4%). One (0.8%) patient experienced an adverse reaction leading to dose reduction (hepatitis B).
Table 3 summarizes the adverse reactions in BGB-3111-206 and BGB-3111-AU-003.
Table 3: Adverse Reactions (≥10%) in Patients Receiving BRUKINSA in BGB-3111-206 and BGB-3111-AU-003 Trials Body System Adverse Reaction Percent of Patients (N=118) All Grades % Grade 3 or Higher % - *
- Upper respiratory tract infection includes upper respiratory tract infection, upper respiratory tract infection viral.
- †
- Pneumonia includes pneumonia, pneumonia fungal, pneumonia cryptococcal, pneumonia streptococcal, atypical pneumonia, lung infection, lower respiratory tract infection, lower respiratory tract infection bacterial, lower respiratory tract infection viral.
- ‡
- Includes fatal adverse reaction.
- §
- Rash includes all related terms containing rash.
- ¶
- Bruising includes all related terms containing bruise, bruising, contusion, ecchymosis.
- #
- Hemorrhage includes all related terms containing hemorrhage, hematoma.
- Þ
- Musculoskeletal pain includes musculoskeletal pain, musculoskeletal discomfort, myalgia, back pain, arthralgia, arthritis.
Infections and infestations Upper respiratory tract infection* 39 0 Pneumonia† 15 10‡ Urinary tract infection 11 0.8 Skin and subcutaneous tissue disorders Rash§ 36 0 Bruising¶ 14 0 Gastrointestinal disorders Diarrhea 23 0.8 Constipation 13 0 Vascular disorders Hypertension 12 3.4 Hemorrhage# 11 3.4‡ Musculoskeletal and connective tissue disorders Musculoskeletal painÞ 14 3.4 Respiratory, thoracic, and mediastinal disorders Cough 12 0 Other clinically significant adverse reactions that occurred in <10% of patients with mantle cell lymphoma include major hemorrhage (defined as ≥ Grade 3 hemorrhage or CNS hemorrhage of any grade) (5%) and headache (4.2%).
Table 4: Selected Laboratory Abnormalities* (>20%) in Patients with MCL in Studies BGB-3111-206 and BGB-3111-AU-003 Laboratory Parameter Percent of Patients (N=118) All Grades (%) Grade 3 or 4 (%) Hematologic abnormalities Neutrophils decreased 45 20 Lymphocytosis† 41 16 Platelets decreased 40 7 Hemoglobin decreased 27 6 Chemistry abnormalities Blood uric acid increased 29 2.6 ALT increased 28 0.9 Bilirubin increased 24 0.9 Waldenström's Macroglobulinemia (WM)
The safety of BRUKINSA was investigated in two cohorts of Study BGB-3111-302 (ASPEN). Cohort 1 included 199 patients with MYD88 mutation (MYD88MUT) WM, randomized to and treated with either BRUKINSA (101 patients) or ibrutinib (98 patients). The trial also included a non-randomized arm, Cohort 2, with 26 wild type MYD88 (MYD88WT) WM patients and 2 patients with unknown MYD88 status [see Clinical Studies (14.2)].
Among patients who received BRUKINSA, 93% were exposed for 6 months or longer, and 89% were exposed for greater than 1 year.
In Cohort 1 of the ASPEN study safety population (N=101), the median age of patients who received BRUKINSA was 70 years (45-87 years old); 67% were male, 86% were White, 4% were Asian, and 10% were not reported (unknown race). In Cohort 2 of the ASPEN study safety population (N=28), the median age of patients who received BRUKINSA was 72 (39-87 years old); 50% were male, 96% were White, and 4% were not reported (unknown race).
In Cohort 1, serious adverse reactions occurred in 44% of patients who received BRUKINSA. Serious adverse reactions in >2% of patients included influenza (3%), pneumonia (4%), neutropenia and neutrophil count decreased (3%), hemorrhage (4%), pyrexia (3%), and febrile neutropenia (3%). In Cohort 2, serious adverse reactions occurred in 39% of patients. Serious adverse reactions in >2 patients included pneumonia (14%).
Permanent discontinuation of BRUKINSA due to an adverse reaction occurred in 2% of patients in Cohort 1 and included hemorrhage (1 patient), neutropenia and neutrophil count decreased (1 patient); in Cohort 2, permanent discontinuation of BRUKINSA due to an adverse reaction occurred in 7% of patients and included subdural hemorrhage (1 patient) and diarrhea (1 patient).
Dosage interruptions of BRUKINSA due to an adverse reaction occurred in 32% of patients in Cohort 1 and in 29% in Cohort 2. Adverse reactions which required dosage interruption in >2% of patients included neutropenia, vomiting, hemorrhage, thrombocytopenia, and pneumonia in Cohort 1. Adverse reactions leading to dosage interruption in >2 patients in Cohort 2 included pneumonia and pyrexia.
Dose reductions of BRUKINSA due to an adverse reaction occurred in 11% of patients in Cohort 1 and in 7% in Cohort 2. Adverse reactions which required dose reductions in >2% of patients included neutropenia in Cohort 1. Adverse reaction leading to dose reduction occurred in 2 patients in Cohort 2 (each with one event: diarrhea and pneumonia).
Table 5 summarizes the adverse reactions in Cohort 1 in ASPEN.
Table 5: Adverse Reactions (≥10%) Occurring in Patients with WM Who Received BRUKINSA in Cohort 1 Body System Adverse Reaction BRUKINSA (N=101) Ibrutinib (N=98) All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- *
- Upper respiratory tract infection includes upper respiratory tract infection, laryngitis, nasopharyngitis, sinusitis, rhinitis, viral upper respiratory tract infection, pharyngitis, rhinovirus infection, upper respiratory tract congestion.
- †
- Pneumonia includes lower respiratory tract infection, lung infiltration, pneumonia, pneumonia aspiration, pneumonia viral.
- ‡
- Fatigue includes asthenia, fatigue, lethargy.
- §
- Bruising includes all related terms containing bruise, contusion, or ecchymosis.
- ¶
- Rash includes all related terms rash, maculo-papular rash, erythema, rash erythematous, drug eruption, dermatitis allergic, dermatitis atopic, rash pruritic, dermatitis, photodermatoses, dermatitis acneiform, stasis dermatitis, vasculitic rash, eyelid rash, urticaria, skin toxicity.
- #
- Musculoskeletal pain includes back pain, arthralgia, pain in extremity, musculoskeletal pain, myalgia, bone pain, spinal pain, musculoskeletal chest pain, neck pain, arthritis, musculoskeletal discomfort.
- Þ
- Hemorrhage includes epistaxis, hematuria, conjunctival hemorrhage, hematoma, rectal hemorrhage, periorbital hemorrhage, mouth hemorrhage, post procedural hemorrhage, hemoptysis, skin hemorrhage, hemorrhoidal hemorrhage, ear hemorrhage, eye hemorrhage, hemorrhagic diathesis, periorbital hematoma, subdural hemorrhage, wound hemorrhage, gastric hemorrhage, lower gastrointestinal hemorrhage, spontaneous hematoma, traumatic hematoma, traumatic intracranial hemorrhage, tumor hemorrhage, retinal hemorrhage, hematochezia, diarrhea hemorrhagic, hemorrhage, melena, post-procedural hematoma, subdural hematoma, anal hemorrhage, hemorrhagic disorder, pericardial hemorrhage, postmenopausal hemorrhage, stoma site hemorrhage, subarachnoid hemorrhage.
Infections and infestations Upper respiratory tract infection* 44 0 40 2 Pneumonia† 12 4 26 10 Urinary tract infection 11 0 13 2 Gastrointestinal disorders Diarrhea 22 3 34 2 Nausea 18 0 13 1 Constipation 16 0 7 0 Vomiting 12 0 14 1 General disorders Fatigue‡ 31 1 25 1 Pyrexia 16 4 13 2 Edema peripheral 12 0 20 0 Skin and subcutaneous tissue disorders Bruising§ 20 0 34 0 Rash¶ 29 0 32 0 Pruritus 11 1 6 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain# 45 9 39 1 Muscle spasms 10 0 28 1 Nervous system disorders Headache 18 1 14 1 Dizziness 13 1 12 0 Respiratory, thoracic, and mediastinal disorders Cough 16 0 18 0 Dyspnea 14 0 7 0 Vascular disorders HemorrhageÞ 42 4 43 9 Hypertension 14 9 19 14 Clinically relevant adverse reactions in <10% of patients who received BRUKINSA included localized infection, atrial fibrillation or atrial flutter, and hematuria.
Table 6 summarizes the laboratory abnormalities in ASPEN.
Table 6: Select Laboratory Abnormalities* (≥20%) that Worsened from Baseline in Patients with WM Who Received BRUKINSA in Cohort 1 Laboratory Abnormality BRUKINSA† Ibrutinib† All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Hematologic abnormalities Neutrophils decreased 50 24 34 9 Platelets decreased 35 8 39 5 Hemoglobin decreased 20 7 20 7 Chemistry abnormalities Glucose increased 45 2.3 33 2.3 Creatinine increased 31 1 21 1 Calcium decreased 27 2 26 0 Potassium increased 24 2 12 0 Phosphate decreased 20 3.1 18 0 Urate increased 16 3.2 34 6 Bilirubin increased 12 1 33 1 Marginal Zone Lymphoma
The safety of BRUKINSA was evaluated in 88 patients with previously treated MZL in two single-arm clinical studies, BGB-3111-214 and BGB-3111-AU-003 [see Clinical Studies (14.3)]. The trials required an absolute neutrophil count ≥1 × 109/L, platelet count ≥50 or ≥75 × 109/L and adequate hepatic function and excluded patients requiring a strong CYP3A inhibitor or inducer. Patients received BRUKINSA 160 mg twice daily (97%) or 320 mg once daily (3%). The median age in both studies combined was 70 years (range: 37 to 95), 52% were male, 64% were White, and 19% were Asian. Most patients (92%) had an ECOG performance status of 0 to 1. Eighty percent received BRUKINSA for 6 months or longer, and 67% received treatment for more than one year.
Two fatal adverse reactions (2.3%) occurred within 30 days of the last dose of BRUKINSA, including myocardial infarction and a Covid-19–related death.
Serious adverse reactions occurred in 40% of patients. The most frequent serious adverse reactions were pyrexia (8%) and pneumonia (7%).
Adverse reactions lead to treatment discontinuation in 6% of patients, dose reduction in 2.3%, and dose interruption in 34%. The leading cause of dose modification was respiratory tract infections (13%).
Table 7 summarizes selected adverse reactions in BGB-3111-214 and BGB-3111-AU-003.
Table 7: Adverse Reactions Occurring in ≥10% Patients with MZL Who Received BRUKINSA Body System Adverse Reaction BRUKINSA (N=88) All Grades
(%)Grade 3 or 4
(%)- *
- Upper respiratory tract infection includes upper respiratory tract infection, nasopharyngitis, sinusitis, tonsillitis, rhinitis, viral upper respiratory tract infection.
- †
- Urinary tract infection includes urinary tract infection, cystitis, Escherichia urinary tract infection, pyelonephritis, cystitis.
- ‡
- Pneumonia includes COVID-19 pneumonia, pneumonia, bronchopulmonary aspergillosis, lower respiratory tract infection, organizing pneumonia.
- §
- Includes 2 fatalities from COVID-19 pneumonia.
- ¶
- Diarrhea includes diarrhea and diarrhea hemorrhagic.
- #
- Abdominal pain includes abdominal pain, abdominal pain upper, abdominal discomfort.
- Þ
- Bruising includes contusion, ecchymosis, increased tendency to bruise, post procedural contusion.
- ß
- Rash includes rash, rash maculo-papular, rash pruritic, dermatitis, dermatitis allergic, dermatitis atopic, dermatitis contact, drug reaction with eosinophilia and systemic symptoms, erythema, photosensitivity reaction, rash erythematous, rash papular, seborrheic dermatitis.
- à
- Musculoskeletal pain includes back pain, arthralgia, musculoskeletal pain, myalgia, pain in extremity, musculoskeletal chest pain, bone pain, musculoskeletal discomfort, neck pain.
- è
- Hemorrhage includes epistaxis, hematuria, hemorrhoidal hemorrhage, hematoma, hemoptysis, conjunctival hemorrhage, diarrhea hemorrhagic, hemorrhage urinary tract, mouth hemorrhage, pulmonary hematoma, subcutaneous hematoma, gingival bleeding, melena, upper gastrointestinal hemorrhage.
- ð
- Fatigue includes fatigue, lethargy, asthenia.
- ø
- Cough includes cough and productive cough.
Infections and infestations Upper respiratory tract infection* 26 3.4 Urinary tract infection† 11 2.3 Pneumonia‡,§ 10 6 Gastrointestinal disorders Diarrhea¶ 25 3.4 Abdominal pain# 14 2.3 Nausea 13 0 Skin and subcutaneous tissue disorders BruisingÞ 24 0 Rashß 21 0 Musculoskeletal and connective tissue disorders Musculoskeletal painà 27 1.1 Vascular disorders Hemorrhageè 23 1.1 General disorders Fatigueð 21 2.3 Respiratory, thoracic, and mediastinal disorders Coughø 10 0 Clinically relevant adverse reactions in <10% of patients who received BRUKINSA included peripheral neuropathy, second primary malignancies, dizziness, edema, headache, petechiae, purpura, and atrial fibrillation or flutter.
Table 8 summarizes select laboratory abnormalities.
Table 8: Select Laboratory Abnormalities (≥20%) that Worsened from Baseline in Patients with MZL Laboratory Abnormality* BRUKINSA All Grades (%) Grade 3 or 4 (%) - *
- The denominator used to calculate the rate varied from 87 to 88 based on the number of patients with a baseline value and at least one post-treatment value.
Hematologic abnormalities Neutrophils decreased 43 15 Platelets decreased 33 10 Lymphocytes decreased 32 8 Hemoglobin decreased 26 6 Chemistry abnormalities Glucose increased 54 4.6 Creatinine increased 34 1.1 Phosphate decreased 27 2.3 Calcium decreased 23 0 ALT increased 22 1.1 Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma
The safety data described below reflect exposure to BRUKINSA (160 mg twice daily) in 675 patients with CLL from two randomized controlled clinical trials [see Clinical Studies (14.4)]. The trials required patients to be unsuitable for fludarabine, cyclophosphamide, and rituximab (FCR) therapy defined as age ≥65 years, or age 18 to <65 years with either a total Cumulative Illness Rating Scale (CIRS) >6, CLcr 30 to 69 mL/min, or history of serious or frequent infections. The trial excluded patients with AST or ALT ≥2 times the upper limit of normal (ULN) or bilirubin ≥3 times (ULN) and patients requiring a strong CYP3A inhibitor or inducer.
SEQUOIA
The safety of BRUKINSA monotherapy in patients with previously untreated CLL/SLL was evaluated in a randomized, multicenter, open-label, actively controlled trial [see Clinical Studies (14.4)]. Patients without deletion of chromosome 17p13.1 (17p deletion) (Cohort 1) received either BRUKINSA 160 mg twice daily until disease progression or unacceptable toxicity (n=240) or bendamustine plus rituximab (BR) for 6 cycles (n=227). Bendamustine was dosed at 90 mg/m2/day intravenously on the first 2 days of each cycle, and rituximab was dosed at 375 mg/m2 on day 1 of Cycle 1 and 500 mg/m2 on day 1 of Cycles 2 to 6.
Additionally, the same BRUKINSA regimen was evaluated in 111 patients with previously untreated CLL/SLL with 17p deletion in a non-randomized single arm (Cohort 2).
Randomized Cohort: Previously Untreated CLL/SLL without 17p Deletion
In patients with previously untreated CLL/SLL without 17p deletion, the median age was 70, 62% were male, 89% were White, 2% were Asian, and 2% were Black. Most patients (93%) had an ECOG performance status of 0 to 1.
The median duration of exposure to BRUKINSA was 26 months, with 71% exposed for more than 2 years.
Serious adverse reactions occurred in 36% of patients who received BRUKINSA. Serious adverse reactions that occurred in ≥5% of patients were COVID-19, pneumonia, and second primary malignancy (5% each). Fatal adverse reactions occurred in 11 (4.6%) patients with the leading cause of death being COVID-19 (2.1%).
Adverse reactions led to permanent discontinuation of BRUKINSA in 8% of patients, dose reduction in 8%, and dose interruption in 46%. The most common adverse reactions leading to permanent discontinuation were second primary malignancy and COVID-19. The leading causes of dose modification (≥5% of all patients) were respiratory infections (COVID-19, pneumonia) and hemorrhage.
Table 9 summarizes select adverse reactions in this randomized cohort.
Table 9: Adverse Reactions in ≥10% Patients with Previously Untreated CLL/SLL without 17p Deletion in SEQUOIA CLL/SLL without 17p deletion BRUKINSA
(N=240)BR
(N=227)System Organ Class
Preferred TermAll Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- *
- Musculoskeletal pain: musculoskeletal pain, arthralgia, back pain, pain in extremity, myalgia, neck pain, spinal pain, musculoskeletal discomfort, bone pain.
- †
- Upper respiratory tract infection: upper respiratory tract infection, nasopharyngitis, sinusitis, rhinitis, pharyngitis, upper respiratory tract congestion, laryngitis, tonsillitis and upper respiratory tract inflammation, and related terms.
- ‡
- Pneumonia: pneumonia, COVID-19 pneumonia, lower respiratory tract infection, lung infiltration, and related terms including specific types of infection.
- §
- Includes 3 fatal outcomes.
- ¶
- Includes 2 fatal outcomes.
- #
- Hemorrhage: all terms containing hematoma, hemorrhage, hemorrhagic, and related terms indicative of bleeding.
- Þ
- Includes multiple similar adverse reaction terms.
- ß
- Rash: Rash, dermatitis, drug eruption, and related terms.
- à
- Bruising: all terms containing bruise, bruising, contusion, or ecchymosis.
- è
- Fatigue: fatigue, asthenia, and lethargy.
- ð
- Second primary malignancy: includes non-melanoma skin cancer, malignant solid tumors (including lung, renal, genitourinary, breast, ovarian, and rectal), and chronic myeloid leukemia.
- ø
- Dizziness: dizziness and vertigo.
Musculoskeletal and connective tissue disorders Musculoskeletal pain* 33 1.7 17 0.4 Infections and infestations Upper respiratory tract infection† 28 1.3 15 0.9 Pneumonia‡ 13§ 5 8¶ 4 Vascular disorders Hemorrhage# 27§ 4 4 0.4 HypertensionÞ 14 7 5 2.6 Skin and subcutaneous tissue disorders Rashß 24 1.3 30 5 Bruisingà 24 0 2.6 0 Respiratory, thoracic, and mediastinal disorders CoughÞ 15 0 10 0 Gastrointestinal disorders Diarrhea 14 0.8 12¶ 0.9 Constipation 10 0.4 18 0 Nausea 10 0 33 1.3 General disorders Fatigueè 14 1.3 21 1.8 Neoplasms Second primary malignancyð 13§ 6 1.3 0.4 Nervous system disorders HeadacheÞ 12 0 8 0 Dizzinessø 11 0.8 5 0 Other clinically significant adverse reactions occurring in <10% of BRUKINSA recipients in this cohort included COVID-19 (9%), edema (8%), abdominal pain (8%), urinary tract infection (7%), and atrial fibrillation or flutter (3.3%).
Table 10 summarizes select laboratory abnormalities in this cohort.
Table 10: Select Laboratory Abnormalities (≥20%) that Worsened from Baseline in Patients with Previously Untreated CLL/SLL without 17p Deletion in SEQUOIA Laboratory Abnormality* BRUKINSA BR All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Hematologic abnormalities Neutrophils decreased 37 15 80 53 Hemoglobin decreased 29 2.5 66 8 Platelets decreased 27 1.7 61 11 Leukocytes increased 21† 21 0.4 0.4 Chemistry abnormalities Glucose increased‡ 55 7 67 10 Creatinine increased 22 0.8 18 0.4 Magnesium increased 22 0 14 0.4 Alanine aminotransferase increased 21 2.1 23 2.2 Single-Arm Cohort: Previously Untreated CLL/SLL and 17p Deletion
In 111 patients with previously untreated, 17p del CLL/SLL, the median age was 70, 71% were male, 95% were White, and 1% were Asian. Most patients (87%) had an ECOG performance status of 0 to 1. The median duration of exposure to BRUKINSA was 30 months.
Fatal adverse reactions occurred in 3 (2.7%) patients, including pneumonia, renal insufficiency, and aortic dissection (1 patient each).
Serious adverse reactions occurred in 41% of patients treated with BRUKINSA. Serious adverse reactions reported in ≥5% of patients were pneumonia (8%) and second primary malignancy (7%).
Adverse reactions led to treatment discontinuation in 5% of patients, dose reduction in 5%, and dose interruption in 51%. The leading causes of dose modification (≥5% of all patients) were pneumonia, neutropenia, second primary malignancy, and diarrhea.
Table 11 summarizes select adverse reactions in this cohort.
Table 11: Adverse Reactions in ≥10% of Patients with Previously Untreated CLL/SLL and 17p Deletion in SEQUOIA CLL/SLL with 17p Deletion BRUKINSA
(N=111)System Organ Class
Preferred TermAll Grades
(%)Grade 3 or 4
(%)- *
- Upper respiratory tract infection: upper respiratory tract infection, nasopharyngitis, sinusitis, rhinitis, pharyngitis, upper respiratory tract congestion, upper respiratory tract inflammation, viral upper respiratory tract infection, and related terms.
- †
- Pneumonia: pneumonia, COVID-19 pneumonia, lower respiratory tract infection, and related terms including specific types of infection.
- ‡
- Includes 1 fatal outcome.
- §
- Musculoskeletal pain: musculoskeletal pain, arthralgia, back pain, pain in extremity, myalgia, neck pain, bone pain.
- ¶
- Rash: Rash, dermatitis, toxic skin eruption, and related terms.
- #
- Bruising: all terms containing bruise, bruising, contusion, or ecchymosis.
- Þ
- Hemorrhage: all terms containing hematoma, hemorrhage, hemorrhagic, and related terms indicative of bleeding.
- ß
- Second primary malignancy: includes non-melanoma skin cancer, malignant solid tumors (including bladder, lung, renal, breast, prostate, ovarian, pelvis, and ureter), and malignant melanoma.
- à
- Includes non-melanoma skin cancer in 13%.
- è
- Includes multiple similar adverse reaction terms.
- ð
- Fatigue: fatigue, asthenia, and lethargy.
Infections and infestations Upper respiratory tract infection* 38 0 Pneumonia† 20‡ 8 Musculoskeletal and connective tissue disorders Musculoskeletal pain§ 38 2.7 Skin and subcutaneous tissue disorders Rash¶ 28 0 Bruising# 26 0.9 Vascular disorders HemorrhageÞ 28 4.5 Hypertensionè 11 5.4 Neoplasms Second primary malignancyß 22à 6 Gastrointestinal disorders Diarrhea 18 0.9 Nausea 16 0 Constipation 15 0 Abdominal painè 12 1.8 Respiratory, thoracic, and mediastinal disorders Coughè 18 0 Dyspneaè 13 0 General disorders and administration site conditions Fatigueð 14 0.9 Nervous system disorders Headache 11 1.8 Clinically significant adverse reactions occurring in <10% of BRUKINSA recipients in this cohort included urinary tract infection (8%), edema (7%), atrial fibrillation or flutter (4.5%), and COVID-19 (3.6%).
Table 12 summarizes select laboratory abnormalities in this cohort.
Table 12: Select Laboratory Abnormalities (≥20%) that Worsened from Baseline in Patients with Previously Untreated CLL/SLL and 17p Deletion in SEQUOIA Laboratory Abnormality* BRUKINSA All Grades (%) Grade 3 or 4 (%) Hematologic abnormalities Neutrophils decreased 42 19† Hemoglobin decreased 26 3.6 Platelets decreased 23 0.9 Chemistry abnormalities Glucose increased‡ 52 6 Magnesium increased 31 0 Creatinine increased 27 0.9 ALPINE
The safety of BRUKINSA monotherapy was evaluated in patients with previously treated CLL/SLL in a randomized, multicenter, open-label, actively controlled trial [see Clinical Studies (14.4)]. In ALPINE, 324 patients received BRUKINSA monotherapy, 160 mg orally twice daily and 324 patients received ibrutinib monotherapy, 420 mg orally daily until disease progression or unacceptable toxicity.
In ALPINE, the median duration of exposure was 24 months for BRUKINSA. Adverse reactions leading to death in the BRUKINSA arm occurred in 24 (7%) patients. Adverse reactions leading to death that occurred in >1% of patients were pneumonia (2.8%) and COVID-19 infection (1.9%).
One hundred and four patients in the BRUKINSA arm (32%) reported ≥1 serious adverse reaction. Serious adverse reactions occurring in ≥5% of patients were pneumonia (10%), COVID-19 (7%), and second primary malignancies (5%).
Adverse reactions led to treatment discontinuation in 13% of patients, dose reduction in 11%, and dose interruption in 42%. The leading cause of treatment discontinuation was pneumonia. The leading causes of dose modification (≥5% of all patients) were respiratory infections (COVID-19, pneumonia) and neutropenia.
Table 13 summarizes select adverse reactions in ALPINE.
Table 13: Adverse Reactions in ≥10% of Patients with Relapsed or Refractory CLL/SLL Who Received BRUKINSA in ALPINE System Organ Class
Preferred TermBRUKINSA
(N=324)Ibrutinib
(N=324)All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- *
- Upper respiratory tract infection: upper respiratory tract infection, sinusitis, pharyngitis, rhinitis, nasopharyngitis, laryngitis, tonsillitis, and related terms.
- †
- Pneumonia: Pneumonia, COVID-19 pneumonia, lower respiratory tract infection, lung infiltration, and related terms including specific types of infection.
- ‡
- Includes fatal outcomes: pneumonia (9 patients), COVID-19 (8 patients), and hemorrhage (1 patient).
- §
- Includes fatal outcomes: pneumonia (10 patients), COVID-19 (9 patients), and hemorrhage (2 patients).
- ¶
- COVID-19: COVID-19, COVID-19 pneumonia, postacute COVID-19 syndrome, SARS-CoV-2 test positive.
- #
- Musculoskeletal pain: musculoskeletal pain, arthralgia, back pain, pain in extremity, myalgia, neck pain, spinal pain, bone pain, and musculoskeletal discomfort.
- Þ
- Hemorrhage: all terms containing hematoma, hemorrhage, hemorrhagic, and related terms indicative of bleeding.
- ß
- Includes multiple similar adverse reaction terms.
- à
- Rash: Rash, Dermatitis, and related terms.
- è
- Bruising: all terms containing bruise, bruising, contusion, or ecchymosis.
- ð
- Fatigue: asthenia, fatigue, lethargy.
Infections and infestations Upper respiratory tract infection* 27 1.2 22 1.2 Pneumonia† 18‡ 9 19§ 11 COVID-19¶ 14‡ 7 10§ 4.6 Musculoskeletal and connective tissue disorders Musculoskeletal pain# 26 0.6 28 0.6 Vascular disorders HemorrhageÞ 24‡ 2.5 26§ 3.7 Hypertensionß 19 13 20 13 Skin and subcutaneous tissue disorders Rashà 20 1.2 21 0.9 Bruisingè 16 0 14 0 Gastrointestinal disorders Diarrhea 14 1.5 22 0.9 General disorders Fatigueð 13 0.9 14 0.9 Respiratory, thoracic, and mediastinal disorders Coughß 11 0.3 11 0 Nervous system disorders Dizzinessß 10 0 7 0 Clinically relevant adverse reactions in <10% of patients who received BRUKINSA included urinary tract infection (9%), supraventricular arrhythmias (9%) including atrial fibrillation or flutter (4.6%), abdominal pain (8%), headache (8%), pruritus (6.2%), constipation (5.9%), and edema (4.6%).
Table 14 summarizes select laboratory abnormalities in ALPINE.
Table 14: Select Laboratory Abnormalities (≥20%) that Worsened from Baseline in Patients Who Received BRUKINSA in ALPINE Laboratory Abnormality* BRUKINSA Ibrutinib All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- *
- The denominator used to calculate the rate was 321 in the BRUKINSA arm, and varied from 320 to 321 in the ibrutinib arm, based on the number of patients with a baseline value and at least one post-treatment value. Grading is based on NCI CTCAE criteria.
Hematologic abnormalities Neutrophils decreased 43 15 33 16 Hemoglobin decreased 28 4 32 3.7 Lymphocytes increased 24 19 26 19 Platelets decreased 22 4 24 3.4 Chemistry abnormalities Glucose increased 52 5 29 2.8 Creatinine increased 26 0 23 0 Phosphate decreased 21 2.5 13 2.2 Calcium decreased 21 0.6 29 0 Follicular Lymphoma
The safety of BRUKINSA in combination with obinutuzumab was evaluated in 143 adult patients with relapsed or refractory follicular lymphoma (FL) in study BGB-3111-212 (ROSEWOOD), a randomized, multicenter, open-label trial [see Clinical Studies (14.5)]. The trial required an absolute neutrophil count ≥1 × 109/L, platelet count ≥50 × 109/L, and CLcr ≥30 mL/min and excluded patients requiring a strong CYP3A inhibitor or inducer.
Patients were randomized to receive either BRUKINSA 160 mg twice daily until disease progression or unacceptable toxicity plus obinutuzumab (n=143) or obinutuzumab monotherapy (n=71). Obinutuzumab was dosed at 1,000 mg intravenously on Days 1, 8, and 15 of Cycle 1; on Day 1 of Cycles 2 to 6; and then every 8 weeks for up to 20 doses. At the discretion of the investigator, obinutuzumab was administered intravenously on Day 1 (100 mg) and on Day 2 (900 mg) of Cycle 1 instead of 1,000 mg on Day 1 of Cycle 1.
In patients who received BRUKINSA in combination with obinutuzumab, the median age was 63, 49% were female, 63% were White, and 21% were Asian. Most patients (97%) had an ECOG performance status of 0 to 1. The median duration of BRUKINSA treatment was 12 months, with 24% of patients treated for at least 2 years.
Serious adverse reactions occurred in 35% of patients who received BRUKINSA in combination with obinutuzumab. Serious adverse reactions in ≥5% of patients included pneumonia (11%) and COVID-19 (10%). Fatal adverse reactions occurred in 4.2% of patients, with the leading cause of death being COVID-19 (2.1%).
Adverse reactions led to permanent discontinuation of BRUKINSA in 17% of patients, dose reduction in 9%, and dose interruption in 40%. Adverse reactions leading to permanent discontinuation in ≥2% of patients were pneumonia, COVID-19, and second primary malignancy. The leading causes of BRUKINSA dosage modification (42% of all patients) were pneumonia, COVID-19, thrombocytopenia, and neutropenia.
Table 15 summarizes adverse reactions in BGB-3111-212.
Table 15: Adverse Reactions in ≥10% of Patients with Relapsed or Refractory FL Who Received BRUKINSA in Study BGB-3111-212 System Organ Class
Preferred TermBGB-3111-212 BRUKINSA + Obinutuzumab
(N=143)Obinutuzumab
(N=71)All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- *
- Includes multiple related terms.
- †
- Fatigue: Fatigue, asthenia, and lethargy.
- ‡
- Musculoskeletal pain: Back pain, musculoskeletal pain, musculoskeletal discomfort, noncardiac chest pain, neck pain, pain in extremity, myalgia, spinal pain, bone pain, arthralgia, and related terms.
- §
- Hemorrhage: All terms containing hematoma, hemorrhage, hemorrhagic, and related terms indicative of bleeding.
- ¶
- Upper respiratory tract infection: Upper respiratory tract infection, sinusitis, pharyngitis, laryngitis, rhinitis, nasopharyngitis, laryngopharyngitis, tonsillitis bacterial, and related terms.
- #
- Pneumonia: Pneumonia, COVID-19 pneumonia, lung infiltration, lung consolidation, and related terms including specific types of infection.
- Þ
- Includes fatal outcomes: COVID-19 (3 patients), pneumonia (2 patients), dyspnea (1 patient).
- ß
- Herpes virus infection: Herpes viral infection, herpes zoster, herpes simplex, herpes simplex reactivation, varicella, and Epstein-Barr viremia.
- à
- Urinary tract infection: Urinary tract infection, cystitis, pyelonephritis, and related terms.
- è
- Rash: Rash, erythema, dermatitis, drug eruption, skin reaction, and related terms.
General disorders and administration site conditions Fatigue*,† 27 1.4 25 1.4 Pyrexia 13 0 20 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain*,‡ 22 3.5 23 1.4 Vascular disorders Hemorrhage*,§ 20 1.4 10 1.4 Gastrointestinal disorders Diarrhea 18 2.8 17 1.4 Constipation 13 0 9 0 Abdominal pain* 11 2.1 11 0 Infections and infestations Upper respiratory tract infection*,¶ 17 2.8 10 0 Pneumonia*,#,Þ 15 13 11 7 COVID-19*,Þ 13 9 11 4.2 Herpes virus infectionß 11 2.1 1.4 0 Urinary tract infectionà 10 1.4 7 0 Respiratory, thoracic, and mediastinal disorders Cough* 14 0 14 0 Dyspnea*,Þ 11 2.1 13 0 Skin and subcutaneous tissue disorders Rash*,è 11 0 14 0 Clinically relevant adverse reactions in <10% of patients who received BRUKINSA in combination with obinutuzumab included bruising, edema, pruritus, petechiae, vomiting, headache, arthralgia, hypertension, sepsis, cardiac arrhythmias, renal insufficiency, febrile neutropenia, transaminase elevation, and pneumonitis.
Table 16: Select Laboratory Abnormalities (≥20%) that Worsened from Baseline in Patients Who Received BRUKINSA in Study BGB-3111-212 Laboratory Abnormality* BGB-3111-212 BRUKINSA + Obinutuzumab Obinutuzumab All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Hematologic abnormalities Platelets decreased 65 11 43 11 Neutrophils decreased 47 17 42 14 Hemoglobin decreased 31 0.8 23 0 Lymphocytes decreased 30 11 51 25 Chemistry Glucose increased† 53 8 41 9 Alanine aminotransferase increased 23 0 28 0 Phosphate decreased 21 0.8 14 0 6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of BRUKINSA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hepatobiliary disorder: drug-induced liver injury
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on BRUKINSA
Table 17: Drug Interactions that Affect Zanubrutinib Moderate and Strong CYP3A Inhibitors Clinical Impact - Coadministration with a moderate or strong CYP3A inhibitor increases zanubrutinib Cmax and AUC [see Clinical Pharmacology (12.3)] which may increase the risk of BRUKINSA toxicities.
Prevention or management - Reduce BRUKINSA dosage when coadministered with moderate or strong CYP3A inhibitors [see Dosage and Administration (2.3)].
Moderate and Strong CYP3A Inducers Clinical Impact - Coadministration with a moderate or strong CYP3A inducer decreases zanubrutinib Cmax and AUC [see Clinical Pharmacology (12.3)] which may reduce BRUKINSA efficacy.
Prevention or management - Avoid coadministration of BRUKINSA with strong CYP3A inducers [see Dosage and Administration (2.3)].
- Avoid coadministration of BRUKINSA with moderate CYP3A inducers [see Dosage and Administration (2.3)]. If these inducers cannot be avoided, increase BRUKINSA dosage to 320 mg twice daily [see Dosage and Administration (2.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animals, BRUKINSA can cause fetal harm when administered to pregnant women. There are no available data on BRUKINSA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, oral administration of zanubrutinib to pregnant rats during the period of organogenesis was associated with fetal heart malformation at approximately 5-fold human exposures (see Data). Women should be advised to avoid pregnancy while taking BRUKINSA. If BRUKINSA is used during pregnancy, or if the patient becomes pregnant while taking BRUKINSA, the patient should be apprised of the potential hazard to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Embryo-fetal development toxicity studies were conducted in both rats and rabbits. Zanubrutinib was administered orally to pregnant rats during the period of organogenesis at doses of 30, 75, and 150 mg/kg/day. Malformations in the heart (2 or 3-chambered hearts) were noted at all dose levels in the absence of maternal toxicity. The dose of 30 mg/kg/day is approximately 5 times the exposure (AUC) in patients receiving the recommended dose of 160 mg twice daily.
Administration of zanubrutinib to pregnant rabbits during the period of organogenesis at 30, 70, and 150 mg/kg/day resulted in postimplantation loss at the highest dose. The dose of 150 mg/kg is approximately 32 times the exposure (AUC) in patients at the recommended dose and was associated with maternal toxicity.
In a pre and postnatal developmental toxicity study, zanubrutinib was administered orally to rats at doses of 30, 75, and 150 mg/kg/day from implantation through weaning. The offspring from the middle and high dose groups had decreased body weights preweaning, and all dose groups had adverse ocular findings (e.g., cataract, protruding eye). The dose of 30 mg/kg/day is approximately 5 times the AUC in patients receiving the recommended dose.
8.2 Lactation
Risk Summary
There are no data on the presence of zanubrutinib or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions from BRUKINSA in a breastfed child, advise lactating women not to breastfeed during treatment with BRUKINSA and for two weeks following the last dose.
8.3 Females and Males of Reproductive Potential
BRUKINSA can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating BRUKINSA therapy.
Contraception
Females
Advise female patients of reproductive potential to use effective contraception during treatment with BRUKINSA and for 1 week following the last dose of BRUKINSA. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be informed of the potential hazard to a fetus.
8.4 Pediatric Use
Safety and effectiveness of BRUKINSA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 1729 patients with MCL, MZL, WM, CLL/SLL, and FL in clinical studies with BRUKINSA, 59% were ≥65 years of age, and 21% were ≥75 years of age. Patients ≥65 years of age had numerically higher rates of Grade 3 or higher adverse reactions and serious adverse reactions (57% and 38%, respectively) than patients <65 years of age (51% and 29%, respectively). No overall differences in effectiveness were observed between younger and older patients.
8.6 Renal Impairment
No dosage modification is recommended in patients with mild, moderate, or severe renal impairment (CLcr ≥15 mL/min, estimated by Cockcroft-Gault). Monitor for BRUKINSA adverse reactions in patients on dialysis [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Dosage modification of BRUKINSA is recommended in patients with severe hepatic impairment [see Dosage and Administration (2.2)]. The safety of BRUKINSA has not been evaluated in patients with severe hepatic impairment. No dosage modification is recommended in patients with mild to moderate hepatic impairment. Monitor for BRUKINSA adverse reactions in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
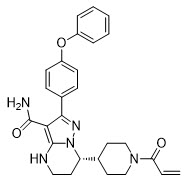
BRUKINSA (zanubrutinib) is a kinase inhibitor. The empirical formula of zanubrutinib is C27H29N5O3 and the chemical name is (S)-7-(1-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamide. Zanubrutinib is a white to off-white powder, with a pH of 7.8 in saturated solution. The aqueous solubility of zanubrutinib is pH dependent, from very slightly soluble to practically insoluble.
The molecular weight of zanubrutinib is 471.55 Daltons.
Zanubrutinib has the following structure:
Each BRUKINSA capsule for oral administration contains 80 mg zanubrutinib and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The capsule shell contains edible black ink, gelatin, and titanium dioxide.
Each BRUKINSA tablet for oral administration contains 160 mg zanubrutinib and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate. The film coating contains FD&C Blue No. 1, FD&C Blue No. 2, hypromellose, titanium dioxide, and triacetin.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Zanubrutinib is a small-molecule inhibitor of Bruton's tyrosine kinase (BTK). Zanubrutinib forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK activity. BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. In B-cells, BTK signaling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis, and adhesion. In nonclinical studies, zanubrutinib inhibited malignant B-cell proliferation and reduced tumor growth.
12.2 Pharmacodynamics
BTK Occupancy in PBMCs and Lymph Nodes
The median steady-state BTK occupancy in peripheral blood mononuclear cells was maintained at 100% over 24 hours at a total daily dose of 320 mg in patients with B-cell malignancies. The median steady-state BTK occupancy in lymph nodes was 94% to 100% following the approved recommended dosage.
Cardiac Electrophysiology
At the approved recommended doses (160 mg twice daily or 320 mg once daily), there were no clinically relevant effects on the QTc interval. The effect of BRUKINSA on the QTc interval above the therapeutic exposure has not been evaluated.
In Vitro Platelet Aggregation
In blood samples from healthy donors, patients on anticoagulant or antiplatelet therapy, and those with severe renal dysfunction, zanubrutinib demonstrated inhibition of platelet aggregation mediated by collagen, CRP-XL, or rhodocytin. Zanubrutinib did not show meaningful inhibition of platelet aggregation for ADP and PAR4-AP.
12.3 Pharmacokinetics
Zanubrutinib maximum plasma concentration (Cmax) and area under the plasma drug concentration over time curve (AUC) increase proportionally over a dosage range from 40 mg to 320 mg (0.13 to 1 time the recommended total daily dose). Limited systemic accumulation of zanubrutinib was observed following repeated administration.
The geometric mean (%CV) zanubrutinib steady-state daily AUC is 2,099 (42%) ng∙h/mL following 160 mg twice daily and 1,917 (59%) ng∙h/mL following 320 mg once daily. The geometric mean (%CV) zanubrutinib steady-state Cmax is 295 (55%) ng/mL following 160 mg twice daily and 537 (55%) ng/mL following 320 mg once daily.
Absorption
The median tmax of zanubrutinib is 2 hours.
Distribution
The geometric mean (%CV) apparent volume of distribution (Vz/F) of zanubrutinib is 537 (73%) L. The plasma protein binding of zanubrutinib is approximately 94% and the blood-to-plasma ratio is 0.7 to 0.8.
Elimination
The mean half-life (t½) of zanubrutinib is approximately 2 to 4 hours following a single oral zanubrutinib dose of 160 mg or 320 mg. The geometric mean (%CV) apparent oral clearance (CL/F) of zanubrutinib is 128 (58%) L/h.
Specific Populations
No clinically significant differences in the pharmacokinetics of zanubrutinib were observed based on age (19 to 90 years), sex, race (Asian, White, and Other), body weight (36 to 144 kg), or mild, moderate or severe renal impairment (CLcr ≥15 mL/min as estimated by Cockcroft-Gault). The effect of dialysis on zanubrutinib pharmacokinetics is unknown.
Hepatic Impairment
The total AUC of zanubrutinib increased by 11% in subjects with mild hepatic impairment (Child-Pugh class A), by 21% in subjects with moderate hepatic impairment (Child-Pugh class B), and by 60% in subjects with severe hepatic impairment (Child-Pugh class C) relative to subjects with normal liver function. The unbound AUC of zanubrutinib increased by 23% in subjects with mild hepatic impairment (Child-Pugh class A), by 43% in subjects with moderate hepatic impairment (Child-Pugh class B) and by 194% in subjects with severe hepatic impairment (Child-Pugh class C) relative to subjects with normal liver function.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
CYP3A Inhibitors: Coadministration of multiple doses of CYP3A inhibitors increases zanubrutinib Cmax and AUC (Table 18).
Table 18: Observed or Predicted Increase in Zanubrutinib Exposure After Coadministration of CYP3A Inhibitors Coadministered CYP3A Inhibitor Increase in Zanubrutinib Cmax Increase in Zanubrutinib AUC - *
- The assessment was conducted in healthy subjects with a single dose of zanubrutinib 20 mg.
- †
- The assessment was conducted in patients with B-cell lymphoma administered multiple doses of zanubrutinib at the respective recommended zanubrutinib dosages in Table 1.
- ‡
- The predicted values were based on simulations with patients administered multiple doses of zanubrutinib at the respective recommended dosages in Table 1.
Observed Itraconazole (200 mg once daily)* 157% 278% Fluconazole (400 mg once daily)† 81% 88% Diltiazem (180 mg once daily)† 62% 62% Voriconazole (200 mg twice daily)† 229% 230% Clarithromycin (250 mg twice daily)† 101% 92% Predicted Posaconazole suspension (100 mg once daily)‡ 169% 180% Posaconazole suspension (100 mg twice daily)‡ 207% 279% Posaconazole delayed-release tablets (300 mg once daily)‡ 232% 407% Posaconazole intravenously (300 mg once daily)‡ 205% 333% Itraconazole (200 mg once daily)‡ 273% 320% CYP3A Inducers: Coadministration of multiple doses of rifampin (strong CYP3A inducer) decreased the zanubrutinib Cmax by 92% and AUC by 93%. Coadministration of multiple doses of rifabutin (moderate CYP3A inducer) decreased the zanubrutinib Cmax by 48% and AUC by 44%.
Coadministration of multiple doses of efavirenz (moderate CYP3A inducer) is predicted to decrease zanubrutinib Cmax by 58% and AUC by 60%.
CYP3A Substrates: Coadministration of multiple doses of zanubrutinib decreased midazolam (CYP3A substrate) Cmax by 30% and AUC by 47%.
CYP2C19 Substrates: Coadministration of multiple doses of zanubrutinib decreased omeprazole (CYP2C19 substrate) Cmax by 20% and AUC by 36%.
Other CYP Substrates: No clinically significant differences were observed with warfarin (CYP2C9 substrate) pharmacokinetics when coadministered with zanubrutinib.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with zanubrutinib.
Zanubrutinib was not mutagenic in a bacterial mutagenicity (Ames) assay, was not clastogenic in a chromosome aberration assay in mammalian (CHO) cells, nor was it clastogenic in an in vivo bone marrow micronucleus assay in rats.
A combined male and female fertility and early embryonic development study was conducted in rats at oral zanubrutinib doses of 30 to 300 mg/kg/day. Male rats were dosed 4 weeks prior to mating and through mating and female rats were dosed 2 weeks prior to mating and to gestation day 7. No effect on male or female fertility was noted at lower doses, but at the highest dose tested, morphological abnormalities in sperm and increased postimplantation loss were noted. The high dose of 300 mg/kg/day is approximately 10 times the human recommended dose, based on body surface area.
-
14 CLINICAL STUDIES
14.1 Mantle Cell Lymphoma
The efficacy of BRUKINSA was assessed in BGB-3111-206 [NCT03206970], a Phase 2, open-label, multicenter, single-arm trial of 86 previously treated patients with MCL who had received at least one prior therapy. BRUKINSA was given orally at a dose of 160 mg twice daily until disease progression or unacceptable toxicity.
The median age of patients was 60.5 years (range: 34 to 75) and the majority were male (78%). The median time since diagnosis to study entry was 30 months (range: 3 to 102) and the median number of prior therapies was 2 (range: 1 to 4). The most common prior regimens were CHOP-based (91%) followed by rituximab-based (74%). The majority of patients had extranodal involvement (71%) and refractory disease (52%). Blastoid variant of MCL was present in 14% of patients. The MIPI score was low in 58%, intermediate in 29%, and high risk in 13%.
The efficacy of BRUKINSA was also assessed in BGB-3111-AU-003 [NCT02343120], a Phase 1/2, open-label, dose-escalation, global, multicenter, single-arm trial of B cell malignancies including 32 previously treated MCL patients treated with BRUKINSA. BRUKINSA was given orally at doses of 160 mg twice daily or 320 mg daily. The median age of patients with previously treated MCL was 70 years (range: 42 to 86) and 38% of patients were ≥75 years old. Most patients were male (69%) and White (78%). The MIPI score was low in 28%, intermediate in 41%, and high risk in 31%.
Tumor response was according to the 2014 Lugano Classification for both studies, and the primary efficacy endpoint was overall response rate as assessed by an Independent Review Committee (IRC).
Table 19: Efficacy Results per IRC in Patients with MCL Study BGB-3111-206
(N=86)Study BGB-3111-AU-003
(N=32)ORR: overall response rate, CR: complete response, PR: partial response, DOR: duration of response, CI: confidence interval, NE: not estimable. - *
- FDG-PET scans were not required for response assessment.
ORR (95% CI) 84% (74, 91) 84% (67, 95) CR 59% 22%* PR 24% 62% Median DOR in months (95% CI) 19.5 (16.6, NE) 18.5 (12.6, NE) 14.2 Waldenström's Macroglobulinemia
The efficacy of BRUKINSA was evaluated in ASPEN [NCT03053440], a randomized, active control, open-label trial, comparing BRUKINSA and ibrutinib in patients with MYD88 L265P mutation (MYD88MUT) WM. Patients in Cohort 1 (n=201) were randomized 1:1 to receive BRUKINSA 160 mg twice daily or ibrutinib 420 mg once daily until disease progression or unacceptable toxicity. Randomization was stratified by number of prior therapies (0 vs 1-3 vs >3) and CXCR4 status (presence or absence of a WHIM-like mutation as measured by Sanger assay).
The major efficacy outcome was the response rate defined as PR or better as assessed by IRC based on standard consensus response criteria from the International Workshop on Waldenström's Macroglobulinemia (IWWM)-6 criteria. An additional efficacy outcome measure was duration of response (DOR).
The median age was 70 years (range: 38 to 90) and 68% were male. Of those enrolled, 2% were Asian, 91% were White, and 7% were of unknown race. ECOG performance status of 0 or 1 was present in 93% patients at baseline and 7% had a baseline ECOG performance status of 2. A total of 82% had relapsed/refractory disease with 85% having received prior alkylating agents and 91% prior anti-CD20 therapy. The median number of prior therapies in those with relapsed/refractory disease was 1 (range: 1 to 8). A total of 91 (45%) patients had International Prognostic Scoring System (IPSS) high WM.
The study did not meet statistical significance for the prespecified efficacy outcome of superior CR+VGPR as assessed by IRC, tested first in patients with R/R disease in ASPEN.
Table 20 shows the response rates in ASPEN based on IRC assessment.
Table 20: Response Rate and Duration of Response Based on IRC Assessment in ASPEN Standard IWWM-6* Modified IWWM-6† Response Category BRUKINSA
(N=102)Ibrutinib
(N=99)BRUKINSA
(N=102)Ibrutinib
(N=99)- *
- IWWM-6 criteria (Owen et al, 2013) require complete resolution of extramedullary disease (EMD) if present at baseline for VGPR to be assessed.
- †
- Modified IWWM-6 criteria (Treon, 2015) require a reduction in EMD if present at baseline for VGPR to be assessed.
- ‡
- 2-sided Clopper-Pearson 95% confidence interval.
- §
- Estimated by Kaplan-Meier method with 95% CIs estimated using the method of Brookmeyer and Crowley.
Response rate (CR+VGPR+PR), n (%) 79 (77.5) 77 (77.8) 79 (77.5) 77 (77.8) 95% CI (%)‡ (68.1, 85.1) (68.3, 85.5) (68.1, 85.1) (68.3, 85.5) Complete Response (CR) 0 (0) 0 (0) 0 (0) 0 (0) Very Good Partial Response (VGPR) 16 (15.7) 7 (7.1) 29 (28.4) 19 (19.2) Partial Response (PR) 63 (61.8) 70 (70.7) 50 (49) 58 (58.6) Duration of response (DOR), Event-free at 12 months (95% CI)§ 94.4%
(85.8, 97.9)87.9%
(77, 93.8)94.4%
(85.8, 97.9)87.9%
(77, 93.8)ASPEN Cohort 2
Cohort 2 enrolled patients with MYD88 wildtype (MYD88WT) or MYD88 mutation unknown WM (N=26 and 2, respectively) and received BRUKINSA 160 mg twice daily. The median age was 72 years (range: 39 to 87) with 43% >75 years, 50% were male, 96% were White, and 4% were not reported (unknown race). Eighty-six percent of patients had a baseline ECOG performance status 0 or 1 and 14% had a baseline performance status of 2. Twenty-three of the 28 patients in Cohort 2 had relapsed or refractory disease.
In Cohort 2, response (CR+VGPR+PR) as assessed by IRC using IWWM-6 or modified IWWM-6 was seen in 50% (13 out of 26 response evaluable patients; 95% CI: 29.9, 70.1).
14.3 Marginal Zone Lymphoma
The efficacy of BRUKINSA was assessed in Study BGB-3111-214 [NCT03846427], an open-label, multicenter, single-arm trial that evaluated 66 patients with MZL who received at least one prior anti–CD20-based therapy. BRUKINSA was given orally at a dosage of 160 mg twice daily until disease progression or unacceptable toxicity. The median age was 70 years (range: 37 to 85); 55% were male; 38% had extranodal MZL, 38% nodal, 18% splenic, and 6% had unknown subtype. The median number of prior systemic therapies was 2 (range: 1 to 6), with 27% having 3 or more lines of systemic therapy; 88% had prior rituximab-based chemotherapy; 32% had refractory disease at study entry.
The efficacy of BRUKINSA was also assessed in BGB-3111-AU-003 [NCT02343120], an open-label, multicenter, single-arm trial that included 20 patients with previously treated MZL (45% having extranodal MZL, 25% nodal, 30% splenic). BRUKINSA was given orally at dosages of 160 mg twice daily or 320 mg once daily. The median age was 70 years (range: 52 to 85); 50% were male. The median number of prior systemic therapies was 2 (range: 1 to 5), with 20% having 3 or more lines of systemic therapy; 95% had prior rituximab-based chemotherapy.
Efficacy was based on overall response rate (ORR) and duration of response as assessed by an Independent Review Committee (IRC) using 2014 Lugano criteria (Table 21).
Table 21: Efficacy Results per IRC in Patients with MZL Parameter Study BGB-3111-214
(N=66)Study BGB-3111-AU-003
(N=20)ORR: overall response rate, CR: complete response, PR: partial response, DOR: duration of response, CI: confidence interval, NE: not estimable. Overall response rate (CT-based)* ORR, n 37 (56%) 16 (80%) (95% CI, %) (43, 68) (56, 94) CR, n 13 (20%) 4 (20%) PR, n 24 (36%) 12 (60%) Time to response Median (range), months 2.9 (1.8, 11.1) 2.9 (2.6, 23.1) Duration of response† Median DOR (95% CI), months NE (NE, NE) NE (8.4, NE) Rate at 12 months (95% CI) 85% (67, 93) 72% (40, 88) In study BGB-3111-214, ORR prioritizing PET-CT when available (55 patients, with the remainder assessed by CT scan) was 67% (95% CI: 54, 78) with a CR rate of 26%.
14.4 Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma
The efficacy of BRUKINSA in patients with CLL/SLL was evaluated in two randomized controlled trials.
SEQUOIA
The efficacy of BRUKINSA in patients with previously untreated CLL/SLL was evaluated in a multicenter, open-label trial (SEQUOIA; NCT03336333). The trial required patients to be unsuitable for FCR therapy defined as either age ≥65 years or age 18 to <65 with a total Cumulative Illness Rating Scale (CIRS) >6, creatinine clearance 30 to 69 mL/min, or history of serious or recurrent infection. Patients without 17p deletion (17p del) were randomized to receive either BRUKINSA 160 mg twice daily until disease progression or unacceptable toxicity (n=241) or bendamustine plus rituximab (BR) for 6 cycles (n=238). Bendamustine was dosed at 90 mg/m2/day intravenously on the first 2 days of each cycle, and rituximab was dosed at 375 mg/m2 on day 1 of Cycle 1 and 500 mg/m2 on day 1 of Cycles 2 to 6 with a 28-day cycle length. Randomization was stratified by age, Binet stage, immunoglobulin variable region heavy chain (IGHV) mutational status, and geographic region.
Additionally, the same BRUKINSA regimen was evaluated in 110 patients with previously untreated 17p del CLL/SLL in a non-randomized cohort.
Efficacy is summarized according to cohort.
Randomized Cohort: Previously Untreated CLL/SLL without 17p Deletion
In the randomized cohort of patients with previously untreated CLL/SLL without 17p deletion, the median age was 70 years; 62% were male, 89% were White, 3% were Asian, and 1% were Black. Fifty-three percent of patients had an unmutated IGHV gene and 29% had Binet Stage C disease. Baseline characteristics were generally similar between treatment arms.
Efficacy in this cohort was based on progression-free survival as assessed by an IRC. Efficacy results are presented in Table 22 and Figure 1.
Table 22: Efficacy Results per IRC in Patients with Previously Untreated CLL/SLL without 17p Deletion in SEQUOIA (Randomized Cohort) Parameter* CLL/SLL without del(17p) BRUKINSA
(N=241)Bendamustine + Rituximab
(N=238)CI=Confidence interval, CR=complete response, CRi=complete response with incomplete hematopoietic recovery, HR=hazard ratio, NE=not estimable, nPR=nodular partial response, ORR=overall response rate, PFS=progression-free survival, PR=partial response. - *
- Efficacy was assessed using the 2008 International Workshop for Chronic Lymphocytic Leukemia (iwCLL) guidelines and 2014 Lugano criteria for SLL.
- †
- Based on Kaplan-Meier estimation. Estimated median follow-up for PFS was 25 months.
- ‡
- Based on a stratified Cox-regression model with bendamustine + rituximab as the reference group.
- §
- Based on a stratified log-rank test, with a 2-sided significance level of 0.0372.
- ¶
- Defined as CR, CRi, PR, and nPR. No patients had CRi as best response.
Progression-free survival Number of Events, n 36 (15%) 71 (30%) Disease Progression 27 (11%) 59 (25%) Death 9 (3.7%) 12 (5%) Median PFS (95% CI), months† NE (NE, NE) 33.7 (28.1, NE) HR (95% CI)‡ 0.42 (0.28, 0.63) P-value§ <0.0001 Overall response rate¶ ORR, n (%) 225 (93) 203 (85) 95% CI, % (89, 96) (80, 90) CR, n (%) 16 (7) 36 (15) nPR, n (%) 3 (1.2) 14 (6) PR, n (%) 206 (85) 153 (64) Figure 1: Kaplan-Meier Plot of IRC-Assessed Progression-Free Survival in Patients with Previously Untreated CLL/SLL without 17p Deletion in SEQUOIA
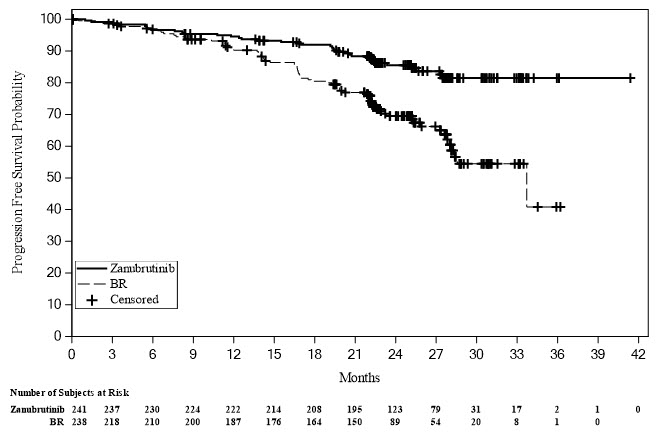
At the time of analysis, overall survival data were immature. With an estimated median follow-up of 25.7 months, median overall survival was not reached in either arm, with fewer than 7% of patients experiencing an event.
Single-Arm Cohort: Previously Untreated CLL/SLL with 17p Deletion
In this cohort, 110 patients with previously untreated CLL/SLL and centrally confirmed 17p deletion received BRUKINSA 160 mg twice daily until disease progression or unacceptable toxicity.
The median age was 70, 71% were male, 95% were White, and 1% were Asian. Sixty percent of patients had an unmutated IGHV gene and 35% had Binet Stage C disease.
Efficacy was based on overall response rate and duration of response as assessed by an IRC. Efficacy results are presented in Table 23.
Table 23: Efficacy Results per IRC in Patients with Previously Untreated CLL/SLL and 17p Deletion in SEQUOIA Parameter* del(17p) CLL/SLL
N=110DOR=duration of response. A + sign indicates a censored observation. Overall response rate† ORR, n (%) 97 (88) (95% CI, %) (81, 94) CR, n (%) 7 (6) nPR, n (%) 2 (1.8) PR, n (%) 88 (80) Time to response Median (range), months 2.9 (1.9 to 13.9) Duration of response Median DOR (95% CI),‡ months NE (NE, NE) Range, months (5.6 to 35.9+) Rate at 12 months, % (95% CI)‡ 96 (89, 98) Rate at 18 months, % (95% CI)‡ 95 (88, 98) ALPINE
The efficacy of BRUKINSA in patients with relapsed or refractory CLL/SLL was evaluated in ALPINE, a randomized, multicenter, open-label, actively controlled trial (NCT03734016). The trial enrolled 652 patients with relapsed or refractory CLL/SLL after at least 1 systemic therapy. The patients were randomized in a 1:1 ratio to receive either BRUKINSA 160 mg orally twice daily (n=327) or ibrutinib 420 mg orally once daily (n=325), each administered until disease progression or unacceptable toxicity.
Randomization was stratified by age, geographic region, refractoriness to last therapy, and 17p deletion/TP53 mutation status.
Baseline characteristics were similar between treatment arms. Overall, the median age was 67 years, 68% were male, 81% were White, 14% were Asian, and 1% were Black. Forty-three percent had advanced stage disease, 73% had an unmutated IGHV gene, and 23% had 17p deletion or TP53 mutation. Patients had a median of one prior line of therapy (range: 1-8), 18% of patients had ≥3 prior lines of therapy, 78% had prior chemoimmunotherapy, and 2.3% had prior BCL2 inhibitor.
Efficacy was based on overall response rate and duration of response as determined by an IRC. For progression-free survival per IRC, the final analysis occurred with a median follow-up of 31 months. Efficacy results are shown in Table 24 and Figure 2.
Table 24: Efficacy Results per IRC in Patients with Relapsed or Refractory CLL/SLL in ALPINE Parameter* BRUKINSA
(N=327)Ibrutinib
(N=325)CI=Confidence interval, CR=complete response, CRi=complete response with incomplete hematopoietic recovery, DOR=duration of response, HR=hazard ratio, NE=not estimable, nPR=nodular partial response, ORR=overall response rate, PR=partial response. A + sign indicates a censored observation. - *
- Efficacy was based on 2008 iwCLL guidelines for CLL and the Lugano criteria for SLL.
- †
- Defined as CR + CRi + nPR + PR. No patients had CRi as best response.
- ‡
- Estimate stratified by randomization stratification factors.
- §
- 2-sided significance level of 0.0469 was allocated for ORR superiority testing.
- ¶
- Based on Kaplan-Meier estimate. Estimated median follow-up for DOR was 14.1 months.
- #
- Based on Kaplan-Meier estimation. Estimated median follow-up for PFS was 30.7 months.
- Þ
- Based on a stratified Cox-regression model with ibrutinib as the reference group.
- ß
- Based on a stratified log-rank test.
Overall response rate† ORR, n (%) 263 (80) 237 (73) (95% CI, %) (76, 85) (68, 78) CR, n (%) 13 (4) 8 (2.5) nPR, n (%) 1 (0.3) 0 (0) PR, n (%) 249 (76) 229 (70) Response Rate Ratio (95% CI)‡ 1.10 (1.01, 1.20) 2-sided p-value§ 0.0264 Duration of response Median DOR (95% CI)¶ NE (NE, NE) NE (NE, NE) Range, months (1.4 to 30.4+) (1.9+ to 30.8+) Rate at 12 months, % (95% CI)¶ 92 (87, 95) 86 (80, 91) Progression-free survival Events, n (%) 88 (27) 120 (37) Median PFS (95% CI), months# NE (34.3, NE) 35 (33.2, 44.3) HR (95% CI)Þ 0.65 (0.49, 0.86) 2-sided p-valueß 0.0024 The median time to response was 5.5 months for BRUKINSA and 5.6 months for ibrutinib.
Figure 2: Kaplan-Meier Plot of Progression-Free Survival by IRC in ALPINE
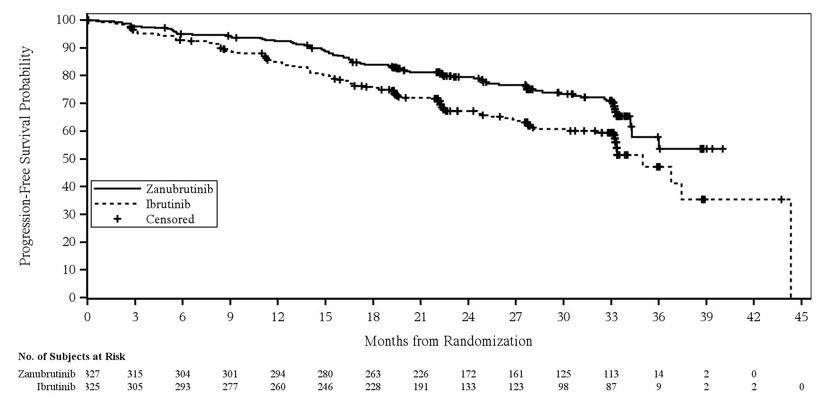
At the time of final analysis, the median overall survival was not reached in either arm. There were a total of 108 deaths: 48 (14.7%) in the zanubrutinib arm and 60 (18.5%) in the ibrutinib arm.
14.5 Follicular Lymphoma
The efficacy of BRUKINSA, in combination with obinutuzumab, was evaluated in Study BGB-3111-212 (ROSEWOOD; NCT03332017), an open-label, multicenter, randomized trial that enrolled 217 adult patients with relapsed or refractory FL after at least 2 prior systemic treatments. The study required prior receipt of an anti-CD20 antibody and an alkylator-based combination therapy, and excluded patients with FL Grade 3b, transformed lymphoma, and prior exposure to a BTK inhibitor.
Patients were randomized in a 2:1 ratio to receive either BRUKINSA 160 mg orally twice daily until disease progression or unacceptable toxicity plus obinutuzumab, or obinutuzumab alone. Obinutuzumab was administered 1,000 mg intravenously on days 1, 8, and 15 of Cycle 1, 1,000 mg on Day 1 of Cycles 2 to 6; and then 1,000 mg every 8 weeks for up to 20 doses. At the discretion of the investigator, obinutuzumab could be administered as 100 mg on Day 1 and 900 mg on Day 2 of Cycle 1.
Randomization was stratified by the number of prior lines of therapy (2 to 3 vs ˃3), rituximab-refractory status (yes vs no), and geographic region.
Of the 217 patients randomized (145 to BRUKINSA plus obinutuzumab, 72 to obinutuzumab monotherapy), the median age was 64 years (range: 31 to 88), 50% were male, 64% were White, and 22% were Asian. In total, 83% had stage 3 or 4 disease and 57% met Groupe d'Etude des Lymphomes Folliculaires (GELF) criteria at enrollment. Patients had a median of 3 prior lines of therapy (range: 2-11), with 27% of patients having more than 3 prior lines of therapy.
In the BRUKINSA in combination with obinutuzumab arm, 5% had received lenalidomide plus rituximab, 21% had received stem cell transplantation, 53% had refractory disease to rituximab, and 37% had progression of disease within 24 months of the first systemic therapy.
Efficacy was based on overall response rate and duration of response, as determined by an IRC. Efficacy results are shown in Table 25. The median time to response in the BRUKINSA combination arm was 2.8 months (range 2 to 23 months).
Table 25: Efficacy Results per IRC in Patients with Relapsed or Refractory Follicular Lymphoma Parameter BRUKINSA + Obinutuzumab
(N=145)Obinutuzumab
(N=72)CI=Confidence interval, CR=complete response, DOR=duration of response, NE=not estimable, ORR=overall response rate, PR=partial response. Overall response rate ORR, n (%) 100 (69) 33 (46) (95% CI)* (61, 76) (34, 58) CR 57 (39) 14 (19) PR 43 (30) 19 (26) Risk difference, % (95% CI)† 22.7 (9, 36.5) 2-sided p-value†,‡ 0.0012 Duration of response Median DOR (95% CI),§ months NE (25.3, NE) 14 (9.2, 25.1) The estimated DOR rate at 18 months was 69% (95% CI: 58, 78) in the BRUKINSA combination arm and 42% (95% CI: 23, 60) in the obinutuzumab monotherapy arm.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Strength Description Package Size NDC Number 80 mg white to off-white opaque capsule, marked with "ZANU 80" in black ink 120-count capsules in a bottle with a child-resistant cap 72579-011-02 160 mg blue, oval, film-coated tablets debossed with "zanu" on one side and functional scoring on the other side 60-count tablets in a bottle with a child-resistant cap 72579-122-01 -
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Hemorrhage
Inform patients to report signs or symptoms of severe bleeding. Inform patients that BRUKINSA may need to be interrupted for major surgeries or procedures [see Warnings and Precautions (5.1)].
Infections
Inform patients to report signs or symptoms suggestive of infection [see Warnings and Precautions (5.2)].
Cytopenias
Inform patients that they will need periodic blood tests to check blood counts during treatment with BRUKINSA [see Warnings and Precautions (5.3)].
Second Primary Malignancies
Inform patients that other malignancies have been reported in patients who have been treated with BRUKINSA, including skin cancer and other solid tumors. Advise patients to use sun protection and have monitoring for development of other cancers [see Warnings and Precautions (5.4)].
Cardiac Arrhythmias
Counsel patients to report any signs of palpitations, lightheadedness, dizziness, fainting, shortness of breath, and chest discomfort [see Warnings and Precautions (5.5)].
Hepatotoxicity, Including Drug-Induced Liver Injury
Inform patients that liver problems, including drug-induced liver injury and abnormalities in liver tests, may develop during BRUKINSA treatment. Advise patients to contact their healthcare provider immediately if they experience abdominal discomfort, dark urine, or jaundice [see Warnings and Precautions (5.6)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential hazard to a fetus and to use effective contraception during treatment and for 1 week after the last dose of BRUKINSA [see Warnings and Precautions (5.7)]. Advise males with female sexual partners of reproductive potential to use effective contraception during BRUKINSA treatment and for 1 week after the last dose of BRUKINSA [see Use in Specific Populations (8.3)].
Lactation
Advise females not to breastfeed during treatment with BRUKINSA and for 2 weeks after the last dose [see Use in Specific Populations (8.2)].
Administration Instructions
- BRUKINSA capsules may be taken with or without food. Advise patients that BRUKINSA capsules should be swallowed whole with a glass of water and not to open, break, or chew the capsules [see Dosage and Administration (2.1)].
- BRUKINSA tablets may be taken with or without food. Advise patients that BRUKINSA tablets should be swallowed whole with a glass of water and not to chew or crush the tablets. Tablets can be split in half as prescribed by the healthcare provider [see Dosage and Administration (2.1)].
Missed Dose
Advise patients that if they miss a dose of BRUKINSA, they may still take it as soon as possible on the same day with a return to the normal schedule the following day [see Dosage and Administration (2.1)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including over-the-counter medications, vitamins, and herbal products [see Drug Interactions (7.1)].
-
SPL UNCLASSIFIED SECTION
BRUKINSA Capsules:
Manufactured for:
BeOne Medicines USA, Inc.
Pennington, NJ 08534BRUKINSA Tablets:
Manufactured for:
BeOne Medicines USA, Inc.
Pennington, NJ 08534
Manufactured by:
Catalent Pharma Solutions, LLC
Winchester, KY 40391BRUKINSA® is a registered trademark owned by BeOne Medicines I GmbH or its affiliates.
© BeOne Medicines I GmbH, 2025. All Rights Reserved. -
PATIENT PACKAGE INSERT
PATIENT INFORMATION BRUKINSA® (BROO-kin-sah)
(zanubrutinib)
capsulesBRUKINSA® (BROO-kin-sah)
(zanubrutinib)
tabletsThis Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 6/2025 What is BRUKINSA?
BRUKINSA is a prescription medicine used to treat adults with:- Mantle cell lymphoma (MCL) who have received at least one prior treatment for their cancer.
- Waldenström's macroglobulinemia (WM).
- Marginal zone lymphoma (MZL) when the disease has come back or did not respond to treatment and who have received at least one certain type of treatment.
- Chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL).
- Follicular lymphoma (FL), in combination with the medicine obinutuzumab, when the disease has come back or did not respond to treatment and who have received at least two prior treatments.
Before taking BRUKINSA, tell your healthcare provider about all of your medical conditions, including if you: - have bleeding problems.
- have had recent surgery or plan to have surgery. Your healthcare provider may stop BRUKINSA for any planned medical, surgical, or dental procedure.
- have an infection or have been advised that you are at increased risk of infection.
- have or had heart rhythm problems.
- have high blood pressure.
- have liver problems, including a history of hepatitis B virus (HBV) infection.
- are pregnant or plan to become pregnant. BRUKINSA can harm your unborn baby.
Females who are able to become pregnant:- You should avoid getting pregnant during treatment and for 1 week after the last dose of BRUKINSA.
- Your healthcare provider may do a pregnancy test before starting treatment with BRUKINSA.
- You should use effective birth control (contraception) during treatment and for 1 week after the last dose of BRUKINSA.
- Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with BRUKINSA.
- You should avoid getting female partners pregnant during treatment and for 1 week after the last dose of BRUKINSA.
- You should use effective birth control (contraception) during treatment and for 1 week after the last dose of BRUKINSA.
- are breastfeeding or plan to breastfeed. It is not known if BRUKINSA passes into your breast milk. Do not breastfeed during treatment with BRUKINSA and for 2 weeks after the last dose of BRUKINSA.
How should I take BRUKINSA? - Take BRUKINSA exactly as your healthcare provider tells you to take it.
- Do not change your dose or stop taking BRUKINSA unless your healthcare provider tells you to.
- Your healthcare provider may tell you to decrease your dose, temporarily stop, or completely stop taking BRUKINSA if you develop certain side effects.
- Take BRUKINSA with or without food.
- Swallow BRUKINSA capsules whole with a glass of water. Do not open, break, or chew the capsules.
- Swallow BRUKINSA tablets whole with a glass of water. Do not chew or crush the tablets.
- BRUKINSA tablets can be split in half as prescribed by your healthcare provider.
- If you miss a dose of BRUKINSA, take it as soon as you remember on the same day. Return to your normal schedule the next day.
What are the possible side effects of BRUKINSA?
BRUKINSA may cause serious side effects, including:- Bleeding problems (hemorrhage). Bleeding problems are common with BRUKINSA, and can be serious and may lead to death. Your risk of bleeding may increase if you are also taking a blood thinner medicine. Tell your healthcare provider if you develop any signs or symptoms of bleeding, including:
- blood in your stools or black stools (looks like tar)
- pink or brown urine
- unexpected bleeding, or bleeding that is severe or you cannot control
- vomit blood or vomit that looks like coffee grounds
- cough up blood or blood clots
- increased bruising
- dizziness
- weakness
- confusion
- change in speech
- headache that lasts a long time
- Infections that can be serious and may lead to death. Tell your healthcare provider right away if you develop fever, chills, or flu-like symptoms during your treatment with BRUKINSA.
- Decrease in blood cell counts (white blood cells, platelets, and red blood cells). Your healthcare provider should do blood tests during treatment with BRUKINSA to check your blood counts.
- Second primary cancers. New cancers have happened in people during treatment with BRUKINSA, including cancers of the skin or other organs. Your healthcare provider will check you for other cancers during treatment with BRUKINSA. Use sun protection when you are outside in sunlight.
- Heart rhythm problems. Serious heart problems, including atrial fibrillation, atrial flutter, and ventricular arrhythmias, have happened in people treated with BRUKINSA and may lead to death. Your risk for heart rhythm problems may be increased if you have high blood pressure or have had heart rhythm problems in the past or have a short-term (acute) infection. Tell your healthcare provider if you develop any of the following signs or symptoms:
- your heartbeat is fast or irregular
- feel lightheaded or dizzy
- pass out (faint)
- shortness of breath
- chest discomfort
- Liver problems. Liver problems, which may be severe or life-threatening, or lead to death, can happen in people treated with BRUKINSA. Your healthcare provider will do blood tests to check your liver before and during treatment with BRUKINSA. Tell your healthcare provider or get medical help right away if you develop any signs of liver problems, including stomach pain or discomfort, dark-colored urine, or yellow skin and eyes.
The most common side effects of BRUKINSA include: - decreased white blood cell count
- decreased platelet count
- cold-like symptoms (upper respiratory tract infection)
- muscle, bone, or joint pain
These are not all the possible side effects of BRUKINSA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store BRUKINSA? - Store BRUKINSA at room temperature between 68°F and 77°F (20°C and 25°C).
- BRUKINSA comes in a bottle with a child-resistant cap.
General information about the safe and effective use of BRUKINSA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use BRUKINSA for a condition for which it was not prescribed. Do not give BRUKINSA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about BRUKINSA that is written for healthcare professionals.What are the ingredients in BRUKINSA?
Active ingredient: zanubrutinib
Capsule inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The capsule shell contains edible black ink, gelatin, and titanium dioxide.
Tablet inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate. The film-coating contains FD&C Blue No. 1, FD&C Blue No. 2, hypromellose, titanium dioxide, and triacetin.
BRUKINSA Capsules:
Manufactured for: BeOne Medicines USA, Inc., Pennington, NJ 08534
BRUKINSA Tablets:
Manufactured for: BeOne Medicines USA, Inc., Pennington, NJ 08534
Manufactured by: Catalent Pharma Solutions, LLC, Winchester, KY 40391
BRUKINSA® is a registered trademark owned by BeOne Medicines I GmbH or its affiliates.
© BeOne Medicines I GmbH, 2025. All Rights Reserved.
For more information, go to www.BRUKINSA.com or call 1-833-969-2463. - PRINCIPAL DISPLAY PANEL - 80 mg Capsule Carton
- PRINCIPAL DISPLAY PANEL - 160 mg Tablet Carton
-
INGREDIENTS AND APPEARANCE
BRUKINSA
zanubrutinib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72579-011 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZANUBRUTINIB (UNII: AG9MHG098Z) (ZANUBRUTINIB - UNII:AG9MHG098Z) ZANUBRUTINIB 80 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM LAURYL SULFATE (UNII: 368GB5141J) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color WHITE (off-white) Score no score Shape CAPSULE Size 22mm Flavor Imprint Code ZANU;80 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72579-011-02 1 in 1 CARTON 11/14/2019 1 120 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 2 NDC:72579-011-01 1 in 1 CARTON 11/14/2019 09/30/2021 2 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213217 11/14/2019 BRUKINSA
zanubrutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72579-122 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZANUBRUTINIB (UNII: AG9MHG098Z) (ZANUBRUTINIB - UNII:AG9MHG098Z) ZANUBRUTINIB 160 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE 102 (UNII: PNR0YF693Y) POVIDONE K30 (UNII: U725QWY32X) SODIUM LAURYL SULFATE (UNII: 368GB5141J) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) Product Characteristics Color blue Score 2 pieces Shape OVAL Size 16mm Flavor Imprint Code zanu Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72579-122-01 1 in 1 CARTON 08/18/2025 1 60 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218785 08/18/2025 Labeler - BeOne Medicines USA, Inc. (119367185) Establishment Name Address ID/FEI Business Operations AndersonBrecon Inc. 053217022 PACK(72579-011, 72579-122) , LABEL(72579-011, 72579-122) Establishment Name Address ID/FEI Business Operations Changzhou SynTheAll Pharmaceutical Co., Ltd 544385021 ANALYSIS(72579-011, 72579-122) , API MANUFACTURE(72579-011, 72579-122) Establishment Name Address ID/FEI Business Operations Catalent CTS, LLC 962674474 ANALYSIS(72579-011) , MANUFACTURE(72579-011) Establishment Name Address ID/FEI Business Operations BeOne Pharmaceutical (Suzhou) Co.,Ltd 543941083 ANALYSIS(72579-011, 72579-122) , MANUFACTURE(72579-011) Establishment Name Address ID/FEI Business Operations Catalent Pharma Solutions, LLC 829672745 ANALYSIS(72579-122) , MANUFACTURE(72579-122) Establishment Name Address ID/FEI Business Operations Allpack Group AG 484572565 PACK(72579-122) , LABEL(72579-122) Establishment Name Address ID/FEI Business Operations Siegfried Evionnaz SA 480093164 ANALYSIS(72579-122) , API MANUFACTURE(72579-122)