Label: HEMLIBRA- emicizumab injection, solution






-
NDC Code(s):
50242-920-01,
50242-921-01,
50242-922-01,
50242-923-01, view more50242-927-01, 50242-930-01
- Packager: Genentech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated July 11, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use HEMLIBRA safely and effectively. See full prescribing information for HEMLIBRA.
HEMLIBRA® (emicizumab-kxwh) injection, for subcutaneous use
Initial U.S. Approval: 2017WARNING: THROMBOTIC MICROANGIOPATHY and THROMBOEMBOLISM
See full prescribing information for complete boxed warning.
Cases of thrombotic microangiopathy and thrombotic events were reported when on average a cumulative amount of >100 U/kg/24 hours of activated prothrombin complex concentrate (aPCC) was administered for 24 hours or more to patients receiving HEMLIBRA prophylaxis. Monitor for the development of thrombotic microangiopathy and thrombotic events if aPCC is administered. Discontinue aPCC and suspend dosing of HEMLIBRA if symptoms occur.
INDICATIONS AND USAGE
HEMLIBRA is a bispecific factor IXa- and factor X-directed antibody indicated for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients ages newborn and older with hemophilia A (congenital factor VIII deficiency) with or without factor VIII inhibitors. (1)
DOSAGE AND ADMINISTRATION
Recommended loading dose is 3 mg/kg by subcutaneous injection once weekly for the first 4 weeks, followed by a maintenance dose of:
- 1.5 mg/kg once every week, or
- 3 mg/kg once every two weeks, or
- 6 mg/kg once every four weeks. (2.1)
See Full Prescribing Information for important preparation and administration instructions. (2.2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Immunogenicity: Anti-emicizumab antibodies (including neutralizing antibodies) have developed in HEMLIBRA-treated patients.
- In case of clinical signs of loss of efficacy, promptly assess the etiology and consider a change in treatment if neutralizing antibodies are suspected. (5.3, 12.6, 14.3)
- Laboratory Coagulation Test Interference: HEMLIBRA interferes with activated clotting time (ACT), activated partial thromboplastin time (aPTT), and coagulation laboratory tests based on aPTT, including one-stage aPTT-based single-factor assays, aPTT-based Activated Protein C Resistance (APC-R), and Bethesda assays (clotting-based) for factor VIII (FVIII) inhibitor titers. Intrinsic pathway clotting-based laboratory tests should not be used. (5.4, 7.2)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 10%) are injection site reactions, headache, and arthralgia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: THROMBOTIC MICROANGIOPATHY AND THROMBOEMBOLISM
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thrombotic Microangiopathy Associated with HEMLIBRA and aPCC
5.2 Thromboembolism Associated with HEMLIBRA and aPCC
5.3 Immunogenicity
5.4 Laboratory Coagulation Test Interference
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Hypercoagulability with Concomitant Use of aPCC
7.2 Drug-Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Hemophilia A without FVIII Inhibitors
14.2 Hemophilia A with FVIII Inhibitors
14.3 Clinical Impact of Immunogenicity
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: THROMBOTIC MICROANGIOPATHY AND THROMBOEMBOLISM
Cases of thrombotic microangiopathy and thrombotic events were reported when on average a cumulative amount of >100 U/kg/24 hours of activated prothrombin complex concentrate was administered for 24 hours or more to patients receiving HEMLIBRA prophylaxis. Monitor for the development of thrombotic microangiopathy and thrombotic events if aPCC is administered. Discontinue aPCC and suspend dosing of HEMLIBRA if symptoms occur.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
For subcutaneous use only.
The recommended loading dose is 3 mg/kg by subcutaneous injection once weekly for the first 4 weeks, followed by a maintenance dose of:
- 1.5 mg/kg once every week, or
- 3 mg/kg once every two weeks, or
- 6 mg/kg once every four weeks.
The selection of a maintenance dose should be based on healthcare provider preference with consideration of regimens that may increase patient adherence.
Discontinue the prophylactic use of bypassing agents the day before starting HEMLIBRA prophylaxis.
The prophylactic use of factor VIII (FVIII) products may be continued during the first week of HEMLIBRA prophylaxis.
2.2 Preparation and Administration
HEMLIBRA is intended for use under the guidance of a healthcare provider. After proper training in subcutaneous injection technique, a patient may self-inject, or the patient's caregiver may administer HEMLIBRA, if a healthcare provider determines that it is appropriate. Self-administration is not recommended for children less than 7 years of age. The HEMLIBRA "Instructions for Use" contains more detailed instructions on the preparation and administration of HEMLIBRA [see Instructions for Use].
- Visually inspect HEMLIBRA for particulate matter and discoloration before administration. HEMLIBRA for subcutaneous administration is a colorless to slightly yellow solution. Do not use if particulate matter is visible or product is discolored.
- A syringe, a transfer needle with filter and an injection needle are needed to withdraw HEMLIBRA solution from the vial and inject it subcutaneously.
- Refer to the HEMLIBRA "Instructions for Use" for handling instructions when combining vials. Do not combine HEMLIBRA vials of different concentrations (i.e. 30 mg/mL and 150 mg/mL) in a single injection.
Please see below the selection criteria for the recommended device options:
- Administer doses of HEMLIBRA up to 1 mL with a 1 mL syringe. A 1 mL syringe fulfilling the following criteria may be used: Transparent polypropylene or polycarbonate syringe with Luer-Lock tip, graduation 0.01 mL, sterile, for injection only, single-use, latex-free and non-pyrogenic, commercially available in the US.
- Administer doses of HEMLIBRA greater than 1 mL and up to 2 mL with a 2 mL or 3 mL syringe. A 2 mL or 3 mL syringe fulfilling the following criteria may be used: Transparent polypropylene or polycarbonate syringe with Luer-Lock tip, graduation 0.1 mL, sterile, for injection only, single-use, latex-free, and non-pyrogenic, commercially available in the US.
- A transfer needle with a filter fulfilling the following criteria should be used: Stainless steel needle with Luer-Lock connection, sterile, 18 gauge, length 1 to 1½ inch, single bevel or semi-blunted tip, single-use, latex-free, containing a 5-micron filter and non-pyrogenic, commercially available in the US.
- An injection needle fulfilling the following criteria may be used: Stainless steel with Luer-Lock connection, sterile, 26 gauge (acceptable range: 25 – 27 gauge), length preferably ⅜ inch or maximal length ½ inch, single-use, latex-free and non-pyrogenic, including needle safety feature, commercially available in the US.
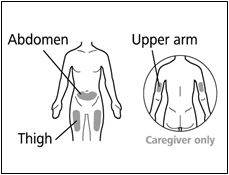
- Administer each injection at a different anatomic location (upper outer arms, thighs, or any quadrant of abdomen) than the previous injection. An injection should never be given into moles, scars, or areas where the skin is tender, bruised, red, hard, or not intact. Administration of HEMLIBRA in the upper outer arm should only be performed by a caregiver or healthcare provider.
- Discard any unused HEMLIBRA remaining in the single-dose vial.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Thrombotic Microangiopathy Associated with HEMLIBRA and aPCC
Cases of thrombotic microangiopathy (TMA) were reported from clinical trials when on average a cumulative amount of >100 U/kg/24 hours of activated prothrombin complex concentrate (aPCC) was administered for 24 hours or more to patients receiving HEMLIBRA prophylaxis. In clinical trials, thrombotic microangiopathy was reported in 0.8% of patients (3/391) and in 8.1% of patients (3/37) who received at least one dose of aPCC. Patients presented with thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney injury, without severe deficiencies in ADAMTS13 activity.
Evidence of improvement was seen within one week following discontinuation of aPCC. One patient resumed HEMLIBRA following resolution of TMA.
Consider the benefits and risks if aPCC must be used in a patient receiving HEMLIBRA prophylaxis. Due to the long half-life of HEMLIBRA, the potential for an interaction with aPCC may persist for up to 6 months after the last dose. Monitor for the development of TMA when administering aPCC. Immediately discontinue aPCC and interrupt HEMLIBRA prophylaxis if clinical symptoms and/or laboratory findings consistent with TMA occur, and manage as clinically indicated. Consider the benefits and risks of resuming HEMLIBRA prophylaxis following complete resolution of TMA on a case-by-case basis.
5.2 Thromboembolism Associated with HEMLIBRA and aPCC
Thrombotic events were reported from clinical trials when on average a cumulative amount of >100 U/kg/24 hours of aPCC was administered for 24 hours or more to patients receiving HEMLIBRA prophylaxis. In clinical trials, thrombotic events were reported in 0.5% of patients (2/391) and in 5.4% of patients (2/37) who received at least one dose of aPCC.
No thrombotic event required anticoagulation therapy. Evidence of improvement or resolution was seen within one month following discontinuation of aPCC. One patient resumed HEMLIBRA following resolution of thrombotic event.
Consider the benefits and risks if aPCC must be used in a patient receiving HEMLIBRA prophylaxis. Due to the long half-life of HEMLIBRA, the potential for an interaction with aPCC may persist for up to 6 months after the last dose. Monitor for the development of thromboembolism when administering aPCC. Immediately discontinue aPCC and interrupt HEMLIBRA prophylaxis if clinical symptoms, imaging, or laboratory findings consistent with thromboembolism occur, and manage as clinically indicated. Consider the benefits and risks of resuming HEMLIBRA prophylaxis following complete resolution of thrombotic events on a case-by-case basis.
5.3 Immunogenicity
Treatment with HEMLIBRA may induce anti-drug antibodies. Anti-emicizumab-kxwh antibodies were reported in 5.1% of patients (34/668) treated with HEMLIBRA in clinical trials. Most patients with anti-emicizumab-kxwh antibodies did not experience a change in HEMLIBRA plasma concentrations or an increase in bleeding events; however, in uncommon cases (incidence < 1%), the presence of neutralizing antibodies with decreasing plasma concentration may be associated with loss of efficacy [see Clinical Pharmacology (12.6), Clinical Studies (14.3)].
Monitor for clinical signs of loss of efficacy (e.g., increase in breakthrough bleeding events) and if observed, promptly assess the etiology and consider a change in treatment if neutralizing anti-emicizumab-kxwh antibodies are suspected.
5.4 Laboratory Coagulation Test Interference
HEMLIBRA affects intrinsic pathway clotting-based laboratory tests, including activated clotting time (ACT), activated partial thromboplastin time (aPTT), and all assays based on aPTT, such as one-stage factor VIII (FVIII) activity (Table 1). Therefore, intrinsic pathway clotting-based laboratory test results in patients treated with HEMLIBRA should not be used to monitor HEMLIBRA activity, determine dosing for factor replacement or anti-coagulation, or measure FVIII inhibitor titers [see Drug Interactions (7.2)]. Laboratory tests affected and unaffected by HEMLIBRA are shown in Table 1.
Table 1 Coagulation Test Results Affected and Unaffected by HEMLIBRA Results Affected by HEMLIBRA Results Unaffected by HEMLIBRA - *
- For important considerations regarding FVIII chromogenic activity assays, see Drug Interactions (7.2).
Activated partial thromboplastin time (aPTT)
Bethesda assays (clotting-based) for FVIII inhibitor titers
One-stage, aPTT-based, single-factor assays
aPTT-based Activated Protein C Resistance (APC-R)
Activated clotting time (ACT)Bethesda assays (bovine chromogenic) for FVIII inhibitor titers
Thrombin time (TT)
One-stage, prothrombin time (PT)-based, single-factor assays
Chromogenic-based single-factor assays other than FVIII*
Immuno-based assays (i.e., ELISA, turbidimetric methods)
Genetic tests of coagulation factors (e.g., Factor V Leiden, Prothrombin 20210) -
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Thrombotic Microangiopathy Associated with HEMLIBRA and aPCC [see Warnings and Precautions (5.1)]
- Thromboembolism Associated with HEMLIBRA and aPCC [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The following adverse reactions are based on pooled data from two randomized trials in adult and adolescent patients (HAVEN 1 and HAVEN 3), one single-arm trial in adult and adolescent patients (HAVEN 4), one single-arm trial in pediatric patients (HAVEN 2), and one dose-finding trial, in which a total of 391 male patients with hemophilia A received at least one dose of HEMLIBRA as routine prophylaxis. Two hundred eighty-one patients (72%) were adults (18 years and older), 50 (13%) were adolescents (12 years up to less than 18 years), 55 (14%) were children (2 years up to less than 12 years), and five (1%) were infants (1 month up to less than 2 years). The median duration of exposure across the studies was 34.1 weeks (0.1 to 224.4 weeks).
The most frequently reported adverse reactions observed in ≥ 10% of patients treated with HEMLIBRA were injection site reactions, headache, and arthralgia.
Four patients (1%) in the clinical trials receiving HEMLIBRA prophylaxis withdrew from treatment due to adverse reactions, which were thrombotic microangiopathy, skin necrosis and superficial thrombophlebitis, headache, and injection site reaction.
Adverse reactions observed in patients who received HEMLIBRA are shown in Table 2.
Table 2 Adverse Reactions Reported in ≥ 5% of Patients from Pooled Clinical Trials with HEMLIBRA Body System Adverse Reaction Number of Patients
n (%)
(N = 391)- *
- Includes injection site bruising, injection site discomfort, injection site erythema, injection site hematoma, injection site induration, injection site pain, injection site pruritus, injection site rash, injection site reaction, injection site swelling, injection site urticaria, and injection site warmth.
General Disorders and Administration Site Conditions Injection site reaction* 85 (22%) Pyrexia 23 (6%) Nervous System Disorders Headache 57 (15%) Gastrointestinal Disorders Diarrhea 22 (6%) Musculoskeletal and Connective Tissue Disorders Arthralgia 59 (15%) Characterization of aPCC treatment in pooled clinical trials
There were 130 instances of aPCC treatment in 37 patients, of which 13 instances (10%) consisted of on average a cumulative amount of >100 U/kg/24 hours of aPCC for 24 hours or more; two of the 13 were associated with thrombotic events and three of the 13 were associated with TMA (Table 3). No TMA or thrombotic events were associated with the remaining instances of aPCC treatment.
Injection Site Reactions
In total, 85 patients (22%) reported injection site reactions (ISRs). All ISRs observed in HEMLIBRA clinical trials were reported as mild to moderate intensity and 93% resolved without treatment. The commonly reported ISR symptoms were injection site erythema (11%), injection site pain (4%), and injection site pruritus (4%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of HEMLIBRA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and subcutaneous tissue disorders: rash, urticaria, angioedema.
Immune system disorders: hypersensitivity.
-
7 DRUG INTERACTIONS
7.1 Hypercoagulability with Concomitant Use of aPCC
Clinical experience suggests that a drug interaction exists with HEMLIBRA and aPCC [see Warnings and Precautions (5.1, 5.2)].
7.2 Drug-Laboratory Test Interactions
HEMLIBRA restores the tenase cofactor activity of missing activated factor VIII (FVIIIa). Coagulation laboratory tests based on intrinsic clotting (i.e., aPTT) measure the total clotting time including time needed for activation of FVIII to FVIIIa by thrombin. Such intrinsic pathway-based tests will yield overly shortened clotting times with HEMLIBRA, which does not require activation by thrombin. The overly shortened intrinsic clotting time will then disturb all single-factor assays based on aPTT, such as the one-stage FVIII activity assay; however, single-factor assays utilizing chromogenic or immuno-based methods are unaffected by HEMLIBRA and may be used to monitor coagulation parameters during treatment, with specific considerations for FVIII chromogenic activity assays as described below.
Chromogenic FVIII activity tests may be manufactured with either human or bovine coagulation proteins. Assays containing human coagulation factors are responsive to HEMLIBRA but may overestimate the clinical hemostatic potential of HEMLIBRA. In contrast, assays containing bovine coagulation factors are insensitive to HEMLIBRA (no activity measured) and can be used to monitor endogenous or infused FVIII activity, or to measure anti-FVIII inhibitors.
HEMLIBRA remains active in the presence of inhibitors against FVIII, so it will produce a false-negative result in clotting-based Bethesda assays for functional inhibition of FVIII. Instead, a chromogenic Bethesda assay utilizing a bovine-based FVIII chromogenic test that is insensitive to HEMLIBRA may be used.
Due to the long half-life of HEMLIBRA, effects on coagulation assays may persist for up to 6 months after the last dose [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on HEMLIBRA use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. Animal reproduction studies have not been conducted with emicizumab-kxwh. It is not known whether HEMLIBRA can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. HEMLIBRA should be used during pregnancy only if the potential benefit for the mother outweighs the risk to the fetus.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2 – 4% and 15 – 20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of emicizumab-kxwh in human milk, the effects on the breastfed child, or the effects on milk production. Human IgG is known to be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for HEMLIBRA and any potential adverse effects on the breastfed child from HEMLIBRA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of HEMLIBRA have been established in pediatric patients. Use of HEMLIBRA in pediatric patients with hemophilia A is supported by two randomized trials (HAVEN 1 and HAVEN 3) and two single-arm trials (HAVEN 2 and HAVEN 4). All clinical trials included pediatric patients in the following age group: 47 adolescents (12 years up to less than 18 years). Only HAVEN 2 included pediatric patients in the following age groups: 55 children (2 years up to less than 12 years) and five infants (1 month up to less than 2 years). No differences in efficacy were observed between the different age groups [see Clinical Studies (14)].
The steady-state plasma trough concentrations of emicizumab-kxwh were comparable in adult and pediatric patients older than 6 months at equivalent weight-based doses. Lower concentrations of emicizumab-kxwh were predicted in pediatric patients less than 6 months old [see Clinical Pharmacology (12.3)].
In general, the adverse reactions in HEMLIBRA-treated pediatric patients were similar in type to those seen in adult patients with hemophilia A [see Adverse Reactions (6.1)].
8.5 Geriatric Use
Clinical studies of HEMLIBRA did not include a sufficient number of patients aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
-
11 DESCRIPTION
Emicizumab-kxwh is a humanized monoclonal modified immunoglobulin G4 (IgG4) bispecific antibody binding factor IXa and factor X. Emicizumab-kxwh has an approximate molecular weight of 145.6 kDa and is produced in genetically engineered mammalian (Chinese hamster ovary) cells. Emicizumab-kxwh has no structural relationship or sequence homology to FVIII and, as such, does not induce or enhance the development of direct inhibitors to FVIII.
HEMLIBRA (emicizumab-kxwh) injection is a sterile, preservative-free, colorless to slightly yellow solution for subcutaneous injection supplied in single-dose vials containing emicizumab-kxwh at 12 mg/0.4 mL, 30 mg/mL, 60 mg/0.4 mL, 105 mg/0.7 mL, 150 mg/mL, or 300 mg/2 mL.
Each single-dose 12 mg vial contains a 0.4 mL solution of emicizumab-kxwh (12 mg), L-arginine (10.5 mg), L-histidine (1.2 mg), and poloxamer 188 (0.2 mg), adjusted to pH 6.0 with L-aspartic acid.
Each single-dose 30 mg vial contains a 1 mL solution of emicizumab-kxwh (30 mg), L-arginine (26.1 mg), L-histidine (3.1 mg), and poloxamer 188 (0.5 mg), adjusted to pH 6.0 with L-aspartic acid.
Each single-dose 60 mg vial contains a 0.4 mL solution of emicizumab-kxwh (60 mg), L-arginine (10.5 mg), L-histidine (1.2 mg), and poloxamer 188 (0.2 mg), adjusted to pH 6.0 with L-aspartic acid.
Each single-dose 105 mg vial contains a 0.7 mL solution of emicizumab-kxwh (105 mg), L-arginine (18.3 mg), L-histidine (2.2 mg), and poloxamer 188 (0.4 mg), adjusted to pH 6.0 with L-aspartic acid.
Each single-dose 150 mg vial contains a 1 mL solution of emicizumab-kxwh (150 mg), L-arginine (26.1 mg), L-histidine (3.1 mg), and poloxamer 188 (0.5 mg), adjusted to pH 6.0 with L-aspartic acid.
Each single-dose 300 mg vial contains a 2 mL solution of emicizumab-kxwh (300 mg), L-arginine (52.3 mg), L-histidine (6.2 mg), and poloxamer 188 (1 mg), adjusted to pH 6.0 with L-aspartic acid.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
HEMLIBRA bridges activated factor IX and factor X to restore the function of missing activated factor VIII that is needed for effective hemostasis.
12.3 Pharmacokinetics
Emicizumab-kxwh exhibited dose-proportional pharmacokinetics over a dose range of 0.3 mg/kg (0.1 times approved recommended starting dosage) to 6 mg/kg following subcutaneous administration. Following multiple subcutaneous administrations of a loading dose of 3 mg/kg emicizumab-kxwh once weekly for the first 4 weeks in hemophilia A patients, mean (± SD) trough plasma concentrations of 52.6 ± 13.6 μg/mL was achieved at Week 5. Sustained mean (± SD) plasma concentrations of emicizumab-kxwh at steady-state with the recommended maintenance doses are shown in Table 4.
Table 4 Mean (± SD) Steady-State Concentrations after emicizumab-kxwh Loading Dose by Maintenance Dose Regimen Maintenance Dose Parameters 1.5 mg/kg once every week 3 mg/kg once every two weeks 6 mg/kg once every four weeks AUCss,τ = area under the concentration time curve at steady-state over the dosing interval (τ = 1, 2, or 4 weeks); Cmax, ss = maximum plasma concentration at steady state; Ctrough, ss = trough concentration at steady state. Cmax, ss (µg/mL) 55.1 ± 15.9 58.3 ± 16.4 67 ± 17.7 AUCss,τ (µg/mL*day) 376 ± 109 752 ± 218 1503 ± 437 Ctrough, ss (µg/mL) 51.2± 15.2 46.9 ± 14.8 38.5 ± 14.2 Cmax/ Ctrough ratio (µg/mL) 1.08 ± 0.03 1.26 ± 0.12 1.85 ± 0.47 Absorption
Following subcutaneous administration, the mean (± SD) absorption half-life was 1.6 ± 1 day.
The absolute bioavailability following subcutaneous administration of 1 mg/kg was between 80.4% and 93.1%. Similar pharmacokinetic profiles were observed following subcutaneous administration in the abdomen, upper arm, and thigh [see Dosage and Administration (2.2)].
Distribution
The mean apparent volume of distribution (% coefficient of variation [%CV]) was 10.4 L (26.0%).
Elimination
The mean apparent clearance (%CV) was 0.27 L/day (28.4%) and the mean elimination apparent half-life (± SD) was 26.9 ± 9.1 days.
Specific Populations
The pharmacokinetics of emicizumab-kxwh are not influenced by age (1 year to 77 years), race (White 62.7%, Asian 22.9%, and Black 8%), inhibitor status (inhibitor present, 50%), mild hepatic impairment (defined as total bilirubin 1× to ≤1.5× the upper limit of normal (ULN) and any aspartate transaminase (AST) level), moderate hepatic impairment (defined as total bilirubin 1.5× to ≤3× the ULN and any AST level), mild renal impairment (defined as creatinine clearance (CrCl) of 60 – 89 mL/min), and moderate renal impairment (defined as CrCl of 30 – 59 mL/min). Emicizumab-kxwh has not been studied in patients with severe hepatic or renal impairment.
In pediatric patients less than 6 months old, the predicted concentrations of emicizumab-kxwh were 19% to 33% lower than the older patients, especially with the 3 mg/kg once every two weeks or 6 mg/kg once every four weeks maintenance dose.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of HEMLIBRA or of other emicizumab products.
In the clinical trials with HEMLIBRA, 5.1% of patients (34/668) tested positive for anti-emicizumab-kxwh antibodies. Participants were exposed to emicizumab-kxwh for a median interquartile range (IQR) of 103.1 (82.4 – 148.1) weeks. Samples testing for anti-drug antibodies were obtained at baseline and at trough periodically throughout the studies duration.
Anti-Drug Antibody Effects on Pharmacokinetics
Antibody positive samples were further evaluated for neutralizing anti-emicizumab-kxwh antibodies using a modified FVIII chromogenic assay. A total of 668 patients were tested for anti-emicizumab-kxwh antibodies. 5.1% of patients (34/668) tested positive for anti-emicizumab-kxwh antibodies and 2.7% of patients (18/668) developed anti-emicizumab-kxwh antibodies that were neutralizing in vitro. Of these, 2.7% of patients (18/668), the neutralizing anti-emicizumab-kxwh antibodies did not have a clinically meaningful impact on the pharmacokinetics of HEMLIBRA in 2.1% of patients (14/668), while decreased emicizumab-kxwh plasma concentrations were observed in 0.6% of patients (4/668).
Anti-Drug Antibody Effects on Pharmacodynamics
One patient (1/668; 0.1%) developed neutralizing anti-emicizumab-kxwh antibodies and had decreased emicizumab-kxwh plasma concentrations, and experienced loss of efficacy (manifest as breakthrough bleeding) after 5 weeks of treatment and discontinued HEMLIBRA treatment. [see Warnings and Precautions (5.3), Clinical Studies (14.3)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Studies in animals investigating the carcinogenic effects of emicizumab-kxwh have not been conducted. In vitro and in vivo testing of emicizumab-kxwh for genotoxicity was not conducted.
Animal fertility studies have not been conducted; however, emicizumab-kxwh did not cause any toxicological changes in the reproductive organs of male or female cynomolgus monkeys at doses of up to 30 mg/kg/week in subcutaneous general toxicity studies of up to 26-week duration and at doses of up to 100 mg/kg/week in a 4-week intravenous general toxicity study.
-
14 CLINICAL STUDIES
14.1 Hemophilia A without FVIII Inhibitors
The efficacy of HEMLIBRA for routine prophylaxis in patients with hemophilia A without FVIII inhibitors was evaluated in two clinical trials [adult and adolescent studies (HAVEN 3 and HAVEN 4)].
HAVEN 3 (Adult and Adolescent Patients)
The HAVEN 3 study (NCT02847637) was a randomized, multicenter, open-label, clinical trial in 152 adult and adolescent males (aged ≥ 12 years and ≥ 40 kg) with hemophilia A without FVIII inhibitors who previously received either episodic (on demand) or prophylactic treatment with FVIII. Patients received HEMLIBRA prophylaxis, 3 mg/kg once weekly for the first 4 weeks followed by either 1.5 mg/kg once every week [Arms A and D] or 3 mg/kg once every two weeks [Arm B] thereafter, or no prophylaxis (Arm C). Patients in Arm C could switch to HEMLIBRA prophylaxis (3 mg/kg once every two weeks) after completing at least 24 weeks without prophylaxis. For Arms A and B, dose up-titration to 3 mg/kg once every week was allowed after 24 weeks on HEMLIBRA prophylaxis for patients who experienced two or more qualified bleeds (i.e., spontaneous and clinically significant bleeds occurring at steady state). For Arm D patients, dose up-titration was allowed after the second qualifying bleed. During the study, five patients underwent up-titration of their maintenance dose; however, this study was not designed to investigate the 3 mg/kg once every week dosing regimen.
Eighty-nine patients previously treated with episodic (on demand) FVIII were randomized in a 2:2:1 ratio to receive HEMLIBRA prophylaxis 1.5 mg/kg once every week (Arm A), 3 mg/kg once every two weeks (Arm B), or no prophylaxis (Arm C), with stratification by prior 24-week bleed rate (< 9 or ≥ 9). Sixty-three patients previously treated with prophylactic FVIII were enrolled into Arm D to receive HEMLIBRA prophylaxis (1.5 mg/kg once every week).
Efficacy was evaluated after a minimum of 24 weeks of follow-up based on the bleed rate for bleeds requiring treatment with coagulation factors among patients previously treated with episodic (on-demand) FVIII who were randomized to HEMLIBRA prophylaxis 1.5 mg/kg once every week (Arm A) or 3 mg/kg once every two weeks (Arm B) compared with those receiving no prophylaxis (Arm C). The study also evaluated the randomized comparison of Arms A and C and Arms B and C for the efficacy of HEMLIBRA prophylaxis in reducing the number of all bleeds, spontaneous bleeds, joint bleeds, and target joint bleeds.
The efficacy of HEMLIBRA prophylaxis compared with previous prophylactic FVIII was also evaluated in patients who had participated in a non-interventional study (NIS) prior to enrollment (Arm D). Only patients from the NIS were included in this comparison, because bleed and treatment data were collected with the same level of granularity as that used in HAVEN 3.
The efficacy results of HEMLIBRA prophylaxis (1.5 mg/kg once every week and 3 mg/kg once every two weeks) compared with no prophylaxis with respect to rate of treated bleeds, all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds are shown in Table 5.
Table 5 Annualized Bleed Rate with HEMLIBRA Prophylaxis versus No Prophylaxis in Patients ≥ 12 Years of Age without Factor VIII Inhibitors Endpoint HEMLIBRA
1.5 mg/kg once every week (N = 36)HEMLIBRA
3 mg/kg once every two weeks (N = 35)No Prophylaxis
(N = 18)ABR = annualized bleed rate; CI = confidence interval; IQR = interquartile range, 25th percentile to 75th percentile. - *
- Based on negative binomial regression model.
Treated Bleeds ABR (95% CI) * 1.5 (0.9, 2.5) 1.3 (0.8, 2.3) 38.2 (22.9, 63.8) % reduction (95% CI)
p-value96% (92.5%, 98%)
< 0.000197% (93.4%, 98.3%)
< 0.0001- % patients with 0 bleeds (95% CI) 55.6 (38.1, 72.1) 60 (42.1, 76.1) 0 (0, 18.5) Median ABR (IQR) 0 (0, 2.5) 0 (0, 1.9) 40.4 (25.3, 56.7) All Bleeds ABR (95% CI) * 2.5 (1.6, 3.9) 2.6 (1.6, 4.3) 47.6 (28.5, 79.6) % reduction (95% CI)
p-value95% (90.1%, 97%)
< 0.000194% (89.7%, 97%)
< 0.0001- % patients with 0 bleeds (95% CI) 50 (32.9, 67.1) 40 (23.9, 57.9) 0 (0, 18.5) Median ABR (IQR) 0.6 (0, 3.9) 1.6 (0, 4) 46.9 (26.1, 73.9) Treated Spontaneous Bleeds ABR (95% CI) * 1.0 (0.5, 1.9) 0.3 (0.1, 0.8) 15.6 (7.6, 31.9) % reduction (95% CI)
p-value94% (84.9%, 97.5%)
< 0.000198% (94.4%, 99.4%)
< 0.0001- % patients with 0 bleeds (95% CI) 66.7 (49.0, 81.4) 88.6 (73.3, 96.8) 22.2 (6.4, 47.6) Median ABR (IQR) 0 (0, 1.3) 0 (0, 0) 10.8 (2.1, 26) Treated Joint Bleeds ABR (95% CI) * 1.1 (0.6, 1.9) 0.9 (0.4, 1.7) 26.5 (14.7, 47.8) % reduction (95% CI)
p-value96% (91.5%, 98.1%)
< 0.000197% (93%, 98.5%)
< 0.0001- % patients with 0 bleeds (95% CI) 58.3 (40.8, 74.5) 74.3 (56.7, 87.5) 0 (0, 18.5) Median ABR (IQR) 0 (0, 1.9) 0 (0, 1.3) 21.3 (14.5, 41.3) Treated Target Joint Bleeds ABR (95% CI) * 0.6 (0.3, 1.4) 0.7 (0.3, 1.6) 13 (5.2, 32.3) % reduction (95% CI)
p-value95% (85.7%, 98.4%)
< 0.000195% (85.3%, 98.2%)
< 0.0001- % patients with 0 bleeds (95% CI) 69.4 (51.9, 83.7) 77.1 (59.9, 89.6) 27.8 (9.7, 53.5) Median ABR (IQR) 0 (0, 1.4) 0 (0, 0) 12.8 (0, 39.1) In the HAVEN 3 intra-patient analysis, HEMLIBRA prophylaxis resulted in a statistically significant (p < 0.0001) reduction (68%) in bleed rate for treated bleeds compared with previous FVIII prophylaxis collected in the NIS prior to enrollment (see Table 6).
Table 6 Intra-Patient Comparison of Annualized Bleed Rate with HEMLIBRA Prophylaxis versus Previous FVIII Prophylaxis Endpoint HEMLIBRA
1.5 mg/kg once every week
(N = 48)Previous FVIII Prophylaxis
(N = 48)ABR = annualized bleed rate; CI = confidence interval; IQR = interquartile range, 25th percentile to 75th percentile. - *
- Based on negative binomial regression model.
Median Observation Period (weeks) 33.7 30.1 Treated Bleeds ABR (95% CI) * 1.5 (1, 2.3) 4.8 (3.2, 7.1) % reduction (95% CI)
p-value68% (48.6%, 80.5%)
< 0.0001% patients with 0 bleeds (95% CI) 54.2 (39.2, 68.6) 39.6 (25.8, 54.7) Median ABR (IQR) 0 (0, 2.1) 1.8 (0, 7.6) HAVEN 4 (Adult and Adolescent Patients)
The HAVEN 4 study (NCT03020160) was a single-arm, multicenter, open-label, clinical trial in 41 adult and adolescent males (aged ≥ 12 years and ≥ 40 kg) with hemophilia A with or without FVIII inhibitors who previously received either episodic (on demand) or prophylactic treatment with FVIII or bypassing agents. Patients received HEMLIBRA prophylaxis at 3 mg/kg once weekly for the first 4 weeks followed by 6 mg/kg once every four weeks thereafter.
Efficacy was evaluated in a subgroup of 36 patients with hemophilia A without FVIII inhibitors based on the bleed rate for bleeds requiring treatment with coagulation factors. The study also evaluated the efficacy of HEMLIBRA prophylaxis on all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds.
The efficacy results of HEMLIBRA prophylaxis 6 mg/kg once every four weeks with respect to rate of treated bleeds, all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds are shown in Table 7. The median observation time was 25.6 weeks (range 24.1 – 29.4 weeks).
Table 7 Annualized Bleed Rate with HEMLIBRA Prophylaxis 6 mg/kg Once Every Four Weeks in Patients ≥ 12 Years of Age without Factor VIII Inhibitors Endpoint ABR* (95% CI)
N = 36Median ABR (IQR)
N = 36% Zero Bleeds (95% CI)
N = 36ABR = annualized bleed rate; CI = confidence interval; IQR = interquartile range, 25th percentile to 75th percentile. - *
- Based on negative binomial regression model.
Treated Bleeds 2.6 (1.5, 4.7) 0 (0, 2.1) 52.8 (35.5, 69.6) All Bleeds 4.8 (3.2, 7.1) 2.1 (0, 6.1) 27.8 (14.2, 45.2) Treated Spontaneous Bleeds 0.6 (0.2, 1.6) 0 (0, 0) 83.3 (67.2, 93.6) Treated Joint Bleeds 1.8 (0.8, 4) 0 (0, 1.9) 69.4 (51.9, 83.7) Treated Target Joint Bleeds 1.1 (0.4, 3.7) 0 (0, 0) 83.3 (67.2, 93.6) 14.2 Hemophilia A with FVIII Inhibitors
The efficacy of HEMLIBRA for routine prophylaxis in patients with hemophilia A with FVIII inhibitors was evaluated in three clinical trials [adult and adolescent studies (HAVEN 1 and HAVEN 4) and a pediatric study (HAVEN 2)].
HAVEN 1 (Adult and Adolescent Patients)
The HAVEN 1 study (NCT02622321) was a randomized, multicenter, open-label, clinical trial in 109 adult and adolescent males (aged ≥ 12 years and ≥ 40 kg) with hemophilia A with FVIII inhibitors who previously received either episodic (on-demand) or prophylactic treatment with bypassing agents. Patients received HEMLIBRA prophylaxis (Arms A, C, and D), 3 mg/kg once weekly for the first 4 weeks followed by 1.5 mg/kg once every week thereafter, or no prophylaxis (Arm B). Patients in Arm B could switch to HEMLIBRA prophylaxis after completing at least 24 weeks without prophylaxis. Dose up-titration to 3 mg/kg once every week was allowed after 24 weeks on HEMLIBRA prophylaxis for patients who experienced two or more qualified bleeds (i.e., spontaneous and clinically significant bleeds occurring at steady state). During the study, two patients underwent up-titration of their maintenance dose; however, this study was not designed to investigate the 3 mg/kg once every week dosing regimen.
Fifty-three patients previously treated with episodic (on-demand) bypassing agents were randomized in a 2:1 ratio to receive HEMLIBRA prophylaxis (Arm A) or no prophylaxis (Arm B), with stratification by prior 24-week bleed rate (< 9 or ≥ 9). Forty-nine patients previously treated with prophylactic bypassing agents were enrolled into Arm C to receive HEMLIBRA prophylaxis. Seven patients previously treated with episodic (on-demand) bypassing agents who had participated in the NIS prior to enrollment, but were unable to enroll into HAVEN 1 prior to the closure of Arms A and B, were enrolled into Arm D to receive HEMLIBRA prophylaxis.
Efficacy was evaluated after a minimum of 24 weeks of follow-up based on the bleed rate for bleeds requiring treatment with coagulation factors among patients previously treated with episodic bypassing agents who were randomized to HEMLIBRA prophylaxis (Arm A) compared with those receiving no prophylaxis (Arm B). The study also evaluated the randomized comparison of Arms A and B for the efficacy of HEMLIBRA prophylaxis in reducing the number of all bleeds, spontaneous bleeds, joint bleeds, and target joint bleeds, as well as patient-reported symptoms and physical functioning.
The efficacy of HEMLIBRA prophylaxis compared with previous prophylactic bypassing agents was also evaluated in patients who had participated in the NIS prior to enrollment (Arm C). Only patients from the NIS were included in this comparison, because bleed and treatment data were collected with the same level of granularity as that used in HAVEN 1.
The efficacy results of HEMLIBRA prophylaxis 1.5 mg/kg once every week compared with no prophylaxis with respect to rate of treated bleeds, all bleeds, treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds are shown in Table 8.
Table 8 Annualized Bleed Rate with HEMLIBRA Prophylaxis versus No Prophylaxis in Patients ≥ 12 Years of Age with Factor VIII Inhibitors Endpoint HEMLIBRA
1.5 mg/kg once every week
(N = 35)No Prophylaxis
(N = 18)ABR = annualized bleed rate; CI = confidence interval; IQR = interquartile range, 25th percentile to 75th percentile. - *
- Based on negative binomial regression model.
Treated Bleeds ABR (95% CI) * 2.9 (1.7, 5.0) 23.3 (12.3, 43.9) % reduction (95% CI)
p-value87% (72.3%, 94.3%)
< 0.0001% patients with 0 bleeds (95% CI) 62.9 (44.9, 78.5) 5.6 (0.1, 27.3) Median ABR (IQR) 0 (0, 3.7) 18.8 (13.0, 35.1) All Bleeds ABR (95% CI) * 5.5 (3.6, 8.6) 28.3 (16.8, 47.8) % reduction (95% CI)
p-value80% (62.5%, 89.8%)
< 0.0001% patients with 0 bleeds (95% CI) 37.1 (21.5, 55.1) 5.6 (0.1, 27.3) Median ABR (IQR) 2 (0, 9.9) 30.2 (18.3, 39.4) Treated Spontaneous Bleeds ABR (95% CI) * 1.3 (0.7, 2.2) 16.8 (9.9, 28.3) % reduction (95% CI)
p-value92% (84.6%, 96.3%)
< 0.0001% patients with 0 bleeds (95% CI) 68.6 (50.7, 83.1) 11.1 (1.4, 34.7) Median ABR (IQR) 0 (0, 3.3) 15.2 (6.6, 30.4) Treated Joint Bleeds ABR (95% CI) * 0.8 (0.3, 2.2) 6.7 (2.0, 22.4) % reduction (95% CI)
p-value89% (48%, 97.5%)
0.0050% patients with 0 bleeds (95% CI) 85.7 (69.7, 95.2) 50.0 (26.0, 74.0) Median ABR (IQR) 0 (0, 0) 1 (0, 14.4) Treated Target Joint Bleeds ABR (95% CI) * 0.1 (0.03, 0.6) 3.0 (1.0, 9.1) % reduction (95% CI)
p-value95% (77.3%, 99.1%)
0.0002% patients with 0 bleeds (95% CI) 94.3 (80.8, 99.3) 50.0 (26.0, 74.0) Median ABR (IQR) 0 (0, 0) 1 (0, 6.5) Descriptive analyses were conducted to assess HEMLIBRA prophylaxis once every week using 12-week treatment intervals up to Week 72. The descriptive mean ABRs for treated bleeds are shown in Table 9.
Table 9 Annualized Bleed Rate with HEMLIBRA Prophylaxis Once Every Week per 12-Week Intervals in Patients ≥ 12 Years of Age with Factor VIII Inhibitors Endpoint Time Interval (Weeks) 1 – 12
(N = 109)13 – 24
(N = 108)25 – 36
(N = 93)37 – 48
(N = 93)49 – 60
(N = 57)61 – 72
(N = 42)ABR = annualized bleed rate; CI = confidence interval based on Poisson distribution; N = number of patients who contributed data for analyses at each time interval. Treated Bleeds Mean ABR (95% CI) 3.9
(1.1, 10.2)2.2
(0.3, 7.6)0.9
(0, 5.5)0.4
(0, 4.4)0.5
(0, 4.7)0.6
(0, 4.9)In the HAVEN 1 intra-patient analysis, HEMLIBRA prophylaxis resulted in a statistically significant (p = 0.0003) reduction (79%) in bleed rate for treated bleeds compared with previous bypassing agent prophylaxis collected in the NIS prior to enrollment (Table 10).
Table 10 Intra-Patient Comparison of Annualized Bleed Rate with HEMLIBRA Prophylaxis versus Previous Bypassing Agent Prophylaxis Endpoint HEMLIBRA
1.5 mg/kg once every week
(N = 24)Previous Bypassing Agent Prophylaxis (N = 24) ABR = annualized bleed rate; CI = confidence interval; IQR = interquartile range, 25th percentile to 75th percentile. - *
- Based on negative binomial regression model.
Median Observation Period (weeks) 30.1 32.1 Treated Bleeds ABR (95% CI) * 3.3 (1.3, 8.1) 15.7 (11.1, 22.3) % reduction (95% CI)
p-value79% (51.4%, 91.1%)
0.0003% patients with 0 bleeds (95% CI) 70.8 (48.9, 87.4) 12.5 (2.7, 32.4) Median ABR (IQR) 0 (0, 2.2) 12 (5.7, 24.2) The HAVEN 1 study evaluated patient-reported hemophilia-related symptoms (painful swellings and presence of joint pain) and physical functioning (pain with movement and difficulty walking far) using the Physical Health Score of the Haemophilia-specific Quality of Life (Haem-A-QoL) questionnaire for patients ≥ 18 years of age. The HEMLIBRA prophylaxis arm (Arm A) showed an improvement compared with the no prophylaxis arm (Arm B) in the Haem-A-QoL Physical Health Subscale score at the Week 25 assessment (Table 11). The improvement in the Physical Health Score was further supported by the Total Score as measured by the Haem-A-QoL at Week 25.
Table 11 Change in Haem-A-QoL Physical Health Score with HEMLIBRA Prophylaxis versus No Prophylaxis in Patients (≥ 18 Years of Age) with Factor VIII Inhibitors at Week 25 Haem-A-QoL Scores at Week 25 HEMLIBRA
1.5 mg/kg once every week
(N = 25*)No Prophylaxis
(N = 14*)Physical Health Score (range 0 to 100)† Adjusted mean ‡ 32.6 54.2 Difference in adjusted means (95% CI) 21.6 (7.9, 35.2) p-value 0.0029 HAVEN 2 (Pediatric Patients)
The HAVEN 2 study (NCT02795767) was a single-arm, multicenter, open-label, clinical trial in pediatric males (age < 12 years, or 12 – 17 years who weigh < 40 kg) with hemophilia A with FVIII inhibitors. Patients received HEMLIBRA prophylaxis at 3 mg/kg once weekly for the first 4 weeks followed by 1.5 mg/kg once every week thereafter.
The study evaluated the efficacy of HEMLIBRA prophylaxis, including the efficacy of HEMLIBRA prophylaxis compared with previous episodic (on-demand) and prophylactic bypassing agent treatment in patients who had participated in a non-interventional study (NIS) prior to enrollment (intra-patient analysis).
At the time of the interim analysis, efficacy was evaluated in 59 pediatric patients who were < 12 years of age and had been receiving HEMLIBRA prophylaxis for at least 12 weeks, including 38 patients age 6 to < 12 years, 17 patients age 2 to < 6 years, and four patients age < 2 years.
Annualized bleed rate (ABR) and percent of patients with zero bleeds were calculated for 59 patients (Table 12). The median observation time for these patients was 29.6 weeks (range 18.4 – 63 weeks).
Table 12 Annualized Bleed Rate with HEMLIBRA Prophylaxis 1.5 mg/kg Once Every Week in Pediatric Patients < 12 Years of Age with Factor VIII Inhibitors (Interim Analysis) Endpoint ABR* (95% CI)
N = 59Median ABR (IQR)
N = 59% Zero Bleeds (95% CI)
N = 59ABR = annualized bleed rate; CI = confidence interval; IQR = interquartile range, 25th percentile to 75th percentile. - *
- Based on negative binomial regression model.
Treated Bleeds 0.3 (0.1, 0.5) 0 (0, 0) 86.4 (75, 94) All Bleeds 3.8 (2.2, 6.5) 0 (0, 3.4) 55.9 (42.4, 68.8) Treated Spontaneous Bleeds 0 (0, 0.2) 0 (0, 0) 98.3 (90.9, 100) Treated Joint Bleeds 0.2 (0.1, 0.4) 0 (0, 0) 89.8 (79.2, 96.2) Treated Target Joint Bleeds 0.1 (0, 0.7) 0 (0, 0) 96.6 (88.3, 99.6) In the intra-patient analysis, 18 pediatric patients who had participated in the NIS had an ABR for treated bleeds of 19.8 (95% CI [15.3, 25.7]) on previous bypassing agent treatment (prophylactic treatment in 15 patients and on-demand treatment for 3 patients). HEMLIBRA prophylaxis resulted in an ABR for treated bleeds of 0.4 (95% CI [0.2, 0.9]) based on negative binomial regression, corresponding to a 98% reduction in bleed rate. On HEMLIBRA prophylaxis, 14 patients (77.8%) had zero treated bleeds.
The HAVEN 2 study evaluated patient-reported hemophilia-related symptoms (painful swellings and presence of joint pain) and physical functioning (pain with movement) using the Physical Health Score of the Hemophilia-specific Quality of Life Short Form (Haemo-QoL-SF) questionnaire for patients ≥ 8 to < 12 years of age. HEMLIBRA prophylaxis showed improvement from baseline in the Haemo-QoL-SF Physical Health Subscale score at the Week 25 assessment.
HAVEN 4 (Adult and Adolescent Patients)
The HAVEN 4 study (NCT03020160) was a single-arm, multicenter, open-label, clinical trial in 41 adult and adolescent males (aged ≥ 12 years and ≥ 40 kg) with hemophilia A with or without FVIII inhibitors who previously received either episodic (on demand) or prophylactic treatment with FVIII or bypassing agents. Patients received HEMLIBRA prophylaxis at 3 mg/kg once weekly for the first 4 weeks followed by 6 mg/kg once every four weeks thereafter.
Efficacy was evaluated in a subgroup of 5 patients with hemophilia A with FVIII inhibitors based on the bleed rate for bleeds requiring treatment with coagulation factors. The median observation time was 26.1 weeks (range 24.4 – 28.6 weeks). HEMLIBRA prophylaxis resulted in an ABR (95% CI) for treated bleeds of 1.2 (0.1, 14.8) based on negative binomial regression. On HEMLIBRA prophylaxis, 4 patients had zero treated bleeds.
The efficacy results of HEMLIBRA prophylaxis (1.5 mg/kg once every week, 3 mg/kg once every two weeks, and 6 mg/kg once every four weeks) with respect to rate of treated bleeds are shown in Table 13.
Table 13 Annualized Bleed Rate (Treated Bleeds) with HEMLIBRA Prophylaxis in Patients with or without Factor VIII Inhibitors Endpoint HAVEN 1 HAVEN 2 HAVEN 3 HAVEN 4 HEMLIBRA 1.5 mg/kg once every week
(N = 35)No Prophylaxis
(N = 18)HEMLIBRA 1.5 mg/kg once every week
(N = 59)HEMLIBRA 1.5 mg/kg once every week
(N = 36)HEMLIBRA 3 mg/kg once every two weeks
(N = 35)No Prophylaxis
(N = 18)HEMLIBRA 6 mg/kg once every four weeks
(N = 41)ABR = annualized bleed rate; CI = confidence interval; IQR = interquartile range, 25th percentile to 75th percentile; HAVEN 1 = adult and adolescent patients with factor VIII inhibitors; HAVEN 2 = pediatric patients with factor VIII inhibitors; HAVEN 3 = adult and adolescent patients without factor VIII inhibitors; HAVEN 4 = adult and adolescent patients with or without factor VIII inhibitors. - *
- Based on negative binomial regression model.
Median Efficacy Period (weeks) 29.3 24 29.6 29.6 31.3 24 25.6 ABR
(95% CI) *2.9
(1.7, 5)23.3
(12.3, 43.9)0.3
(0.1, 0.5)1.5
(0.9, 2.5)1.3
(0.8, 2.3)38.2
(22.9, 63.8)2.4
(1.4, 4.3)% reduction vs no prophylaxis (95% CI),
p-value87%
(72.3%, 94.3%)
< 0.0001- - 96%
(92.5%, 98%)
< 0.000197%
(93.4%, 98.3%)
< 0.0001- - % patients with 0 bleeds
(95% CI)62.9
(44.9, 78.5)5.6
(0.1, 27.3)86.4
(75, 94)55.6
(38.1, 72.1)60
(42.1, 76.1)0
(0, 18.5)56.1
(39.7, 71.5)% patients with 0 - 3 bleeds (95% CI) 85.7
(69.7, 95.2)11.1
(1.4, 34.7)100
(93.9, 100)91.7
(77.5, 98.2)94.3
(80.8, 99.3)5.6
(0.1, 27.3)90.2
(76.9, 97.3)Median ABR (IQR) 0
(0, 3.7)18.8
(13, 35.1)0
(0, 0)0
(0, 2.5)0
(0, 1.9)40.4
(25.3, 56.7)0
(0, 2.1)14.3 Clinical Impact of Immunogenicity
One patient (1/668, 0.1%) with neutralizing anti-emicizumab-kxwh antibodies in the HEMLIBRA clinical trials had decreased emicizumab-kxwh plasma concentrations and loss of efficacy (manifest as breakthrough bleeding) after 5 weeks of treatment and discontinued HEMLIBRA treatment [see Warnings and Precautions (5.3), Clinical Pharmacology (12.6)].
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
HEMLIBRA (emicizumab-kxwh) injection is available as a sterile, preservative-free, colorless to slightly yellow solution in single-dose vials in the following dosage strengths:
Strength Nominal Volume Concentration Package Size (per carton) Cap Color NDC 12 mg 0.4 mL 30 mg/mL 1 vial Grey 50242-927-01 30 mg 1 mL 30 mg/mL 1 vial Sky Blue 50242-920-01 60 mg 0.4 mL 150 mg/mL 1 vial Purple 50242-921-01 105 mg 0.7 mL 150 mg/mL 1 vial Turquoise 50242-922-01 150 mg 1 mL 150 mg/mL 1 vial Brown 50242-923-01 300 mg 2 mL 150 mg/mL 1 vial Yellow 50242-930-01 Storage and Handling
- Store HEMLIBRA vials in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze. Do not shake.
- Prior to administration, if needed, unopened vials of HEMLIBRA may be stored out of and then returned to refrigeration. The temperature and total combined time out of refrigeration should not exceed 30°C (86°F) and 7 days (at a temperature below 30°C [86°F]), respectively.
- Once removed from the vial, discard HEMLIBRA if not used immediately.
- Discard any unused HEMLIBRA.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Use of Bypassing Agents or FVIII
Inform the patient and/or caregiver that HEMLIBRA increases coagulation potential. Advise the patient and/or caregiver to discontinue prophylactic use of bypassing agents the day before starting HEMLIBRA prophylaxis. Advise the patient and/or caregiver that prophylactic use of FVIII may be continued for the first week of HEMLIBRA prophylaxis. Discuss the appropriate dosing of concomitant agents such as bypassing agents or FVIII with the patient and/or caregiver prior to starting HEMLIBRA prophylaxis [see Warnings and Precautions (5.1, 5.2) and Drug Interactions (7.1)].
Thrombotic Microangiopathy Associated with HEMLIBRA and aPCC
Inform the patient and/or caregiver of the potential risk of thrombotic microangiopathy if aPCC is administered while receiving HEMLIBRA prophylaxis. Instruct the patient and/or caregiver to consult their healthcare provider if aPCC is required in cumulative doses exceeding 100 U/kg. Advise the patient and/or caregiver to seek immediate medical attention if any signs or symptoms of thrombotic microangiopathy occur [see Warnings and Precautions (5.1)].
Thromboembolism Associated with HEMLIBRA and aPCC
Inform the patient and/or caregiver of the potential risk of thromboembolism if aPCC is administered while receiving HEMLIBRA prophylaxis. Instruct the patient and/or caregiver to consult their healthcare provider if aPCC is required in cumulative doses exceeding 100 U/kg. Advise the patient and/or caregiver to seek immediate medical attention if any signs or symptoms of thromboembolism occur [see Warnings and Precautions (5.2)].
Immunogenicity
Inform the patient and/or caregiver of the uncommon occurrence (incidence < 1%) of loss of efficacy while receiving HEMLIBRA prophylaxis due to immunogenicity (neutralizing anti-emicizumab-kxwh antibodies). Instruct the patient and/or caregiver to promptly report clinical signs of loss of efficacy (e.g., increase in breakthrough bleeding events) [see Warnings and Precautions (5.3)].
Laboratory Coagulation Test Interference
Inform the patient and/or caregiver that HEMLIBRA interferes with some laboratory tests that measure blood clotting and may cause a false reading. Advise the patient and/or caregiver that they should notify any healthcare provider about this possibility prior to any blood tests or medical procedures [see Warnings and Precautions (5.4)].
Instruction on Injection Technique
HEMLIBRA is intended for use under the guidance of a healthcare provider. If a patient or caregiver is to administer subcutaneous HEMLIBRA, instruct him/her in injection techniques and assess his/her ability to inject subcutaneously to ensure proper administration of subcutaneous HEMLIBRA and the suitability for home use [see Instructions for Use].
Advise the patient to follow the recommendations in the FDA-approved patient labeling regarding proper sharps disposal.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: 07/2025 Medication Guide
HEMLIBRA® (hem-lee-bruh)
(emicizumab-kxwh)
injection, for subcutaneous useWhat is the most important information I should know about HEMLIBRA?
HEMLIBRA increases the potential for your blood to clot. Carefully follow your healthcare provider's instructions regarding when to use an on-demand bypassing agent or factor VIII (FVIII) and the recommended dose and schedule to use for breakthrough bleed treatment.
HEMLIBRA may cause the following serious side effects when used with activated prothrombin complex concentrate (aPCC; FEIBA®), including:- Thrombotic microangiopathy (TMA). This is a condition involving blood clots and injury to small blood vessels that may cause harm to your kidneys, brain, and other organs. Get medical help right away if you have any of the following signs or symptoms during or after treatment with HEMLIBRA:
- confusion
- weakness
- swelling of arms and legs
- yellowing of skin and eyes
- stomach (abdomen) or back pain
- nausea or vomiting
- feeling sick
- decreased urination
- Blood clots (thrombotic events). Blood clots may form in blood vessels in your arm, leg, lung, or head. Get medical help right away if you have any of these signs or symptoms of blood clots during or after treatment with HEMLIBRA:
- swelling in arms or legs
- pain or redness in your arms or legs
- shortness of breath
- chest pain or tightness
- fast heart rate
- cough up blood
- feel faint
- headache
- numbness in your face
- eye pain or swelling
- trouble seeing
If aPCC (FEIBA®) is needed, talk to your healthcare provider in case you feel you need more than 100 U/kg of aPCC (FEIBA®) total.
Your body may make antibodies against HEMLIBRA, which may stop HEMLIBRA from working properly. Contact your healthcare provider immediately if you notice that HEMLIBRA has stopped working for you (e.g., increase in bleeds).
See "What are the possible side effects of HEMLIBRA?" for more information about side effects.What is HEMLIBRA?
HEMLIBRA is a prescription medicine used for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children, ages newborn and older, with hemophilia A with or without factor VIII inhibitors.
Hemophilia A is a bleeding condition people can be born with where a missing or faulty blood clotting factor (factor VIII) prevents blood from clotting normally.
HEMLIBRA is a therapeutic antibody that bridges clotting factors to help your blood clot.Before using HEMLIBRA, tell your healthcare provider about all of your medical conditions, including if you: - are pregnant or plan to become pregnant. It is not known if HEMLIBRA may harm your unborn baby. Females who are able to become pregnant should use birth control (contraception) during treatment with HEMLIBRA.
- are breastfeeding or plan to breastfeed. It is not known if HEMLIBRA passes into your breast milk.
How should I use HEMLIBRA?
See the detailed "Instructions for Use" that comes with your HEMLIBRA for information on how to prepare and inject a dose of HEMLIBRA, and how to properly throw away (dispose of) used needles and syringes.- Use HEMLIBRA exactly as prescribed by your healthcare provider.
- Stop (discontinue) prophylactic use of bypassing agents the day before starting HEMLIBRA prophylaxis.
- You may continue prophylactic use of FVIII for the first week of HEMLIBRA prophylaxis.
- HEMLIBRA is given as an injection under your skin (subcutaneous injection) by you or a caregiver.
- Your healthcare provider should show you or your caregiver how to prepare, measure, and inject your dose of HEMLIBRA before you inject yourself for the first time.
- Do not attempt to inject yourself or another person unless you have been taught how to do so by a healthcare provider.
- Your healthcare provider will prescribe your dose based on your weight. If your weight changes, tell your healthcare provider.
- You will receive HEMLIBRA 1 time a week for the first four weeks. Then you will receive a maintenance dose as prescribed by your healthcare provider.
- If you miss a dose of HEMLIBRA on your scheduled day, you should give the dose as soon as you remember. You must give the missed dose as soon as possible before the next scheduled dose, and then continue with your normal dosing schedule. Do not give two doses on the same day to make up for a missed dose.
- HEMLIBRA may interfere with laboratory tests that measure how well your blood is clotting and may cause a false reading. Talk to your healthcare provider about how this may affect your care.
What are the possible side effects of HEMLIBRA? The most common side effects of HEMLIBRA include: - redness, tenderness, warmth, or itching at the site of injection
- headache
- joint pain
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store HEMLIBRA? - Store HEMLIBRA in the refrigerator at 36°F to 46°F (2°C to 8°C). Do not freeze.
- Store HEMLIBRA in the original carton to protect the vials from light.
- Do not shake HEMLIBRA.
- If needed, unopened vials of HEMLIBRA can be stored out of the refrigerator and then returned to the refrigerator. HEMLIBRA should not be stored out of the refrigerator for more than a total of 7 days or at a temperature greater than 86°F (30°C).
- After HEMLIBRA is transferred from the vial to the syringe, HEMLIBRA should be used right away.
- Throw away (dispose of) any unused HEMLIBRA left in the vial.
General information about the safe and effective use of HEMLIBRA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use HEMLIBRA for a condition for which it was not prescribed. Do not give HEMLIBRA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about HEMLIBRA that is written for health professionals.What are the ingredients in HEMLIBRA?
Active ingredient: emicizumab-kxwh
Inactive ingredients: L-arginine, L-histidine, poloxamer 188, and L-aspartic acid.
Manufactured by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990
U.S. License No. 1048
HEMLIBRA® is a registered trademark of Chugai Pharmaceutical Co., Ltd., Tokyo, Japan
©2025 Genentech, Inc. All rights reserved.
For more information, go to www.HEMLIBRA.com or call 1-866-HEMLIBRA. -
Instructions for UseHEMLIBRA® (hem-lee-bruh)(emicizumab-kxwh)injection, for subcutaneous use
Be sure that you read, understand, and follow this Instructions for Use before injecting HEMLIBRA. Your healthcare provider should show you or your caregiver how to prepare, measure, and inject HEMLIBRA properly before you use it for the first time. Ask your healthcare provider if you have any questions.
Important Information:
- Do not inject yourself or someone else unless you have been shown how to by your healthcare provider.
- Make sure the name HEMLIBRA appears on the box and vial label.
- Before opening the vial, read the vial label to make sure you have the medicine strength(s) needed to give the dose prescribed by your healthcare provider.
- Your healthcare provider will determine your dose in milliliters (mL) that you will need to give based on your body weight.
- HEMLIBRA comes in multiple strengths. Depending on your dose, you may need to use more than one vial to give your total prescribed dose. Do not combine HEMLIBRA vials of different concentrations in one injection to give the prescribed dose.
- Check the expiration date on the box and vial label. Do not use if the expiration date has passed.
- Only use the vial one time. After you inject your dose, dispose of (throw away) any unused HEMLIBRA left in the vial. Do not save unused HEMLIBRA in the vial for later use.
- Only use the syringes, transfer needles, and injection needles that your healthcare provider prescribes.
- Only use the syringes, transfer needles and injection needles one time. Dispose of (throw away) any used syringes and needles in a sharps disposal container.
- If your prescribed dose is more than 2 mL, you will need to give more than one injection of HEMLIBRA.
Storing HEMLIBRA:
- Store HEMLIBRA in the refrigerator at 36°F to 46°F (2°C to 8°C). Do not freeze.
- Store HEMLIBRA in the original carton to protect the vials from light.
- Do not shake HEMLIBRA.
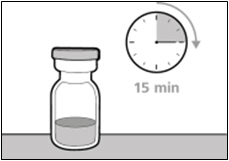
- Take the vial out of the refrigerator 15 minutes before use and allow it to reach room temperature before preparing an injection.
- Before giving the injection, unopened vials of HEMLIBRA may be stored out of the refrigerator and then returned to the refrigerator. HEMLIBRA should not be stored out of the refrigerator:
- for more than a total of 7 days or
- at a temperature greater than 86°F (30°C).
Keep HEMLIBRA and all medicines out of the reach of children.
Inspecting the HEMLIBRA vial and your supplies:
- Collect all supplies listed below to prepare and give your injection.
- Check the expiration date on the box, on the vial label, and on the supplies listed below. Do not use if the expiration date has passed.
- Inspect the supplies for damage. Do not use if they appear damaged or if they have been dropped.
- Place the supplies on a clean, well-lit flat work surface.
HEMLIBRA is colorless to slightly yellow in color. Do not use the vial if:
- the medicine is cloudy, hazy, or colored.
- the medicine contains particles.
- the cap covering the stopper is missing.
Included in the box: 
- Vial containing HEMLIBRA

- HEMLIBRA Instructions for Use
Not included in the box: 
-
Alcohol wipes
Note: If you need to use more than one vial to inject your prescribed dose, you must use a new alcohol wipe for each vial. - Gauze
- Cotton Ball
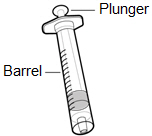
-
Syringe
Note: For injection amount up to 1 mL, use a 1 mL syringe.
For injection amount between 1 mL and 2 mL, use a 2 mL or 3 mL syringe.
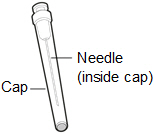
-
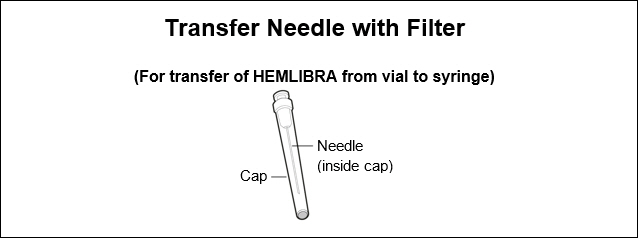
18 gauge Transfer Needle with 5 micrometer filter.
Note: If you need to use more than one vial to inject your prescribed dose, you must use a new transfer needle for each vial.
Do not use the transfer needle to inject HEMLIBRA.
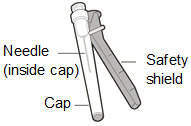
-
Injection Needle with safety shield. You may use a 25, 26 or 27 gauge needle.
Do not use the injection needle to withdraw HEMLIBRA from vial.
- Sharps disposal container
Get ready: - Before use, allow the vial(s) to warm up to room temperature for about 15 minutes on a clean flat surface away from direct sunlight.
- Do not try to warm the vial by any other way.
- Wash your hands well with soap and water.
Selecting and preparing an injection site: - Clean the chosen injection site area using an alcohol wipe.
- Let the skin dry for about 10 seconds. Do not touch, fan, or blow on the cleaned area before your injection.
-
You can use your:
- Thigh (front and middle).
- Stomach area (abdomen), except for 2 inches around the navel (belly button).
- Outer area of the upper arm (only if a caregiver is giving the injection).
- You should use a different injection site each time you give an injection, at least 1 inch away from the area you used for your previous injection.
- Do not inject into areas that could be irritated by a belt or waistband. Do not inject into moles, scars, bruises, or areas where the skin is tender, red, hard, or the skin is broken.
Preparing the syringe for injection:
- HEMLIBRA must not be stored in the syringe.
- HEMLIBRA in the syringe must be injected under the skin (subcutaneous injection) immediately.
- Dispose of (throw away) any used vial(s), needles, vial and injection needle caps, and used syringes in a sharps disposal container.
Important information after the injection:
- Do not rub the injection site after an injection.
- If you see drops of blood at the injection site, you can press a sterile cotton ball or gauze over the injection site for at least 10 seconds, until bleeding has stopped.
- If you have bruising (small area of bleeding under the skin), an ice pack can also be applied with gentle pressure to the site. If bleeding does not stop, please contact your healthcare provider.
Disposing of used HEMLIBRA vial(s), needles, and syringes:
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. Do not dispose of (throw away) any loose needles and syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic.
- can be closed with a tight-fitting, puncture resistant lid, without sharps being able to come out.
- upright and stable during use.
- leak-resistant.
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of (throw away) any used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Important: Always keep the sharps disposal container out of reach of children.
1. PREPARATION
Step 1. Remove vial cap and clean top 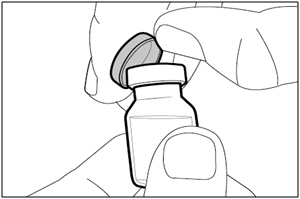
- Take the cap off the vial(s).
- Clean the top of the vial(s) stopper with an alcohol wipe.
- Dispose of (throw away) the vial cap(s) into the sharps disposal container.
Step 2. Attach transfer needle with filter to syringe 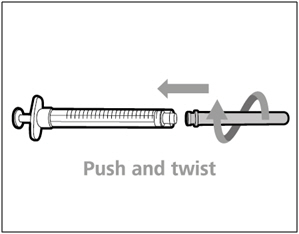
- Push and twist the transfer needle with filter clockwise on to the syringe until it is fully attached.
- Slowly pull back on the plunger and draw air into the syringe that is the same amount for your prescribed dose.
Step 3. Uncap transfer needle 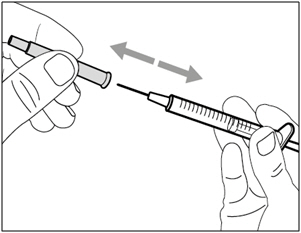
- Hold the syringe by the barrel with the transfer needle pointing up.
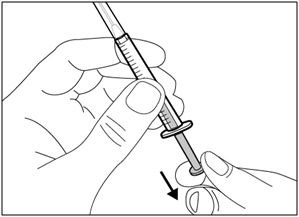
- Carefully pull the transfer needle cap straight off and away from your body. Do not throw the cap away. Place the transfer needle cap down on a clean flat surface. You will need to recap the transfer needle after transferring the medicine.
- Do not touch the needle tip or place it on a surface after the needle cap has been removed.
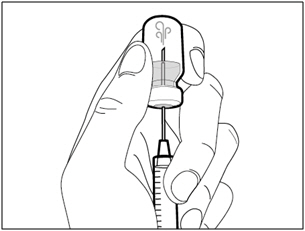
Step 4. Inject air into vial 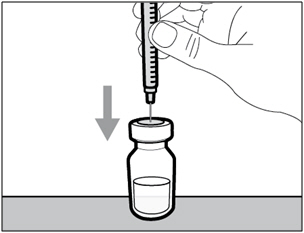
- Keep the vial on the flat working surface and insert the transfer needle and syringe straight down into the center of the vial stopper.
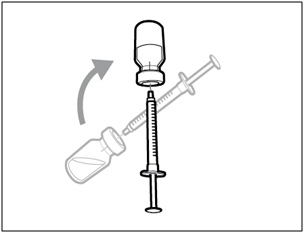
- Keep the needle in the vial and turn the vial upside down.
- With the needle pointing upwards, push on the plunger to inject the air from the syringe above the medicine.
- Keep your finger pressed down on the syringe plunger.
- Do not inject air into the medicine as this could create air bubbles or foam in the medicine.
Step 5. Transfer HEMLIBRA to syringe 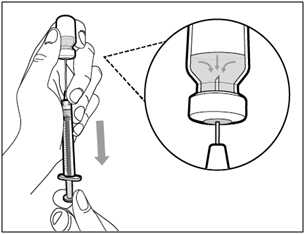
- Slide the tip of the needle down so that it is within the medicine.
- Slowly pull back the plunger to prevent air bubbles/foam.
Fill the syringe with more than the amount of HEMLIBRA needed for your prescribed dose. - Be careful not to pull the plunger out of the syringe.
Important: If your prescribed dose is more than the amount of HEMLIBRA in the vial, withdraw all HEMLIBRA and go to the "Combining Vials" section now. Step 6. Remove air bubbles 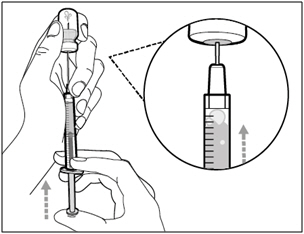
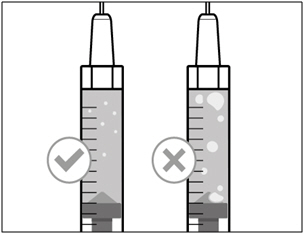
- Keep the needle in the vial and check the syringe for larger air bubbles. Too large an air bubble can reduce the dose you receive.
- Remove the larger air bubbles by gently tapping the syringe barrel with your fingers until the air bubbles rise to the top of the syringe. Move the tip of the needle above the medicine and slowly push the plunger up to push the air bubbles out of the syringe.
- If the amount of HEMLIBRA in the syringe is now at or below your prescribed dose, move the tip of the needle to within the medicine and slowly pull back the plunger until you have more than the amount of HEMLIBRA needed for your prescribed dose.
- Be careful not to pull the plunger out of the syringe.
- Repeat the steps above until you have removed the larger air bubbles.
Note: Ensure you have enough HEMLIBRA in the syringe to complete your dose before moving on to the next step. If you cannot remove all of HEMLIBRA, turn the vial upright to reach the remaining amount.  Do not use the transfer needle to inject HEMLIBRA as this may cause harm such as pain and bleeding.
Do not use the transfer needle to inject HEMLIBRA as this may cause harm such as pain and bleeding.2. INJECTION
Step 7. Recap transfer needle 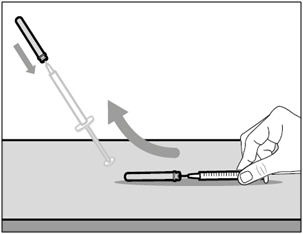
- Remove the syringe and transfer needle from the vial.
- Using one hand, slide the transfer needle into the cap and scoop upwards to cover the needle.
- Once the needle is covered, push the transfer needle cap towards the syringe to fully attach it with one hand to prevent accidentally sticking yourself with the needle.
Step 8. Clean injection site 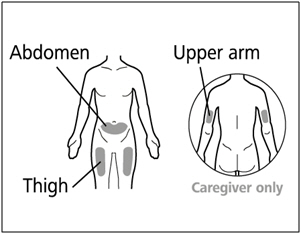
- Select and clean your injection site with an alcohol wipe.
- Let the skin dry for about 10 seconds. Do not touch, fan, or blow on the cleaned area before your injection.
Step 9. Remove transfer needle 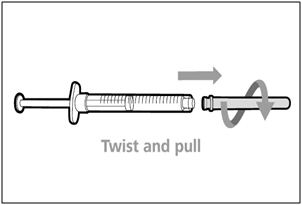
- Remove the transfer needle from the syringe by twisting counter-clockwise and gently pulling.
- Dispose of (throw away) the used transfer needle into a sharps disposal container.
Step 10. Attach injection needle to syringe 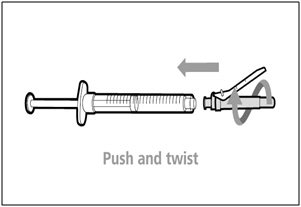
- Push and twist the injection needle clockwise on to the syringe until it is fully attached.
Step 11. Move safety shield 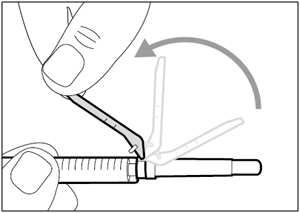
- Move the safety shield away from the needle and towards the syringe barrel.
Step 12. Uncap injection needle 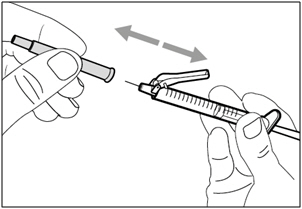
- Carefully pull the injection needle cap away from the syringe.
- Dispose of (throw away) the cap into a sharps disposal container.
- Do not touch the needle tip or allow it to touch any surface.
- After the injection needle cap has been removed, HEMLIBRA in the syringe must be injected right away.
Step 13. Adjust plunger to prescribed dose 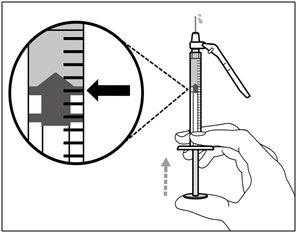
- Slowly push the plunger to your prescribed dose.
- Ensure the top rim of the plunger is in line with the mark on the syringe for your prescribed dose.
Step 14. Subcutaneous (under the skin) injection 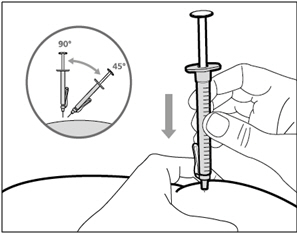
- Pinch the selected injection site and fully insert the needle at a 45° to 90° angle with a quick, firm action. Do not hold or push on the plunger while inserting the needle.
- Hold the position of the syringe and let go of the pinched injection site.
Step 15. Inject HEMLIBRA 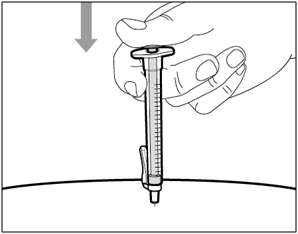
- Slowly inject all of HEMLIBRA by gently pushing the plunger all the way down.
- Remove the needle and syringe from the injection site at the same angle as inserted.
3. DISPOSAL
Step 16. Cover needle with safety shield 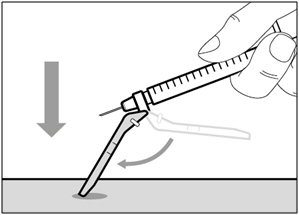
- Move the safety shield forward 90°, away from the syringe barrel.
- Holding the syringe with one hand, press the safety shield down against a flat surface with a firm, quick motion until you hear a "click".
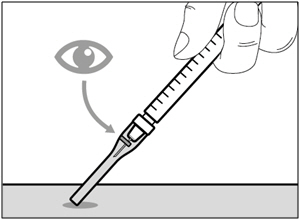
- If you do not hear a click, look to see that the needle is fully covered by the safety shield.
- Keep your fingers behind the safety shield and away from the needle at all times.
- Do not remove the injection needle from the syringe.
Step 17. Dispose of (throw away) the used HEMLIBRA vial(s), needle, and syringe
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. For further information, refer to the section "Disposing of used HEMLIBRA vial(s), needles, and syringes" above.
- Do not try to remove the used injection needle from the used syringe.
- Do not recap the injection needle with the cap.
- Important: Always keep the sharps disposal container out of reach of children.
- Dispose of (throw away) any used vial(s), needles, vial and injection needle caps, and used syringes in a sharps disposal container.
Combining Vials If you need to use more than one vial to get to your total prescribed dose, follow these steps after you have drawn up HEMLIBRA from the first vial:
Step A. Recap transfer needle 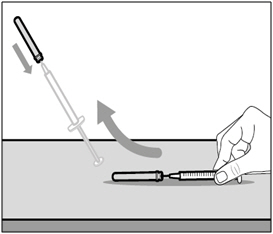
- Remove the syringe and transfer needle from the first vial.
- Using one hand, slide the transfer needle into the cap and scoop upwards to cover the needle.
- Once the needle is covered, push the transfer needle cap toward the syringe to fully attach it with one hand to prevent accidentally sticking yourself with the needle.
Step B. Remove transfer needle 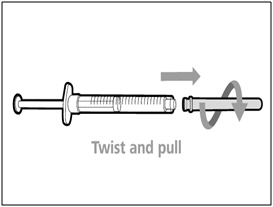
- Remove the transfer needle from the syringe by twisting counter- clockwise and gently pulling.
- Dispose of (throw away) the used transfer needle into a sharps disposal container.
Step C. Attach a new transfer needle with filter to syringe 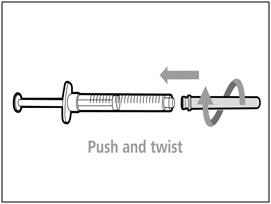
Note: You must use a new transfer needle with filter each time you withdraw HEMLIBRA from a new vial.- Push and twist a new transfer needle clockwise on to the syringe until it is fully attached.
- Slowly pull back the plunger and draw some air into the syringe.
Step D. Uncap transfer needle 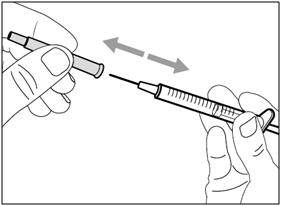
- Hold the syringe by the barrel with the transfer needle cap pointing up.
- Carefully pull the transfer needle cap straight off and away from your body. Do not throw the cap away. You will need to recap the transfer needle after drawing up the medicine.
- Do not touch the needle tip.
Step E. Inject air into vial 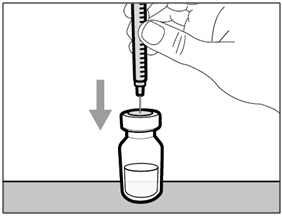
- With the new vial on the flat working surface, insert the new transfer needle and syringe, straight down into the center of the vial stopper.
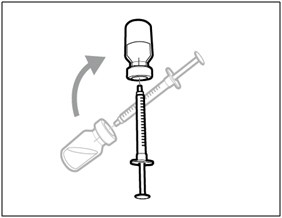
- Keep the transfer needle in the vial and turn the vial upside down.
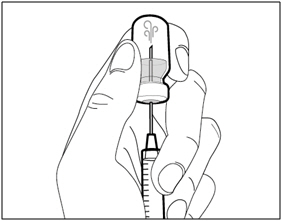
- With the needle pointing upwards, inject the air from the syringe above the medicine.
- Keep your finger pressed down on the syringe plunger.
- Do not inject air into the medicine as this could create air bubbles or foam in the medicine.
Step F. Transfer HEMLIBRA to syringe 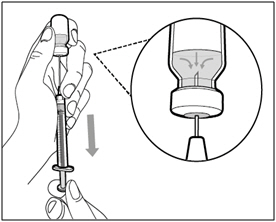
- Slide the tip of the needle down so that it is within the medicine.
- Slowly pull back the plunger to prevent air bubbles/foam.
Fill the syringe barrel with more than the amount of HEMLIBRA needed for your prescribed dose. - Be careful not to pull the plunger out of the syringe.
Note: Ensure you have enough HEMLIBRA in the syringe to complete your dose before moving onto the next steps. If you cannot remove all of HEMLIBRA, turn the vial upright to reach the remaining amount.  Do not use the transfer needle to inject HEMLIBRA as this may cause harm such as pain and bleeding.
Do not use the transfer needle to inject HEMLIBRA as this may cause harm such as pain and bleeding.Repeat steps A to F with each additional vial until you have more than the amount of HEMLIBRA needed for your prescribed dose. Once completed, keep the transfer needle inserted in the vial and return to Step 6. Continue with the remaining steps. For more information, go to www.HEMLIBRA.com or call 1-866-HEMLIBRA.
HEMLIBRA® [emicizumab-kxwh]
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990HEMLIBRA® is a registered trademark of Chugai Pharmaceutical Co., Ltd., Tokyo, Japan
©2023 Genentech, Inc. All rights reserved.U.S. License No. 1048
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 07/2023 -
SPL UNCLASSIFIED SECTION
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
- PRINCIPAL DISPLAY PANEL - 30 mg Vial Carton
- PRINCIPAL DISPLAY PANEL - 60 mg Vial Carton
- PRINCIPAL DISPLAY PANEL - 105 mg Vial Carton
- PRINCIPAL DISPLAY PANEL - 150 mg Vial Carton
- PRINCIPAL DISPLAY PANEL - 300 mg Vial Carton
- PRINCIPAL DISPLAY PANEL - 12 mg./0.4 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
HEMLIBRA
emicizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-920 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EMICIZUMAB (UNII: 7NL2E3F6K3) (EMICIZUMAB - UNII:7NL2E3F6K3) EMICIZUMAB 30 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) ASPARTIC ACID (UNII: 30KYC7MIAI) ARGININE (UNII: 94ZLA3W45F) POLOXAMER 188 (UNII: LQA7B6G8JG) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-920-01 1 in 1 CARTON 11/16/2017 1 1 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761083 11/16/2017 HEMLIBRA
emicizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-921 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EMICIZUMAB (UNII: 7NL2E3F6K3) (EMICIZUMAB - UNII:7NL2E3F6K3) EMICIZUMAB 60 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) ASPARTIC ACID (UNII: 30KYC7MIAI) ARGININE (UNII: 94ZLA3W45F) POLOXAMER 188 (UNII: LQA7B6G8JG) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-921-01 1 in 1 CARTON 11/16/2017 1 0.4 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761083 11/16/2017 HEMLIBRA
emicizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-922 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EMICIZUMAB (UNII: 7NL2E3F6K3) (EMICIZUMAB - UNII:7NL2E3F6K3) EMICIZUMAB 105 mg in 0.7 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) ASPARTIC ACID (UNII: 30KYC7MIAI) ARGININE (UNII: 94ZLA3W45F) POLOXAMER 188 (UNII: LQA7B6G8JG) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-922-01 1 in 1 CARTON 11/16/2017 1 0.7 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761083 11/16/2017 HEMLIBRA
emicizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-923 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EMICIZUMAB (UNII: 7NL2E3F6K3) (EMICIZUMAB - UNII:7NL2E3F6K3) EMICIZUMAB 150 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) ASPARTIC ACID (UNII: 30KYC7MIAI) ARGININE (UNII: 94ZLA3W45F) POLOXAMER 188 (UNII: LQA7B6G8JG) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-923-01 1 in 1 CARTON 11/16/2017 1 1 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761083 11/16/2017 HEMLIBRA
emicizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-930 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EMICIZUMAB (UNII: 7NL2E3F6K3) (EMICIZUMAB - UNII:7NL2E3F6K3) EMICIZUMAB 300 mg in 2 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) ASPARTIC ACID (UNII: 30KYC7MIAI) ARGININE (UNII: 94ZLA3W45F) POLOXAMER 188 (UNII: LQA7B6G8JG) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-930-01 1 in 1 CARTON 03/16/2023 1 2 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761083 03/16/2023 HEMLIBRA
emicizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-927 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EMICIZUMAB (UNII: 7NL2E3F6K3) (EMICIZUMAB - UNII:7NL2E3F6K3) EMICIZUMAB 12 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) ASPARTIC ACID (UNII: 30KYC7MIAI) ARGININE (UNII: 94ZLA3W45F) POLOXAMER 188 (UNII: LQA7B6G8JG) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-927-01 1 in 1 CARTON 01/31/2024 1 0.4 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761083 01/31/2024 Labeler - Genentech, Inc. (080129000) Registrant - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd. 485244961 ANALYSIS(50242-920, 50242-921, 50242-922, 50242-923, 50242-927, 50242-930) , LABEL(50242-920, 50242-921, 50242-922, 50242-923, 50242-927, 50242-930) , PACK(50242-920, 50242-921, 50242-922, 50242-923, 50242-927, 50242-930) Establishment Name Address ID/FEI Business Operations Roche Diagnostics 323105205 ANALYSIS(50242-920, 50242-921, 50242-922, 50242-923, 50242-927, 50242-930) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche AG 482242971 API MANUFACTURE(50242-920, 50242-921, 50242-922, 50242-923, 50242-930) , ANALYSIS(50242-920, 50242-921, 50242-922, 50242-923, 50242-930)