Label: RYBREVANT- amivantamab injection

- NDC Code(s): 57894-501-00, 57894-501-01
- Packager: Janssen Biotech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated November 11, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RYBREVANT safely and effectively. See full prescribing information for RYBREVANT.
RYBREVANT® (amivantamab-vmjw) injection, for intravenous use
Initial U.S. Approval: 2021RECENT MAJOR CHANGES
INDICATIONS AND USAGE
RYBREVANT is a bispecific EGF receptor-directed and MET receptor-directed antibody indicated:
- in combination with lazertinib for the first-line treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R substitution mutations, as detected by an FDA-approved test. ( 1, 2.2)
- in combination with carboplatin and pemetrexed for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations, whose disease has progressed on or after treatment with an EGFR tyrosine kinase inhibitor. ( 1, 2.2)
- in combination with carboplatin and pemetrexed for the first-line treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations, as detected by an FDA-approved test. ( 1, 2.2)
- as a single agent for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy. ( 1, 2.2)
DOSAGE AND ADMINISTRATION
- The recommended dosage of RYBREVANT is based on baseline body weight and administered as an intravenous infusion after dilution. ( 2.3, 2.4)
- Administer prophylactic and concomitant medications as recommended to reduce the risk of dermatologic adverse reactions. ( 2.6)
- Administer via a peripheral line on Week 1 and Week 2 to reduce the risk of infusion-related reactions. ( 2.10)
- Administer RYBREVANT in combination with lazertinib or RYBREVANT as a single agent weekly for 5 weeks, with the initial dose as a split infusion in Week 1 on Day 1 and Day 2, then administer every 2 weeks starting at Week 7. ( 2.3)
- Administer RYBREVANT in combination with chemotherapy weekly for 4 weeks, with the initial dose as a split infusion in Week 1 on Day 1 and Day 2, then administer every 3 weeks starting at Week 7. (2.4)
- When administering RYBREVANT in combination with lazertinib, administer anticoagulant prophylaxis to reduce the risk of venous thromboembolic (VTE) events for the first four months of treatment. ( 2.7)
- Administer diluted RYBREVANT intravenously according to the infusion rates in Tables 8 and 9. ( 2.9, 2.10)
Body Weight (at Baseline) Dosage Recommended Dose RYBREVANT in Combination with Lazertinib or RYBREVANT as a Single Agent Less than 80 kg Weeks 1–5
Week 7 onwards1,050 mg Greater than or equal to 80 kg Weeks 1–5
Week 7 onwards1,400 mg RYBREVANT in Combination with Carboplatin and Pemetrexed Less than 80 kg Weeks 1–4 1,400 mg Week 7 onwards 1,750 mg
Greater than or equal to 80 kg Weeks 1–4 1,750 mg Week 7 onwards 2,100 mg
DOSAGE FORMS AND STRENGTHS
Injection: 350 mg/7 mL (50 mg/mL) solution in a single-dose vial ( 3)
CONTRAINDICATIONS
None. ( 4)
WARNINGS AND PRECAUTIONS
- Infusion-Related Reactions (IRR): Interrupt infusion at the first sign of IRRs. Reduce the infusion rate or permanently discontinue RYBREVANT based on severity. ( 2.5, 2.8, 5.1)
- Interstitial Lung Disease (ILD)/Pneumonitis: Monitor for new or worsening symptoms indicative of ILD. Immediately withhold RYBREVANT in patients with suspected ILD/pneumonitis and permanently discontinue if ILD/pneumonitis is confirmed. ( 2.8, 5.2)
- Venous Thromboembolic (VTE) Events with Concomitant Use with Lazertinib: Prophylactic anticoagulation is recommended for the first four months of treatment. Monitor for signs and symptoms of VTE and treat as medically appropriate. Withhold RYBREVANT and lazertinib based on severity. Once anticoagulant treatment has been initiated, resume RYBREVANT and lazertinib at the same dose at the discretion of the healthcare provider. Permanently discontinue RYBREVANT and continue lazertinib for recurrent VTE despite therapeutic anticoagulation. ( 2.7, 2.8, 5.3)
- Dermatologic Adverse Reactions: Can cause severe rash including toxic epidermal necrolysis (TEN) and acneiform dermatitis. At treatment initiation, prophylactic and concomitant medications are recommended. Withhold, reduce the dose, or permanently discontinue RYBREVANT based on severity. ( 2.6, 2.8, 5.4)
- Ocular Toxicity: Promptly refer patients with worsening eye symptoms to an ophthalmologist. Withhold, reduce the dose, or permanently discontinue RYBREVANT based on severity. ( 2.8, 5.5)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception. ( 5.6, 8.1, 8.3)
ADVERSE REACTIONS
RYBREVANT in Combination with Lazertinib
- The most common adverse reactions (≥ 20%) were rash, nail toxicity, infusion-related reaction, musculoskeletal pain, stomatitis, edema, VTE, paresthesia, fatigue, diarrhea, constipation, COVID-19, hemorrhage, dry skin, decreased appetite, pruritus, and nausea. ( 6.1)
- The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were decreased albumin, decreased sodium, increased ALT, decreased potassium, decreased hemoglobin, increased AST, increased GGT, and increased magnesium. ( 6.1)
RYBREVANT in Combination with Carboplatin and Pemetrexed
- The most common adverse reactions (≥ 20%) were rash, nail toxicity, infusion-related reaction, fatigue, nausea, stomatitis, constipation, edema, decreased appetite, musculoskeletal pain, vomiting, and COVID-19. ( 6.1)
- The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were decreased neutrophils, decreased leukocytes, decreased platelets, decreased hemoglobin, decreased potassium, decreased sodium, increased alanine aminotransferase, increased gamma glutamyl transferase, and decreased albumin. ( 6.1)
RYBREVANT as a Single Agent
- The most common adverse reactions (≥ 20%) were rash, IRR, paronychia, musculoskeletal pain, dyspnea, nausea, fatigue, edema, stomatitis, cough, constipation, and vomiting. ( 6.1)
- The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were decreased lymphocytes, decreased albumin, decreased phosphate, decreased potassium, increased alkaline phosphatase, increased glucose, increased gamma-glutamyl transferase, and decreased sodium. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-526-7736 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 First-Line Treatment of NSCLC with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations
1.2 Previously Treated NSCLC with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations
1.3 First-Line Treatment of NSCLC with EGFR Exon 20 Insertion Mutations
1.4 Previously Treated NSCLC with EGFR Exon 20 Insertion Mutations
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage Information
2.2 Patient Selection
2.3 Recommended Dosage of RYBREVANT in Combination with Lazertinib or RYBREVANT as a Single Agent - Every 2-week dosing
2.4 Recommended Dosage of RYBREVANT in Combination with Carboplatin and Pemetrexed for the Treatment of NSCLC – Every 3-week dosing
2.5 Recommended Premedications to Reduce the Risk of Infusion-Related Reactions
2.6 Prophylactic and Concomitant Medications to Reduce the Risk of Dermatologic Adverse Reactions
2.7 RYBREVANT in Combination with Lazertinib: Concomitant Medications to Reduce the Risk of Venous Thromboembolic Events
2.8 Dosage Modifications for Adverse Reactions
2.9 Preparation
2.10 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions
5.2 Interstitial Lung Disease/Pneumonitis
5.3 Venous Thromboembolic (VTE) Events with Concomitant Use of RYBREVANT and Lazertinib
5.4 Dermatologic Adverse Reactions
5.5 Ocular Toxicity
5.6 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 First Line Treatment of NSCLC with Exon 19 deletion or Exon 21 L858R Substitution Mutation - MARIPOSA
14.2 Previously Treated NSCLC Patients with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations - MARIPOSA-2
14.3 First Line Treatment of NSCLC with Exon 20 Insertion Mutations - PAPILLON
14.4 Previously Treated NSCLC with Exon 20 Insertion Mutations - CHRYSALIS
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 First-Line Treatment of NSCLC with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations
RYBREVANT, in combination with lazertinib, is indicated for the first-line treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R substitution mutations, as detected by an FDA-approved test [see Dosage and Administration (2.2)] .
1.2 Previously Treated NSCLC with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations
RYBREVANT, in combination with carboplatin and pemetrexed, is indicated for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations, whose disease has progressed on or after treatment with an EGFR tyrosine kinase inhibitor [see Dosage and Administration (2.2)] .
1.3 First-Line Treatment of NSCLC with EGFR Exon 20 Insertion Mutations
RYBREVANT, in combination with carboplatin and pemetrexed, is indicated for the first-line treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations, as detected by an FDA-approved test [see Dosage and Administration (2.2)] .
1.4 Previously Treated NSCLC with EGFR Exon 20 Insertion Mutations
RYBREVANT is indicated as a single agent for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations, as detected by an FDA-approved test [see Dosage and Administration (2.2)] , whose disease has progressed on or after platinum-based chemotherapy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage Information
- To reduce the risk of infusion-related reactions, administer premedications before each RYBREVANT infusion as recommended [see Dosage and Administration (2.5)].
- To reduce the risk of infusion-related reactions, administer RYBREVANT via peripheral line for Week 1 Day 1 and 2 and Week 2 [see Dosage and Administration (2.10)].
- To reduce the risk and severity of dermatologic adverse reactions with RYBREVANT, prophylactic and concomitant medications are recommended [see Dosage and Administration (2.6)].
- To reduce the risk of venous thromboembolic (VTE) events when administering RYBREVANT in combination with lazertinib, administer anticoagulant prophylaxis for the first four months of treatment [see Dosage and Administration (2.7)].
- Administer diluted RYBREVANT intravenously according to the infusion rates in Tables 8 and 9, with the initial dose as a split infusion on Week 1 on Day 1 and Day 2 [see Dosage and Administration (2.10)].
- When administering RYBREVANT in combination with lazertinib, administer lazertinib orally any time before the RYBREVANT infusion [see Dosage and Administration (2.10)].
- When administering RYBREVANT in combination with carboplatin and pemetrexed, infuse pemetrexed first, carboplatin second, and RYBREVANT last [see Dosage and Administration (2.10)].
2.2 Patient Selection
Select patients for treatment with RYBREVANT based on the presence of a mutation as detected by an FDA-approved test.
Table 1: Patient Selection Indication Treatment Regimen Source for Testing Information on FDA approved tests is available at: http://www.fda.gov/CompanionDiagnostics. First-Line Treatment of NSCLC with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations [see Indications and Usage (1.1)] RYBREVANT in combination with lazertinib - Tumor or plasma specimens.
- Testing may be performed at any time from initial diagnosis.
- Testing does not need to be repeated once EGFR mutation status has been established.
Previously treated locally advanced or metastatic NSCLC with EGFR Exon 19 deletions or Exon 21 L858R substitution mutations (progressive disease on an EGFR tyrosine kinase inhibitor) [see Indications and Usage (1.2)] RYBREVANT in combination with carboplatin and pemetrexed First-Line Treatment of NSCLC with EGFR Exon 20 Insertion Mutations [see Indications and Usage (1.3)] RYBREVANT in combination with carboplatin and pemetrexed Previously Treated NSCLC with EGFR Exon 20 Insertion Mutations [see Indications and Usage (1.4)] RYBREVANT as a single agent 2.3 Recommended Dosage of RYBREVANT in Combination with Lazertinib or RYBREVANT as a Single Agent - Every 2-week dosing
The recommended dosage of RYBREVANT in combination with lazertinib or RYBREVANT as a single agent, based on baseline body weight, are provided in Table 2. Administer RYBREVANT until disease progression or unacceptable toxicity.
Table 2: Recommended Dosage Schedule for RYBREVANT in Combination with Lazertinib or RYBREVANT as a Single Agent Body weight at Baseline* Recommended Dose Dosing Schedule - *
- Dose adjustment is not required for subsequent body weight changes.
Less than 80 kg 1,050 mg Weekly (total of 5 doses) from Weeks 1 to 5 - Week 1 - split infusion on Day 1 and Day 2
- Weeks 2 to 5 - infusion on Day 1
- Week 6 – no dose
Every 2 weeks starting at Week 7 onwards Greater than or equal to 80 kg 1,400 mg Weekly (total of 5 doses) from Weeks 1 to 5 - Week 1 - split infusion on Day 1 and Day 2
- Weeks 2 to 5 - infusion on Day 1
- Week 6 – no dose
Every 2 weeks starting at Week 7 onwards RYBREVANT in Combination with Lazertinib
Order of Administration
When given in combination with lazertinib, administer RYBREVANT any time after lazertinib when given on the same day. Refer to the lazertinib prescribing information for recommended lazertinib dosing information. Administer RYBREVANT in combination with lazertinib until disease progression or unacceptable toxicity.
2.4 Recommended Dosage of RYBREVANT in Combination with Carboplatin and Pemetrexed for the Treatment of NSCLC – Every 3-week dosing
The recommended dosage of RYBREVANT, administered in combination with carboplatin and pemetrexed is based on baseline body weight is provided in Table 3.
Table 3: Recommended Dosage for RYBREVANT in Combination with Carboplatin and Pemetrexed Body weight at Baseline * Recommended Dose Dosing Schedule - *
- Dose adjustment is not required for subsequent body weight changes.
Less than 80 kg 1,400 mg Weekly (total of 4 doses) from Weeks 1 to 4 - Week 1 - split infusion on Day 1 and Day 2
- Weeks 2 to 4 - infusion on Day 1
- Weeks 5 and 6 – no dose
1,750 mg
Every 3 weeks starting at Week 7 onwards Greater than or equal to 80 kg 1,750 mg Weekly (total of 4 doses) from Weeks 1 to 4 - Week 1 - split infusion on Day 1 and Day 2
- Weeks 2 to 4 - infusion on Day 1
- Weeks 5 and 6 – no dose
2,100 mg
Every 3 weeks starting at Week 7 onwards The recommended order of administration and regimen for RYBREVANT in combination with carboplatin and pemetrexed are provided in Table 4.
Table 4: Order of Administration and Regimen for RYBREVANT in Combination with Carboplatin and Pemetrexed RYBREVANT in Combination with Carboplatin and Pemetrexed Administer the regimen in the following order: pemetrexed first, carboplatin second, and RYBREVANT last. Drug Dose Duration/Timing of Treatment Pemetrexed Pemetrexed 500 mg/m2 intravenously
Refer to the pemetrexed Full Prescribing Information for complete information.Every 3 weeks, continue until disease progression or unacceptable toxicity. Carboplatin Carboplatin AUC 5 intravenously
Refer to the carboplatin Full Prescribing Information for complete information.Every 3 weeks for up to 12 weeks. RYBREVANT RYBREVANT intravenously
See Table 3.Every 3 weeks, continue until disease progression or unacceptable toxicity. 2.5 Recommended Premedications to Reduce the Risk of Infusion-Related Reactions
Administer premedications as described in Table 5 .
After prolonged dose interruptions, restart the following Week 1 Day 1 premedications upon re-initiation: intravenous dexamethasone, diphenhydramine, and acetaminophen.
Table 5: Premedications Premedication Schedule Medication and Frequency Route of Administration Dosing Window Prior to RYBREVANT Administration Initial dose as a split infusion Dexamethasone
8 mg
(or equivalent)Oral 48 hours Week 1 Day -2 twice daily Initial dose as a split infusion Dexamethasone
8 mg
(or equivalent)Oral 24 hours Week 1 Day -1 twice daily Initial dose as a split infusion Dexamethasone
8 mg
(or equivalent)Oral One hour Week 1 Day 1 one dose Dexamethasone
20 mg
(or equivalent)
one doseIntravenous 45 to 60 minutes Diphenhydramine
25 mg to 50 mg
(or equivalent)
one doseOral 30 to 60 minutes Intravenous 15 to 30 minutes Acetaminophen
650 mg to 1,000 mg
one doseOral 30 to 60 minutes Intravenous 15 to 30 minutes Initial dose as a split infusion Dexamethasone
10 mg
(or equivalent)Intravenous 45 to 60 minutes Week 1 Day 2 one dose Diphenhydramine
25 mg to 50 mg
(or equivalent)
one doseOral 30 to 60 minutes Intravenous 15 to 30 minutes Acetaminophen
650 mg to 1,000 mg
one doseOral 30 to 60 minutes Intravenous 15 to 30 minutes All subsequent infusions Diphenhydramine
25 mg to 50 mg
(or equivalent)
one doseOral 30 to 60 minutes Intravenous 15 to 30 minutes Acetaminophen
650 mg to 1,000 mg
one doseOral 30 to 60 minutes Intravenous 15 to 30 minutes Optional:
Dexamethasone
10 mg
(or equivalent)
one doseIntravenous 45 to 60 minutes 2.6 Prophylactic and Concomitant Medications to Reduce the Risk of Dermatologic Adverse Reactions
When initiating treatment with RYBREVANT, prophylactic and concomitant medications are recommended to reduce the risk and severity of dermatologic adverse reactions [see Warnings and Precautions (5.4)].
- Administer an oral antibiotic (doxycycline or minocycline, 100 mg orally twice daily) starting on Day 1 for the first 12 weeks of treatment.
- After completion of oral antibiotic treatment, administer antibiotic lotion to the scalp (clindamycin 1% topical once daily) for the next 9 months of treatment.
- Administer non-comedogenic skin moisturizer (ceramide-based or other formulations that provide long-lasting skin hydration and exclude drying agents) on the face and whole body (except scalp).
- Wash hands and feet with 4% chlorhexidine solution once daily.
- Limit sun exposure during and for 2 months after treatment. Advise patients to wear protective clothing and use broad-spectrum UVA/UVB sunscreen to reduce the risk of dermatologic adverse reactions.
2.7 RYBREVANT in Combination with Lazertinib: Concomitant Medications to Reduce the Risk of Venous Thromboembolic Events
When initiating treatment with RYBREVANT in combination with lazertinib, administer anticoagulant prophylaxis to reduce the risk of venous thromboembolic (VTE) events for the first four months of treatment [see Warnings and Precautions (5.3)]. If there are no signs or symptoms of VTE during the first four months of treatment, consider discontinuation of anticoagulant prophylaxis at the discretion of the healthcare provider. Refer to the lazertinib prescribing information for information about concomitant medications.
2.8 Dosage Modifications for Adverse Reactions
The recommended dose reductions for adverse reactions for RYBREVANT are listed in Table 6.
Table 6: Dose Reductions for Adverse Reactions for RYBREVANT Dose at which the adverse reaction occurred 1st Dose Reduction 2nd Dose Reduction 3rd Dose Reduction 1,050 mg 700 mg 350 mg Discontinue RYBREVANT 1,400 mg 1,050 mg 700 mg 1,750 mg 1,400 mg 1,050 mg 2,100 mg 1,750 mg 1,400 mg The recommended dosage modifications and management for adverse reactions for RYBREVANT are provided in Table 7.
Table 7: Recommended Dosage Modifications and Management for Adverse Reactions for RYBREVANT Adverse Reaction Severity Dosage Modifications Infusion-related reactions (IRR) [see Warnings and Precautions (5.1)] Grade 1 to 2 - Interrupt RYBREVANT infusion if IRR is suspected and monitor patient until reaction symptoms resolve.
- Resume the infusion at 50% of the infusion rate at which the reaction occurred.
- If there are no additional symptoms after 30 minutes, the infusion rate may be escalated (see Tables 8 and 9).
- Include corticosteroid with premedications for subsequent dose (see Table 5).
Grade 3 - Interrupt RYBREVANT infusion and administer supportive care medications. Continuously monitor patient until reaction symptoms resolve.
- Resume the infusion at 50% of the infusion rate at which the reaction occurred.
- If there are no additional symptoms after 30 minutes, the infusion rate may be escalated (see Tables 8 and 9).
- Include corticosteroid with premedications for subsequent dose (see Table 5). For recurrent Grade 3, permanently discontinue RYBREVANT.
Grade 4 or any Grade anaphylaxis / anaphylactic reactions - Permanently discontinue RYBREVANT.
Interstitial Lung Disease (ILD)/pneumonitis [see Warnings and Precautions (5.2)] Any Grade - Withhold RYBREVANT if ILD/pneumonitis is suspected.
- Permanently discontinue RYBREVANT if ILD/pneumonitis is confirmed.
Venous Thromboembolic (VTE) Events [Applies to the combination with lazertinib, see Warnings and Precautions (5.3)] Grade 2 or 3 - Withhold RYBREVANT and lazertinib.
- Administer anticoagulant treatment as clinically indicated.
- Once anticoagulant treatment has been initiated, resume RYBREVANT and lazertinib at the same dose level, at the discretion of the healthcare provider.
Grade 4 or recurrent Grade 2 or 3 despite therapeutic level anticoagulation - Withhold lazertinib and permanently discontinue RYBREVANT.
- Administer anticoagulant treatment as clinically indicated.
- Once anticoagulant treatment has been initiated, treatment can continue with lazertinib at the same dose level at the discretion of the healthcare provider.
Dermatologic Adverse Reactions (including dermatitis acneiform, pruritus, dry skin) [see Warnings and Precautions (5.4)] Grade 1 or Grade 2 - Initiate supportive care management as clinically indicated.
- Reassess after 2 weeks; if rash does not improve, consider dose reduction.
Grade 3 - Withhold RYBREVANT and initiate supportive care management as clinically indicated.
- Upon recovery to ≤ Grade 2, resume RYBREVANT at reduced dose.
- If no improvement within 2 weeks, permanently discontinue treatment.
Grade 4
or
Severe bullous, blistering or exfoliating skin conditions (including toxic epidermal necrolysis (TEN))- Permanently discontinue RYBREVANT.
Other Adverse Reactions [see Adverse Reactions (6.1)] Grade 3 - Withhold RYBREVANT until recovery to ≤ Grade 1 or baseline.
- Resume at the same dose if recovery occurs within 1 week.
- Resume at reduced dose if recovery occurs after 1 week but within 4 weeks.
- Permanently discontinue if recovery does not occur within 4 weeks.
Grade 4 - Withhold RYBREVANT until recovery to ≤ Grade 1 or baseline.
- Resume at reduced dose if recovery occurs within 4 weeks.
- Permanently discontinue if recovery does not occur within 4 weeks.
- Permanently discontinue for recurrent Grade 4 reactions.
Recommended Dosage Modifications for Adverse Reactions for RYBREVANT in Combination with Lazertinib
When administering RYBREVANT in combination with lazertinib, if there is an adverse reaction requiring dose reduction after withholding treatment and resolution, reduce the dose of RYBREVANT first.
Refer to the lazertinib prescribing information for information about dosage modifications for lazertinib.
Recommended Dosage Modifications for Adverse Reactions for RYBREVANT in Combination with Carboplatin and Pemetrexed
When administering RYBREVANT in combination with carboplatin and pemetrexed, modify the dosage of one or more drugs. Withhold or discontinue RYBREVANT as shown in Table 7. Refer to prescribing information for carboplatin and pemetrexed for additional dosage modification information.
2.9 Preparation
Dilute and prepare RYBREVANT for intravenous infusion before administration.
- Check that the RYBREVANT solution is colorless to pale yellow. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if discoloration or visible particles are present.
- Determine the dose required and number of RYBREVANT vials needed based on patient's baseline weight [see Dosage and Administration (2.3, 2.4)] . Each vial of RYBREVANT contains 350 mg of amivantamab-vmjw.
- Withdraw and then discard a volume of either 5% Dextrose Injection or 0.9% Sodium Chloride Injection from the 250 mL infusion bag equal to the volume of RYBREVANT to be added (i.e., discard 7 mL diluent from the infusion bag for each RYBREVANT vial). Only use infusion bags made of polyvinylchloride (PVC), polypropylene (PP), polyethylene (PE), or polyolefin blend (PP+PE).
- Withdraw 7 mL of RYBREVANT from each vial and add it to the infusion bag. The final volume in the infusion bag should be 250 mL. Discard any unused portion left in the vial.
- Gently invert the bag to mix the solution. Do not shake.
- Diluted solutions should be administered within 10 hours (including infusion time) at room temperature 15°C to 25°C (59°F to 77°F).
2.10 Administration
- Administer the diluted RYBREVANT solution [see Dosage and Administration (2.9)] by intravenous infusion using an infusion set fitted with a flow regulator and with an in-line, sterile, non-pyrogenic, low protein-binding polyethersulfone (PES) filter (pore size 0.2 micrometer).
- Administration sets must be made of either polyurethane (PU), polybutadiene (PBD), PVC, PP, or PE.
- The administration set with filter, must be primed with either 5% Dextrose Injection or 0.9% Sodium Chloride Injection prior to the initiation of each RYBREVANT infusion.
- Do not infuse RYBREVANT concomitantly in the same intravenous line with other agents.
RYBREVANT in Combination with Lazertinib or RYBREVANT as a Single Agent
- Administer RYBREVANT as a single agent infusion every 2 weeks intravenously until disease progression or unacceptable toxicity according to the infusion rates in Table 8.
- Administer RYBREVANT via a peripheral line on Week 1 and Week 2, to reduce the risk of infusion-related reactions during initial treatment [see Warnings and Precautions (5.1)].
- RYBREVANT may be administered via central line for subsequent weeks.
- For the initial infusion, prepare RYBREVANT as close to administration time as possible to allow for the possibility of extended infusion time in the event of an infusion-related reaction.
- When given in combination with lazertinib, administer RYBREVANT any time after lazertinib when given on the same day.
Table 8: Infusion Rates of RYBREVANT in Combination with Lazertinib or RYBREVANT as Single Agent Body Weight Less Than 80 kg Week Dose
(per 250 mL bag)Initial Infusion Rate
(mL/hr)Subsequent Infusion Rate*
(mL/hr)- *
- In the absence of infusion-related reactions, increase the initial infusion rate to the subsequent infusion rate after 2 hours based on patient tolerance. Total infusion time is approximately 4–6 hours for day 1 and 6–8 hours for day 2. Subsequent infusion time is approximately 2 hours.
Week 1 (split dose infusion) Week 1 Day 1 350 mg 50 75 Week 1 Day 2 700 mg 50 75 Week 2 1,050 mg 85 Week 3 1,050 mg 125 Week 4 1,050 mg 125 Week 5 1,050 mg 125 Week 6 No dose Week 7 and every 2 weeks thereafter 1,050 mg 125 Body Weight Greater Than or Equal to 80 kg Week Dose
(per 250 mL bag)Initial Infusion Rate
(mL/hr)Subsequent Infusion Rate*
(mL/hr)Week 1 (split dose infusion) Week 1 Day 1 350 mg 50 75 Week 1 Day 2 1,050 mg 35 50 Week 2 1,400 mg 65 Week 3 1,400 mg 85 Week 4 1,400 mg 125 Week 5 1,400 mg 125 Week 6 No dose Week 7 and every 2 weeks thereafter 1,400 mg 125 RYBREVANT in Combination with Carboplatin and Pemetrexed
- Administer RYBREVANT in combination with carboplatin and pemetrexed infusions every 3 weeks intravenously until disease progression or unacceptable toxicity according to the infusion rates in Table 9.
- Administer RYBREVANT via a peripheral line on Week 1 and Week 2 to reduce the risk of infusion-related reactions during initial treatment [see Warnings and Precautions (5.1)].
- RYBREVANT may be administered via central line for subsequent weeks.
- For the initial infusion, prepare RYBREVANT as close to administration time as possible to allow for the possibility of extended infusion time in the event of an infusion-related reaction.
- Administer the pemetrexed infusion first, carboplatin infusion second, and the RYBREVANT infusion last.
Table 9: Infusion Rates of RYBREVANT in Combination with Carboplatin and Pemetrexed for Treatment of NSCLC - *
- In the absence of infusion-related reactions, increase the initial infusion rate to the subsequent infusion rate after 2 hours based on patient tolerance. Total infusion time is approximately 4–6 hours for day 1 and 6–8 hours for day 2. Subsequent infusion time is approximately 2 hours.
Body Weight Less Than 80 kg Week Dose
(per 250 mL bag)Initial Infusion Rate
(mL/hr)Subsequent Infusion Rate*
(mL/hr)Week 1 (split dose infusion) Week 1 Day 1 350 mg 50 75 Week 1 Day 2 1,050 mg 33 50 Week 2 1,400 mg 65 Week 3 1,400 mg 85 Week 4 1,400 mg 125 Weeks 5 and 6 No dose Week 7 and every 3 weeks thereafter 1,750 mg 125 Body Weight Greater Than or Equal to 80 kg Week Dose
(per 250 mL bag)Initial Infusion Rate
(mL/hr)Subsequent Infusion Rate *
(mL/hr)Week 1 (split dose infusion) Week 1 Day 1 350 mg 50 75 Week 1 Day 2 1,400 mg 25 50 Week 2 1,750 mg 65 Week 3 1,750 mg 85 Week 4 1,750 mg 125 Week 5 and 6 No dose Week 7 and every 3 weeks thereafter 2,100 mg 125 - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions
RYBREVANT can cause infusion-related reactions (IRR) including anaphylaxis; signs and symptoms of IRR include dyspnea, flushing, fever, chills, nausea, chest discomfort, hypotension, and vomiting. The median time to IRR onset is approximately 1 hour.
RYBREVANT with Lazertinib
RYBREVANT in combination with lazertinib can cause infusion-related reactions. In MARIPOSA, [see Adverse Reactions (6.1)] , IRRs occurred in 63% of patients treated with RYBREVANT in combination with lazertinib, including Grade 3 in 5% and Grade 4 in 1% of patients. The incidence of infusion modifications due to IRR was 54%, and IRRs leading to dose reduction of RYBREVANT occurred in 0.7% of patients. Infusion-related reactions leading to permanent discontinuation of RYBREVANT occurred in 4.5% of patients receiving RYBREVANT in combination with lazertinib.
RYBREVANT with Carboplatin and Pemetrexed
Based on the pooled safety population [see Adverse Reactions (6.1)] , IRR occurred in 50% of patients treated with RYBREVANT in combination with carboplatin and pemetrexed, including Grade 3 (3.2%) adverse reactions. The incidence of infusion modifications due to IRR was 46%, and 2.8% of patients permanently discontinued RYBREVANT due to IRR.
RYBREVANT as a Single Agent
In CHRYSALIS, [see Adverse Reactions (6.1)], IRR occurred in 66% of patients treated with RYBREVANT as a single agent. Among patients receiving treatment on Week 1 Day 1, 65% experienced an IRR, while the incidence of IRR was 3.4% with the Day 2 infusion, 0.4% with the Week 2 infusion, and cumulatively 1.1% with subsequent infusions. Of the reported IRRs, 97% were Grade 1–2, 2.2% were Grade 3, and 0.4% were Grade 4. The median time to onset was 1 hour (range 0.1 to 18 hours) after start of infusion. The incidence of infusion modifications due to IRR was 62%, and 1.3% of patients permanently discontinued RYBREVANT due to IRR.
Premedicate with antihistamines, antipyretics, and glucocorticoids and infuse RYBREVANT as recommended [see Dosage and Administration (2.5)] . Administer RYBREVANT via a peripheral line on Week 1 and Week 2 to reduce the risk of infusion-related reactions [see Dosage and Administration (2.10)] .
Monitor patients for signs and symptoms of infusion reactions during RYBREVANT infusion in a setting where cardiopulmonary resuscitation medication and equipment are available. Interrupt infusion if IRR is suspected. Reduce the infusion rate or permanently discontinue RYBREVANT based on severity [see Dosage and Administration (2.8)]. If an anaphylactic reaction occurs, permanently discontinue RYBREVANT.
5.2 Interstitial Lung Disease/Pneumonitis
RYBREVANT can cause severe and fatal interstitial lung disease (ILD)/pneumonitis.
RYBREVANT with Lazertinib
In MARIPOSA [see Adverse Reactions (6.1)] , ILD/pneumonitis occurred in 3.1% of patients treated with RYBREVANT in combination with lazertinib, including Grade 3 in 1% and Grade 4 in 0.2% of patients. There was one fatal case of ILD/pneumonitis and 2.9% of patients permanently discontinued RYBREVANT and lazertinib due to ILD/pneumonitis [see Adverse Reactions (6.1)].
RYBREVANT with Carboplatin and Pemetrexed
Based on the pooled safety population [see Adverse Reactions (6.1)], ILD/pneumonitis occurred in 2.1% treated with RYBREVANT in combination with carboplatin and pemetrexed with 1.8% of patients experiencing Grade 3 ILD/pneumonitis. 2.1% discontinued RYBREVANT due to ILD/pneumonitis.
RYBREVANT as a Single Agent
In CHRYSALIS, [see Adverse Reactions (6.1)] , ILD/pneumonitis occurred in 3.3% of patients treated with RYBREVANT as a single agent, with 0.7% of patients experiencing Grade 3 ILD/pneumonitis. Three patients (1%) permanently discontinued RYBREVANT due to ILD/pneumonitis.
Monitor patients for new or worsening symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, fever). Immediately withhold RYBREVANT in patients with suspected ILD/pneumonitis and permanently discontinue if ILD/pneumonitis is confirmed [see Dosage and Administration (2.8)] .
5.3 Venous Thromboembolic (VTE) Events with Concomitant Use of RYBREVANT and Lazertinib
RYBREVANT in combination with lazertinib can cause serious and fatal venous thromboembolic (VTE) events, including deep vein thrombosis and pulmonary embolism. The majority of these events occurred during the first four months of treatment [see Adverse Reactions (6.1)] .
In MARIPOSA [see Adverse Reactions (6.1)], VTEs occurred in 36% of patients receiving RYBREVANT in combination with lazertinib, including Grade 3 in 10% and Grade 4 in 0.5% of patients. On-study VTEs occurred in 1.2% of patients (n=5) while receiving anticoagulation therapy. There were two fatal cases of VTE (0.5%), 9% of patients had VTE leading to dose interruptions of RYBREVANT, 1% of patients had VTE leading to dose reductions of RYBREVANT, and 3.1% of patients had VTE leading to permanent discontinuation of RYBREVANT. The median time to onset of VTEs was 84 days (range: 6 to 777). Administer prophylactic anticoagulation for the first four months of treatment [see Dosage and Administration (2.7)]. The use of Vitamin K antagonists is not recommended. Monitor for signs and symptoms of VTE events and treat as medically appropriate.
Withhold RYBREVANT and lazertinib based on severity [see Dosage and Administration (2.8)] . Once anticoagulant treatment has been initiated, resume RYBREVANT and lazertinib at the same dose level at the discretion of the healthcare provider. In the event of VTE recurrence despite therapeutic anticoagulation, permanently discontinue RYBREVANT. Treatment can continue with lazertinib at the same dose level at the discretion of the healthcare provider [see Dosage and Administration (2.8)]. Refer to the lazertinib prescribing information for recommended lazertinib dosage modification.
5.4 Dermatologic Adverse Reactions
RYBREVANT can cause severe rash including toxic epidermal necrolysis (TEN), dermatitis acneiform, pruritus, and dry skin.
RYBREVANT with Lazertinib
In MARIPOSA, [see Adverse Reactions (6.1)] , rash occurred in 86% of patients treated with RYBREVANT in combination with lazertinib, including Grade 3 in 26% of patients. The median time to onset of rash was 14 days (range: 1 to 556 days). Rash leading to dose interruptions of RYBREVANT occurred in 37% of patients, rash leading to dose reductions of RYBREVANT occurred in 23% of patients, and rash leading to permanent discontinuation of RYBREVANT occurred in 5% of patients.
RYBREVANT with Carboplatin and Pemetrexed
Based on the pooled safety population [see Adverse Reactions (6.1)] , rash occurred in 82% of patients treated with RYBREVANT in combination with carboplatin and pemetrexed, including Grade 3 (15%) adverse reactions. Rash leading to dose reductions occurred in 14% of patients, and 2.5% permanently discontinued RYBREVANT and 3.1% discontinued pemetrexed.
RYBREVANT as a Single Agent
In CHRYSALIS, [see Adverse Reactions (6.1)] , rash occurred in 74% of patients treated with RYBREVANT as a single agent, including Grade 3 rash in 3.3% of patients. The median time to onset of rash was 14 days (range: 1 to 276 days). Rash leading to dose reduction occurred in 5% of patients, and RYBREVANT was permanently discontinued due to rash in 0.7% of patients [see Adverse Reactions (6.1)].
Toxic epidermal necrolysis (TEN) occurred in one patient (0.3%) treated with RYBREVANT as a single agent.
When initiating treatment with RYBREVANT, prophylactic and concomitant medications are recommended to reduce the risk and severity of dermatologic adverse reactions [see Dosage and Administration (2.6)]. Instruct patients to limit sun exposure during and for 2 months after treatment with RYBREVANT. Advise patients to wear protective clothing and use broad-spectrum UVA/UVB sunscreen.
If skin reactions develop, administer supportive care including topical corticosteroids and topical and/or oral antibiotics. For Grade 3 reactions, add oral steroids and consider dermatologic consultation. Promptly refer patients presenting with severe rash, atypical appearance or distribution, or lack of improvement within 2 weeks to a dermatologist. Withhold, reduce the dose, or permanently discontinue RYBREVANT based on severity [see Dosage and Administration (2.8)].
5.5 Ocular Toxicity
RYBREVANT can cause ocular toxicity including keratitis, blepharitis, dry eye symptoms, conjunctival redness, blurred vision, visual impairment, ocular itching, eye pruritus, and uveitis.
RYBREVANT with Lazertinib
In MARIPOSA [see Adverse Reactions (6.1)] , ocular toxicity occurred in 16% of patients treated with RYBREVANT in combination with lazertinib, including Grade 3 or 4 ocular toxicity in 0.7% of patients. Withhold, reduce the dose, or permanently discontinue RYBREVANT and continue lazertinib based on severity [see Dosage and Administration (2.8)] .
RYBREVANT with Carboplatin and Pemetrexed
Based on the pooled safety population [see Adverse Reactions (6.1)], ocular toxicity occurred in 16% of patients treated with RYBREVANT in combination with carboplatin and pemetrexed. All events were Grade 1 or 2.
RYBREVANT as a Single Agent
In CHRYSALIS, [see Adverse Reactions (6.1)] , keratitis occurred in 0.7% and uveitis occurred in 0.3% of patients treated with RYBREVANT. All events were Grade 1–2.
Promptly refer patients with new or worsening eye symptoms to an ophthalmologist. Withhold, reduce the dose, or permanently discontinue RYBREVANT based on severity [see Dosage and Administration (2.8)] .
5.6 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal models, RYBREVANT can cause fetal harm when administered to a pregnant woman. Administration of other EGFR inhibitor molecules to pregnant animals has resulted in an increased incidence of impairment of embryo-fetal development, embryo lethality, and abortion. Advise females of reproductive potential of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of RYBREVANT. [see Use in Specific Populations (8.1, 8.3)] .
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed elsewhere in the labeling:
- Infusion-Related Reactions [see Warnings and Precautions (5.1)]
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.2)]
- Venous Thromboembolic Events [see Warnings and Precautions (5.3)]
- Dermatologic Adverse Reactions [see Warnings and Precautions (5.4)]
- Ocular Toxicity [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
RYBREVANT in Combination with Lazertinib
The data described in the WARNINGS AND PRECAUTIONS reflect exposure to RYBREVANT in combination with lazertinib in the MARIPOSA study in 421 patients with previously untreated locally advanced or metastatic NSCLC whose tumors have EGFR exon 19 deletions or exon 21 L858R substitution mutations [see Clinical Studies (14.1)] . Patients received RYBREVANT intravenously at 1,050 mg (for patients < 80 kg) or 1,400 mg (for patients ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5 in combination with lazertinib, 240 mg orally once daily, until disease progression or unacceptable toxicity. Among 421 patients who received RYBREVANT in combination with lazertinib, 73% were exposed for 6 months or longer and 59% were exposed for greater than one year. The most common adverse reactions (≥ 20%) were rash, nail toxicity, infusion-related reaction, edema, musculoskeletal pain, stomatitis, VTE, paresthesia, fatigue, diarrhea, constipation, COVID-19, dry skin, hemorrhage, decreased appetite, pruritus, and nausea. The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were decreased albumin, increased ALT, decreased sodium, decreased hemoglobin, increased AST, increased GGT and increased magnesium.
RYBREVANT in Combination with Carboplatin and Pemetrexed
The pooled safety population described in the WARNINGS AND PRECAUTIONS also reflect exposure to RYBREVANT in combination with carboplatin and pemetrexed in 281 patients in two studies:
- MARIPOSA-2 [see Clinical Studies (14.2)] in 130 patients with previously treated locally advanced or metastatic NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations whose disease has progressed on or after treatment with osimertinib.
- PAPILLON [see Clinical Studies (14.3)] in 151 patients with previously untreated, locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations.
Patients received RYBREVANT intravenously at 1,400 mg (for patients < 80 kg) or 1,750 mg (for patients ≥ 80 kg) once weekly through 4 weeks, then every 3 weeks with a dose of 1,750 mg (for patients < 80 kg) or 2,100 mg (for patients ≥ 80 kg) starting at Week 7 until disease progression or unacceptable toxicity, in combination with carboplatin at area under the curve AUC 5 once every 3 weeks, for up to 12 weeks, and pemetrexed at 500 mg/m2 once every 3 weeks until disease progression or unacceptable toxicity. Among 281 patients who received RYBREVANT in combination with carboplatin and pemetrexed, 65% were exposed for 6 months or longer and 24% were exposed for greater than one year. In the safety population, the most common (≥ 20%) adverse reactions were rash, nail toxicity, infusion-related reaction, fatigue, nausea, stomatitis, constipation, edema, decreased appetite, musculoskeletal pain, vomiting, and COVID-19. The most common Grade 3 to 4 laboratory abnormalities (≥ 2%) were decreased neutrophils, decreased leukocytes, decreased platelets, decreased hemoglobin, decreased potassium, decreased sodium, increased alanine aminotransferase, increased gamma-glutamyl transferase, and decreased albumin.
RYBREVANT as a Single Agent
The data in the WARNINGS AND PRECAUTIONS also reflect exposure to RYBREVANT as a single agent in CHRYSALIS [see Clinical Studies (14.4)] in 302 patients with locally advanced or metastatic NSCLC. Patients received RYBREVANT at 1,050 mg (for patient baseline body weight < 80 kg) or 1,400 mg (for patient baseline body weight ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter until disease progression or unacceptable toxicity. Among 302 patients who received RYBREVANT as a single agent, 36% were exposed for 6 months or longer and 12% were exposed for greater than one year. In the safety population, the most common (≥ 20%) adverse reactions were rash, infusion-related reaction, paronychia, musculoskeletal pain, dyspnea, nausea, edema, cough, fatigue, stomatitis, constipation, vomiting and pruritus. The most common Grade 3 to 4 laboratory abnormalities (≥ 2%) were increased gamma glutamyl transference, decreased sodium, decreased potassium and increased alkaline phosphatase.
First-line Treatment of NSCLC with Exon 19 deletions or Exon 21 L858R substitution mutations
The safety data described below reflect exposure to RYBREVANT in combination with lazertinib in 421 previously untreated patients with locally advanced or metastatic NSCLC whose tumors have EGFR exon 19 deletions or exon 21 L858R substitution mutation in the MARIPOSA [see Clinical Studies (14.1)]. Patients received RYBREVANT intravenously at 1,050 mg (for patients < 80 kg) or 1,400 mg (for patients ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5 in combination with lazertinib, 240 mg orally once daily. Among the 421 patients who received RYBREVANT in combination with lazertinib, 73% were exposed to RYBREVANT for ≥ 6 months and 59% were exposed to RYBREVANT for > 1 year.
The median age of patients who received RYBREVANT in combination with lazertinib was 64 years (range: 25 to 88); 64% were female; 59% were Asian, 38% were White, 1.7% were American Indian or Alaska Native, 0.7% were Black or African American, 1% were of unknown or other races; and 13% were Hispanic or Latino, 67% had Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 1, 33% had ECOG PS of 0, 60% had EGFR exon 19 deletions, and 40% had EGFR exon 21 L858R substitution mutations.
Serious adverse reactions occurred in 49% of patients who received RYBREVANT in combination with lazertinib. Serious adverse reactions occurring in ≥ 2% of patients included VTE (11%), pneumonia (4%), rash, and ILD/pneumonitis (2.9% each), COVID-19 (2.4%), pleural effusion and infusion-related reaction (2.1% each). Fatal adverse reactions occurred in 7% of patients who received RYBREVANT in combination with lazertinib due to death not otherwise specified (1.2%); sepsis and respiratory failure (1% each); pneumonia, myocardial infarction and sudden death (0.7% each); cerebral infarction, pulmonary embolism (PE), and COVID-19 infection (0.5% each); and ILD/pneumonitis, acute respiratory distress syndrome (ARDS), and cardiopulmonary arrest (0.2% each).
Permanent discontinuation of RYBREVANT due to an adverse reaction occurred in 34% of patients. Adverse reactions which resulted in permanent discontinuation in ≥ 1% of patients included rash, infusion-related reactions, nail toxicity, VTE, ILD/pneumonitis, pneumonia, edema, hypoalbuminemia, fatigue, paresthesia and dyspnea.
Dosage interruption of RYBREVANT due to an adverse reaction occurred in 88% of patients. Adverse reactions which required dosage interruption in ≥ 5% of patients were infusion-related reactions, rash, nail toxicity, COVID-19, VTE, increased ALT, edema, and hypoalbuminemia.
Dose reductions of RYBREVANT due to an adverse reaction occurred in 46% of patients. Adverse reactions requiring dose reductions in ≥ 5% of patients were rash and nail toxicity.
The most common adverse reactions (≥ 20%) were rash, nail toxicity, infusion-related reaction, musculoskeletal pain, stomatitis, edema, VTE, paresthesia, fatigue, diarrhea, constipation, COVID-19, hemorrhage, dry skin, decreased appetite, pruritus, and nausea. The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were decreased albumin, decreased sodium, increased ALT, decreased potassium, decreased hemoglobin, increased AST, increased GGT, and increased magnesium.
Table 10 summarizes the adverse reactions (≥ 10%) in MARIPOSA.
Table 10: Adverse Reactions (≥ 10%) in Patients with NSCLC with Exon 19 Deletion or Exon 21 L858R Substitution Mutations in MARIPOSA Adverse Reaction RYBREVANT in combination with lazertinib
(N=421)Osimertinib
(N=428)All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Skin and subcutaneous tissue disorders Rash * 86 26 48 1.2 Nail toxicity * 71 11 34 0.7 Dry skin * 25 1 18 0.2 Pruritus 24 0.5 17 0.2 Injury, poisoning and procedural complications Infusion-related reaction † 63 6 0 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain * 47 2.1 39 1.9 Gastrointestinal disorders Stomatitis * 43 2.4 27 0.5 Diarrhea * 31 2.6 45 0.9 Constipation 29 0 13 0 Nausea 21 1.2 14 0.2 Vomiting 12 0.5 5 0 Abdominal pain * 11 0 10 0 Hemorrhoids 10 0.2 2.1 0.2 General disorders and administration site conditions Edema * 43 2.6 8 0 Fatigue * 32 3.8 20 1.9 Pyrexia 12 0 9 0 Vascular disorders Venous thromboembolism * 36 11 8 2.8 Hemorrhage * 25 1 13 1.2 Nervous system disorders Paresthesia * 35 1.7 10 0.2 Dizziness * 14 0 10 0 Headache * 13 0.2 13 0 Infections and infestations COVID-19 26 1.7 24 1.4 Conjunctivitis 11 0.2 1.6 0 Metabolism and nutrition disorders Decreased appetite 24 1 18 1.4 Respiratory, thoracic and mediastinal disorders Cough * 19 0 23 0 Dyspnea * 14 1.7 17 3.5 Eye disorders Ocular toxicity * 16 0.7 7 0 Psychiatric disorders Insomnia 10 0 11 0 Clinically relevant adverse reactions in < 10% of patients who received RYBREVANT in combination with lazertinib included skin ulcer and ILD/pneumonitis.
Table 11 summarizes the laboratory abnormalities in MARIPOSA.
Table 11: Select Laboratory Abnormalities (≥ 20%) That Worsened from Baseline in Patients with NSCLC with EGFR Exon 19 Deletion or Exon 21 L858R Substitution Mutations in MARIPOSA * RYBREVANT in combination with lazertinib
(N=421)Osimertinib
(N=428)Laboratory Abnormality All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- *
- The denominator used to calculate the rate is the number of patients with a baseline value and at least one post-treatment value for the specific lab test.
Chemistry Decreased albumin 89 8 22 0.2 Increased ALT 65 7 29 2.6 Increased AST 52 3.8 36 1.9 Increased alkaline phosphatase 45 0.5 15 0.5 Decreased calcium (corrected) 41 1.4 27 0.7 Increased GGT 39 2.6 24 1.9 Decreased sodium 38 7 35 5 Decreased potassium 30 5 15 1.2 Increased creatinine 26 0.7 35 0.7 Decreased magnesium 25 0.7 10 0.2 Increased magnesium 12 2.6 20 4.8 Hematology Decreased platelet count 52 0.7 57 1.4 Decreased hemoglobin 47 3.8 56 1.9 Decreased white blood cell 38 1 66 0.7 Decreased neutrophils 15 1.4 33 1.4 Previously Treated Non-Small Cell Lung Cancer (NSCLC) with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations
The safety data described below reflect exposure to RYBREVANT in combination with carboplatin and pemetrexed was evaluated in MARIPOSA-2 [see Clinical Studies (14.2)]. Eligible patients had locally advanced or metastatic NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations with progressive disease on or after treatment with osimertinib . Patients with asymptomatic or previously treated and stable intracranial metastases were eligible. Patients received RYBREVANT intravenously at 1,400 mg (for patients < 80 kg) or 1,750 mg (for patients ≥ 80 kg) once weekly through 4 weeks, then every 3 weeks with a dose of 1,750 mg (for patients < 80 kg) or 2,100 mg (for patients ≥ 80 kg) starting at Week 7 until disease progression or unacceptable toxicity, in combination with carboplatin at area under the curve AUC 5 once every 3 weeks, for up to 12 weeks, and pemetrexed at 500 mg/m2 once every 3 weeks until disease progression or unacceptable toxicity. Among patients who received RYBREVANT (n=130), 52% were exposed for 6 months or longer and 7% were exposed for greater than one year. The median treatment duration was 6.3 months (range: 0 to 14.7 months).
The median age was 62 years (range: 36 to 84 years); 62% were female; 48% were Asian, 46% were White, 2.3% Black or African American, 1.5% race not reported, 1.5% race unknown, 0.8% Alaska native; 7% were Hispanic or Latino; and 87% had baseline body weight < 80 kg.
Serious adverse reactions occurred in 32% of patients who received RYBREVANT in combination with carboplatin and pemetrexed. Serious adverse reactions in > 2% of patients included dyspnea (3.1%), thrombocytopenia (3.1%), sepsis (2.3%), and pulmonary embolism (2.3%). Fatal adverse reactions occurred in 2.3% of patients who received RYBREVANT in combination with carboplatin and pemetrexed; these included respiratory failure, sepsis, and ventricular fibrillation (0.8% each).
Permanent discontinuation of RYBREVANT due to adverse reactions occurred in 11% of patients. The most frequent adverse reactions leading to discontinuation of RYBREVANT in ≥ 5% of patients were infusion-related reactions.
Dose interruptions of RYBREVANT due to an adverse reaction occurred in 60% of patients. Infusion-related reactions (IRR) requiring infusion interruptions occurred in 52% of patients. Adverse reactions requiring dose interruption in ≥ 5% of patients included infusion-related reaction, rash and fatigue.
Dose reductions of RYBREVANT due to an adverse reaction occurred in 17% of patients. Adverse reactions requiring dose reductions in ≥ 2% of patients included rash.
The most common adverse reactions ≥ 20% were rash, infusion-related reactions, fatigue, nail toxicity, nausea, constipation, edema, stomatitis, decreased appetite, musculoskeletal pain, vomiting, and COVID-19.
Table 12 summarizes the adverse reactions in MARIPOSA-2.
Table 12: Adverse Reactions (≥ 10%) in Previously Treated Patients with NSCLC with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations Treated with RYBREVANT in Combination with Carboplatin and Pemetrexed in MARIPOSA-2 Adverse Reaction RYBREVANT + Carboplatin + Pemetrexed Carboplatin + Pemetrexed (N=130) (N=243) All Grades (%) Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- *
- Grouped term
Skin and subcutaneous tissue disorders Rash * 72 11 12 0 Nail toxicity * 45 2.3 0.4 0 Pruritus 15 0 7 0 Dry skin * 15 0 2.5 0 General disorders and administration site conditions Infusion-related reaction 59 5.4 0.4 0 Fatigue * 51 3.8 35 3.7 Edema * 36 1.5 11 0.4 Pyrexia 12 0 10 0 Gastrointestinal disorders Nausea 45 0.8 37 0.8 Constipation 39 0.8 30 0 Stomatitis * 35 2.3 11 0 Vomiting 25 0.8 17 0.4 Diarrhea * 15 1.5 7 0.8 Metabolism and nutrition disorders Decreased appetite 31 0 21 1.2 Musculoskeletal and connective tissue disorders Musculoskeletal pain * 30 3.1 19 0.8 Infections and infestations COVID-19 21 1.5 10 0 Eye disorders Ocular toxicity * 17 0 3.7 0 Vascular disorders Hemorrhage * 14 0.8 4.9 0 Venous Thromboembolism *(VTE) 10 2.3 4.5 2.9 Respiratory, thoracic and mediastinal disorders Cough * 14 0 16 0.4 Dyspnea * 13 1.5 8 1.2 Clinically relevant adverse reactions in < 10% of patients who received RYBREVANT in combination with carboplatin and pemetrexed include: abdominal pain, hemorrhoids, dizziness, visual impairment, trichomegaly, keratitis, interstitial lung disease, and skin ulcer.
Table 13 summarizes the laboratory abnormalities in MARIPOSA-2.
Table 13: Select Laboratory Abnormalities (≥ 20%) That Worsened from Baseline in Patients with NSCLC with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations Treated with RYBREVANT in Combination with Carboplatin and Pemetrexed in MARIPOSA-2 RYBREVANT + Carboplatin + Pemetrexed
(N=130)Carboplatin + Pemetrexed
(N=243)Laboratory Abnormality All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Hematology Decreased white blood cells 91 42 85 19 Decreased neutrophils 74 49 64 25 Decreased platelets 74 17 58 9 Decreased hemoglobin 71 12 77 9 Decreased lymphocytes 69 28 58 18 Chemistry Decreased albumin 73 3.8 26 0.4 Decreased sodium 49 11 30 6 Increased aspartate aminotransferase 47 0.8 52 0.9 Increased alkaline phosphatase 42 0 29 0.4 Increased alanine aminotransferase 39 3.9 56 6 Decreased magnesium 38 0.8 17 0.4 Decreased potassium 37 11 12 3.4 Increased gamma-glutamyl transferase 30 3.1 41 1.3 Decreased calcium (corrected) 25 0 11 0.9 First-line Treatment of Non-Small Cell Lung Cancer (NSCLC) with Exon 20 Insertion Mutations
The safety data described below reflect exposure to RYBREVANT in combination with carboplatin and pemetrexed at the recommended dosage in the PAPILLON trial [see Clinical Studies (14.3)] in 151 patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. Among patients who received RYBREVANT in combination with carboplatin and pemetrexed the median exposure was 9.7 months (range: 0.0 to 26.9 months). In patients that received carboplatin and pemetrexed alone, the median exposure was 6.7 months (range 0.0 to 25.3).
The median age was 61 years (range: 27 to 86 years); 56% were female; 64% were Asian, 32% were White, 1.3% were Black or African American, race was not reported in 1.3% of patients; 89% were not Hispanic or Latino; 86% had baseline body weight < 80 kg.
Serious adverse reactions occurred in 37% of patients who received RYBREVANT in combination with carboplatin and pemetrexed. Serious adverse reactions in ≥ 2% of patients included rash, pneumonia, interstitial lung disease (ILD), pulmonary embolism, vomiting, and COVID-19. Fatal adverse reactions occurred in 7 patients (4.6%) due to pneumonia, cerebrovascular accident, cardio-respiratory arrest, COVID-19, sepsis, and death not otherwise specified.
Permanent discontinuation of RYBREVANT due to an adverse reaction occurred in 11% of patients. Adverse reactions resulting in permanent discontinuation of RYBREVANT in ≥ 1% of patients were rash and ILD.
Dose interruptions of RYBREVANT due to an adverse reaction occurred in 64% of patients. Infusion-related reactions (IRR) requiring infusion interruptions occurred in 38% of patients. Adverse reactions requiring dose interruption in ≥ 5% of patients included rash and nail toxicity.
Dose reductions of RYBREVANT due to an adverse reaction occurred in 36% of patients. Adverse reactions requiring dose reductions in ≥ 5% of patients included rash and nail toxicity.
The most common adverse reactions (≥ 20%) were rash, nail toxicity, stomatitis, infusion-related reaction, fatigue, edema, constipation, decreased appetite, nausea, COVID-19, diarrhea, and vomiting. The most common Grade 3 to 4 laboratory abnormalities (≥ 2%) were decreased albumin, increased alanine aminotransferase, increased gamma-glutamyl transferase, decreased sodium, decreased potassium, decreased magnesium, and decreases in white blood cells, hemoglobin, neutrophils, platelets, and lymphocytes.
Table 14 summarizes the adverse reactions in PAPILLON.
Table 14: Adverse Reactions (≥ 10%) in Patients with Metastatic NSCLC with Exon 20 Insertion Mutations Who Received RYBREVANT in Combination with Carboplatin and Pemetrexed in PAPILLON RYBREVANT in Combination with Carboplatin and Pemetrexed
(n=151)Carboplatin and Pemetrexed
(n=155)Adverse Reaction * All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Skin and subcutaneous tissue disorders Rash † 90 19 19 0 Nail toxicity † 62 7 3 0 Dry skin † 17 0 6 0 Gastrointestinal disorders Stomatitis † 43 4 11 0 Constipation 40 0 30 0.7 Nausea 36 0.7 42 0 Vomiting 21 3.3 19 0.7 Diarrhea 21 3 13 1.3 Hemorrhoids 12 1 1.3 0 Abdominal pain † 11 0.7 8 0 General disorders and administration site conditions Infusion-related reaction 42 1.3 1.3 0 Fatigue † 42 6 45 3.9 Edema † 40 1.3 19 0 Pyrexia † 17 0 6 0 Metabolism and nutrition disorders Decreased appetite 36 2.6 28 1.3 Infections and infestations COVID-19 24 2 14 0.6 Pneumonia † 13 5 6 1.9 Vascular disorders Hemorrhage † 18 0.7 11 1.9 Respiratory, thoracic, and mediastinal disorders Cough † 17 0 16 0 Dyspnea † 11 1.3 16 3.2 Investigations Weight decreased 14 0.7 8 0 Nervous system disorders Dizziness † 11 0 12 0 Psychiatric disorders Insomnia 11 0 13 0 Clinically relevant adverse reactions in < 10% of patients who received RYBREVANT in combination with carboplatin and pemetrexed included pulmonary embolism, deep vein thrombosis, skin ulcer, conjunctivitis, and interstitial lung disease (ILD)/pneumonitis.
Table 15 summarizes the laboratory abnormalities in PAPILLON.
Table 15: Select Laboratory Abnormalities (≥ 20%) That Worsened from Baseline in Patients with Metastatic NSCLC with EGFR Exon 20 Insertion Mutations Who Received RYBREVANT in Combination with Carboplatin and Pemetrexed in PAPILLON RYBREVANT in Combination with Carboplatin and Pemetrexed * Carboplatin in Combination with Pemetrexed † Laboratory Abnormality ‡ All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- *
- The denominator used to calculate the rate varied from 113 to 150 based on the number of patients with a baseline value and at least one post-treatment value.
- †
- The denominator used to calculate the rate varied from 119 to 154 based on the number of patients with a baseline value and at least one post-treatment value.
- ‡
- Adverse reactions were graded using CTCAE version 5.0
Hematology Decreased white blood cells 89 17 76 10 Decreased hemoglobin 79 11 85 13 Decreased neutrophils 76 36 61 23 Decreased platelets 70 10 54 12 Decreased lymphocytes 61 11 49 13 Chemistry Decreased albumin 87 7 34 1 Increased aspartate aminotransferase 60 1 61 1 Increased alanine aminotransferase 57 4 54 1 Decreased sodium 55 7 39 4 Increased alkaline phosphatase 51 1 28 0 Decreased potassium 44 11 17 1 Decreased magnesium 39 2 30 1 Increased gamma-glutamyl transferase 38 4 43 4 Decreased calcium (corrected) 27 1 18 1 Previously Treated NSCLC Exon 20 Insertion Mutations
The safety data described below reflect exposure to RYBREVANT at the recommended dosage in 129 patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations in the CHRYSALIS trial [see Clinical Studies (14.4)], whose disease had progressed on or after platinum-based chemotherapy. Among patients who received RYBREVANT, 44% were exposed for 6 months or longer and 12% were exposed for greater than one year.
The median age was 62 years (range: 36 to 84 years); 61% were female; 55% were Asian, 35% were White, and 2.3% were Black; and 82% had baseline body weight < 80 kg.
Serious adverse reactions occurred in 30% of patients who received RYBREVANT. Serious adverse reactions in ≥ 2% of patients included pulmonary embolism, pneumonitis/ILD, dyspnea, musculoskeletal pain, pneumonia, and muscular weakness. Fatal adverse reactions occurred in 2 patients (1.5%) due to pneumonia and 1 patient (0.8%) due to sudden death.
Permanent discontinuation of RYBREVANT due to an adverse reaction occurred in 11% of patients. Adverse reactions resulting in permanent discontinuation of RYBREVANT in ≥ 1% of patients were pneumonia, IRR, pneumonitis/ILD, dyspnea, pleural effusion, and rash.
Dose interruptions of RYBREVANT due to an adverse reaction occurred in 78% of patients. Infusion-related reactions (IRR) requiring infusion interruptions occurred in 59% of patients. Adverse reactions requiring dose interruption in ≥ 5% of patients included dyspnea, nausea, rash, vomiting, fatigue, and diarrhea.
Dose reductions of RYBREVANT due to an adverse reaction occurred in 15% of patients. Adverse reactions requiring dose reductions in ≥ 2% of patients included rash and paronychia.
The most common adverse reactions (≥ 20%) were rash, IRR, paronychia, musculoskeletal pain, dyspnea, nausea, fatigue, edema, stomatitis, cough, constipation, and vomiting. The most common Grade 3 to 4 laboratory abnormalities (≥ 2%) were decreased lymphocytes, decreased albumin, decreased phosphate, decreased potassium, increased glucose, increased alkaline phosphatase, increased gamma-glutamyl transferase, and decreased sodium.
Table 16 summarizes the adverse reactions in CHRYSALIS.
Table 16: Adverse Reactions (≥ 10%) in Patients with NSCLC with Exon 20 Insertion Mutations Whose Disease Has Progressed on or after Platinum-based Chemotherapy and Received RYBREVANT in CHRYSALIS Adverse Reactions RYBREVANT *
(N=129)All Grades (%) Grades 3 or 4 (%) Skin and subcutaneous tissue disorders Rash † 84 3.9 Pruritus 18 0 Dry skin 14 0 General disorders and administration site conditions Infusion-related reaction 64 3.1 Fatigue † 33 2.3 Edema † 27 0.8 Pyrexia 13 0 Infections and infestations Paronychia 50 3.1 Pneumonia † 10 0.8 Musculoskeletal and connective tissue disorders Musculoskeletal pain † 47 0 Respiratory, thoracic, and mediastinal disorders Dyspnea † 37 2.3 Cough † 25 0 Gastrointestinal disorders Nausea 36 0 Stomatitis † 26 0.8 Constipation 23 0 Vomiting 22 0 Diarrhea 16 3.1 Abdominal Pain † 11 0.8 Vascular disorders Hemorrhage † 19 0 Metabolism and nutrition disorders Decreased appetite 15 0 Nervous system disorders Peripheral neuropathy † 13 0 Dizziness 12 0.8 Headache † 10 0.8 Clinically relevant adverse reactions in < 10% of patients who received RYBREVANT included ocular toxicity, ILD/pneumonitis, and toxic epidermal necrolysis (TEN).
Table 17 summarizes the laboratory abnormalities in CHRYSALIS.
Table 17: Select Laboratory Abnormalities (≥ 20%) That Worsened from Baseline in Patients With Metastatic NSCLC with EGFR Exon 20 Insertion Mutations Whose Disease Has Progressed on or After Platinum-based Chemotherapy and Who Received RYBREVANT in CHRYSALIS Laboratory Abnormality RYBREVANT *
(N=129)All Grades
(%)Grades 3 or 4
(%)- *
- The denominator used to calculate the rate was 126 based on the number of patients with a baseline value and at least one post-treatment value.
Chemistry Decreased albumin 79 8 Increased glucose 56 4 Increased alkaline phosphatase 53 4.8 Increased creatinine 46 0 Increased alanine aminotransferase 38 1.6 Decreased phosphate 33 8 Increased aspartate aminotransferase 33 0 Decreased magnesium 27 0 Increased gamma-glutamyl transferase 27 4 Decreased sodium 27 4 Decreased potassium 26 6 Hematology Decreased lymphocytes 36 8 6.2 Postmarketing Experience
The following adverse reactions associated with the use of RYBREVANT were identified in clinical studies or postmarketing reports. Because some of these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune system disorders: Infusion-related reactions, including anaphylaxis/anaphylactic reactions
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on the mechanism of action and findings in animal models, RYBREVANT can cause fetal harm when administered to a pregnant woman. There are no available data on the use of RYBREVANT in pregnant women or animal data to assess the risk of RYBREVANT in pregnancy. Disruption or depletion of EGFR in animal models resulted in impairment of embryo-fetal development including effects on placental, lung, cardiac, skin, and neural development. The absence of EGFR or MET signaling has resulted in embryo lethality, malformations, and post-natal death in animals (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
No animal studies have been conducted to evaluate the effects of amivantamab-vmjw on reproduction and fetal development; however, based on its mechanism of action, RYBREVANT can cause fetal harm or developmental anomalies. In mice, EGFR is critically important in reproductive and developmental processes including blastocyst implantation, placental development, and embryo-fetal/postnatal survival and development. Reduction or elimination of embryo-fetal or maternal EGFR signaling can prevent implantation, can cause embryo-fetal loss during various stages of gestation (through effects on placental development) and can cause developmental anomalies and early death in surviving fetuses. Adverse developmental outcomes were observed in multiple organs in embryos/neonates of mice with disrupted EGFR signaling. Similarly, knock out of MET or its ligand HGF was embryonic lethal due to severe defects in placental development, and fetuses displayed defects in muscle development in multiple organs. Human IgG1 is known to cross the placenta; therefore, amivantamab-vmjw has the potential to be transmitted from the mother to the developing fetus.
8.2 Lactation
Risk Summary
There are no data on the presence of amivantamab-vmjw in human milk, the effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions from RYBREVANT in breast-fed children, advise women not to breastfeed during treatment with RYBREVANT and for 3 months after the last dose.
8.3 Females and Males of Reproductive Potential
RYBREVANT can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and efficacy of RYBREVANT have not been established in pediatric patients.
8.5 Geriatric Use
- Of the 421 patients with locally advanced or metastatic NSCLC treated with RYBREVANT in combination with lazertinib in the MARIPOSA study, 45% were ≥ 65 years of age and 12% were ≥ 75 years of age.
- Of the 130 patients with locally advanced or metastatic NSCLC treated with RYBREVANT in combination with carboplatin and pemetrexed in the MARIPOSA-2 study, 40% were ≥ 65 years of age and 10% were ≥ 75 years of age.
- Of the 151 patients with locally advanced or metastatic NSCLC treated with RYBREVANT in combination with carboplatin and pemetrexed in the PAPILLON study, 37% were ≥ 65 years of age and 8% were ≥ 75 years of age.
- Of the 302 patients with locally advanced or metastatic NSCLC treated with RYBREVANT as a single agent in the CHRYSALIS study, 39% were ≥ 65 years of age and 11% were ≥ 75 years of age.
No clinically important differences in safety or efficacy were observed between patients who were ≥ 65 years of age and younger patients.
-
11 DESCRIPTION
Amivantamab-vmjw is a low-fucose human immunoglobulin G1-based bispecific antibody directed against the EGF and MET receptors, produced by mammalian cell line (Chinese Hamster Ovary [CHO]) using recombinant DNA technology that has a molecular weight of approximately 148 kDa. RYBREVANT® (amivantamab-vmjw) injection for intravenous infusion is a sterile, preservative-free, colorless to pale yellow solution in single-dose vials. The pH is 5.7.
Each RYBREVANT vial contains 350 mg (50 mg/mL) amivantamab-vmjw, EDTA disodium salt dihydrate (0.14 mg), L-histidine (2.3 mg), L-histidine hydrochloride monohydrate (8.6 mg), L-methionine (7 mg), polysorbate 80 (4.2 mg), sucrose (595 mg), and water for injection, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Amivantamab-vmjw is a bispecific antibody that binds to the extracellular domains of EGFR and MET.
In in vitro and in vivo studies amivantamab-vmjw was able to disrupt EGFR and MET signaling functions in mutation models of exon 19 deletions, exon 21 L858R substitutions, and exon 20 insertions through blocking ligand binding or degradation of EGFR and MET. The presence of EGFR and MET on the surface of tumor cells also allows for targeting of these cells for destruction by immune effector cells, such as natural killer cells and macrophages, through antibody-dependent cellular cytotoxicity (ADCC) and trogocytosis mechanisms, respectively. Treatment with amivantamab in combination with lazertinib increased in vivo anti-tumor activity compared to either agent alone in a mouse xenograft model of human NSCLC with an EGFR L858R mutation.
12.2 Pharmacodynamics
The exposure-response relationship and time-course of pharmacodynamic response of amivantamab-vmjw have not been fully characterized in patients with NSCLC with EGFR mutations.
12.3 Pharmacokinetics
Amivantamab-vmjw exposures increased proportionally over a dosage range from 350 to 1,750 mg (0.33 to 1.7 times the lowest approved recommended dosage) when RYBREVANT was administered as a single agent. Steady-state concentrations of RYBREVANT were reached by week 13 for both the 3-week and 2-week dosing regimen and the systemic accumulation was 1.9-fold.
Elimination
The mean (% CV) linear clearance (CL) is 0.26 L/day (30%) and mean terminal half-life is 14 days (33%).
Specific Populations
No clinically meaningful differences in the pharmacokinetics of amivantamab-vmjw were observed based on age (range: 21–88 years), body weight (31 to 140 kg), sex, race (White, Asian or Black or African American) and ethnicity (Hispanic/Latino or not Hispanic/Latino), mild or moderate renal impairment (eGFR 30 to 89 mL/min) or mild hepatic impairment [(total bilirubin ≤ ULN and AST > ULN) or (ULN < total bilirubin ≤ 1.5 times ULN)]. The effect of severe renal impairment (eGFR 15 to 29 mL/min), end-stage renal disease (eGFR < 15 mL/min) or moderate to severe hepatic impairment (total bilirubin > 1.5 times ULN and any AST) on amivantamab-vmjw pharmacokinetics has not been studied.
Body Weight
Increases in body weight increased the volume of distribution and clearance of amivantamab-vmjw. Amivantamab-vmjw exposures are 30 to 40% lower in patients who weighed ≥ 80 kg compared to patients with body weight < 80 kg at the same dose. Exposures of amivantamab-vmjw were comparable between patients who weighed < 80 kg and received 1,050 mg dose and patients who weighed ≥ 80 kg and received 1,400 mg dose.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies (ADA) in the studies described below with the incidence of anti-drug antibodies in other studies, including those of amivantamab-vmjw or amivantamab products.
During treatment in studies CHRYSALIS, CHRYSALIS-2, PAPILLON, MARIPOSA, and MARIPOSA-2 (up to 39 months), 4 of the 1,862 (0.2%) patients who received RYBREVANT as a single agent or in combination developed a treatment-emergent anti-amivantamab-vmjw antibodies. Given the low occurrence of anti-drug antibodies, the effect of these antibodies on the pharmacokinetics, safety or efficacy of RYBREVANT is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been performed to assess the potential of amivantamab-vmjw for carcinogenicity or genotoxicity. Fertility studies have not been performed to evaluate the potential effects of amivantamab-vmjw. In 6-week and 3-month repeat-dose toxicology studies in monkeys, there were no notable effects in the male and female reproductive organs.
-
14 CLINICAL STUDIES
14.1 First Line Treatment of NSCLC with Exon 19 deletion or Exon 21 L858R Substitution Mutation - MARIPOSA
The efficacy of RYBREVANT, in combination with lazertinib, was evaluated in MARIPOSA [NCT04487080], a randomized, active-controlled, multicenter trial. Eligible patients were required to have untreated locally advanced or metastatic NSCLC with either exon 19 deletions or exon 21 L858R substitution EGFR mutations identified by local testing, not amenable to curative therapy. Patients with asymptomatic or previously treated and stable intracranial metastases were eligible to enroll.
Patients were randomized (2:2:1) to receive RYBREVANT in combination with lazertinib (N=429), osimertinib monotherapy (N=429), or lazertinib monotherapy (an unapproved regimen for NSCLC) until disease progression or unacceptable toxicity. The evaluation of efficacy for the treatment of untreated metastatic NSCLC relied upon comparison between:
- RYBREVANT administered intravenously at 1,050 mg (for patients < 80 kg) or 1,400 mg (for patients ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5 in combination with lazertinib administered at 240 mg orally once daily.
- Osimertinib administered at a dose of 80 mg orally once daily.
Randomization was stratified by EGFR mutation type (exon 19 deletion or exon 21 L858R substitution mutation), Asian race (yes or no), and history of brain metastasis (yes or no). Tumor assessments were performed every 8 weeks for 30 months, and then every 12 weeks until disease progression.
The major efficacy outcome measure was progression-free survival (PFS) as assessed by blinded independent central review (BICR). Additional efficacy outcome measures included overall survival (OS), overall response rate (ORR), and duration of response (DOR).
A total of 858 patients were randomized between the two study arms, 429 to the RYBREVANT in combination with lazertinib arm and 429 to the osimertinib arm. The median age was 63 (range: 25–88) years; 61% were female; 58% were Asian, 38% were White, 1.6% were American Indian or Alaska Native, 0.8% were Black or African American, 0.2% were Native Hawaiian or other Pacific Islander, 0.6% were unknown race or multiple races; and 12% were Hispanic or Latino. Eastern Cooperative Oncology Group (ECOG) performance status was 0 (34%) or 1 (66%); 69% never smoked; 41% had prior brain metastases; and 89% had Stage IV cancer at initial diagnosis. Sixty percent of patients had tumors harboring exon 19 deletions and the remaining 40% had exon 21 L858R substitution mutations.
Among the 858 patients with EGFR exon 19 deletion or L858R substitution mutations that were randomized between the RYBREVANT plus lazertinib arm versus the osimertinib arm, available tissue samples from 544 (63%) patients had evaluable results when tested retrospectively using the cobas EGFR Mutation Test v2. Of the 544 patients with evaluable results, 527 (97%) patients were positive for EGFR exon 19 deletion or L858R substitution mutations, while 17 (3%) patients were negative. Available plasma samples from patients were retrospectively tested using an FDA-approved test to confirm the biomarker status.
The trial demonstrated a statistically significant improvement in PFS by BICR assessment and OS for RYBREVANT in combination with lazertinib compared to osimertinib (see Table 18 and Figures 1 and 2).
Efficacy results for RYBREVANT in combination with lazertinib are provided in Table 18.
Table 18: Efficacy Results in MARIPOSA by BICR Assessment RYBREVANT in combination with lazertinib
(N=429)Osimertinib
(N=429)CI = confidence interval; NR = not reached; NE = not estimable - *
- Stratified by mutation type (Exon 19del or Exon 21 L858R), prior brain metastases (yes or no), and Asian race (yes or no).
- †
- Stratified Cox proportional hazards regression.
- ‡
- Stratified log-rank test.
- §
- Confirmed responses based on the ITT population.
- ¶
- In confirmed responders.
- #
- Based on observed rates.
Progression-free survival (PFS) Number of events (%) 192 (45) 252 (59) Median, months (95% CI) 23.7 (19.1, 27.7) 16.6 (14.8, 18.5) HR *,†(95% CI); p-value *,‡ 0.70 (0.58, 0.85); p=0.0002 Overall survival (OS) Number of events (%) 173 (40) 217 (51) Median, months (95% CI) NR (42.9, NE) 36.7 (33.4, 41.0) HR *,†(95% CI); p-value *,‡ 0.75 (0.61, 0.92); p=0.0048 Overall response rate (ORR)§ ORR, % (95% CI) 78 (74, 82) 73 (69, 78) Complete response, % 5.4 3.5 Partial response, % 73 70 Duration of response (DOR)¶ Median (95% CI), months 25.8 (20.1, NE) 16.7 (14.8, 18.5) Patients with DOR ≥ 6 months #, % 86 85 Patients with DOR ≥ 12 months #, % 68 57 Figure 1: Kaplan-Meier Curves of PFS by BICR Assessment in Patients with Previously Untreated NSCLC
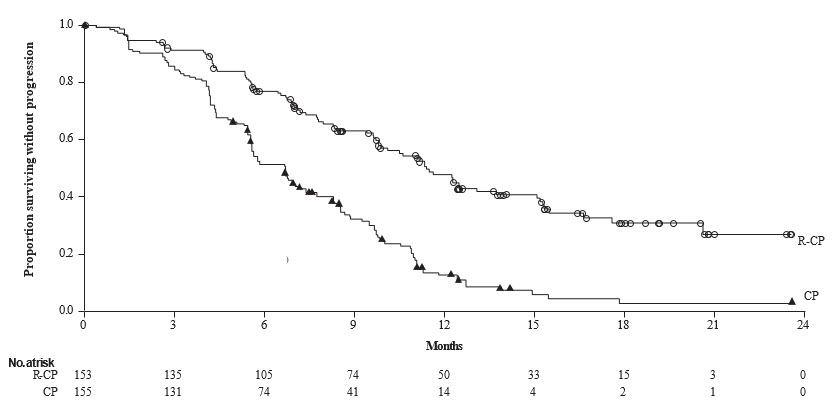
Figure 2: Kaplan-Meier Curves of OS in Patients with Previously Untreated NSCLC

Out of all randomized patients (n=858), 367 (43%) had baseline intracranial lesions assessed by BICR using modified RECIST. Results of pre-specified analyses of intracranial ORR and DOR by BICR in the subset of patients with intracranial lesions at baseline for the RYBREVANT in combination with lazertinib arm and the osimertinib arm are summarized in Table 19.
Table 19: Exploratory Analysis of Intracranial ORR and DOR by BICR Assessment in Subjects with Intracranial Lesions at Baseline RYBREVANT in combination with lazertinib
(N=180)Osimertinib
(N=187)CI = confidence interval Intracranial tumor response assessment Intracranial ORR *, % (95% CI) 68 (60, 75) 69 (62, 76) Complete response % 55 52 Intracranial DOR† Number of responders 122 129 Patients with DOR ≥ 12 months ‡, % 66 59 Patients with DOR ≥ 18 months ‡, % 35 23 14.2 Previously Treated NSCLC Patients with EGFR Exon 19 Deletions or Exon 21 L858R Substitution Mutations - MARIPOSA-2
The efficacy of RYBREVANT in combination with carboplatin and pemetrexed was evaluated in MARIPOSA-2 (NCT04988295), a randomized, open-label, multicenter trial. Eligible patients were required to have locally advanced or metastatic NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations and progressive disease on or after receiving osimertinib. Patients with asymptomatic or previously treated and stable intracranial metastases were eligible to enroll. Patients were randomized (1:2:2) to receive RYBREVANT in combination with carboplatin and pemetrexed (RYBREVANT-CP, N=131), carboplatin and pemetrexed (CP, N=263), or RYBREVANT as part of another combination regimen. The evaluation of efficacy for metastatic NSCLC relied upon comparison between:
- RYBREVANT in combination with carboplatin and pemetrexed. RYBREVANT was administered intravenously at 1,400 mg (for patients < 80 kg) or 1,750 mg (for patients ≥ 80 kg) once weekly through 4 weeks, then every 3 weeks with a dose of 1,750 mg (for patients < 80 kg) or 2,100 mg (for patients ≥ 80 kg) starting at Week 7 until disease progression or unacceptable toxicity.
- Platinum-based chemotherapy with carboplatin and pemetrexed.
For both arms, carboplatin was administered intravenously at area under the concentration-time curve 5 mg/mL per minute (AUC 5) once every 3 weeks, for up to 12 weeks and pemetrexed was administered intravenously at 500 mg/m2 once every 3 weeks until disease progression or unacceptable toxicity.
Randomization was stratified by osimertinib line of therapy (first-line or second-line), prior brain metastases (yes or no), and Asian race (yes or no). Tumor assessments were performed every 6 weeks for the first 12 months and every 12 weeks thereafter.
The major efficacy outcome measure was progression-free survival (PFS) as assessed by blinded independent central review (BICR). Overall survival (OS) and overall response rate (ORR) as assessed by BICR were key secondary outcome measures.
A total of 394 patients were randomized between the two arms, 131 to the RYBREVANT-CP arm and 263 to the CP arm. The median age was 62 (range: 31 to 85) years, with 38% of patients ≥ 65 years of age; 60% were female; and 48% were Asian and 46% were White, 1% were American Indian or Alaska Native, 1% were Black or African American, 0.5% were multiple races and 2.8% were race not reported or race unknown; 8% were Hispanic or Latino. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (40%) or 1 (60%); 65% never smoked; 45% had history of brain metastasis, and 99.7% had Stage IV cancer at study enrollment.
The trial demonstrated a statistically significant improvement in PFS by BICR for RYBREVANT in combination with carboplatin and pemetrexed compared to carboplatin and pemetrexed.
Efficacy results are summarized in Table 20.
Table 20: Efficacy results in MARIPOSA-2 RYBREVANT + carboplatin+ pemetrexed
(N=131)carboplatin+ pemetrexed
(N=263)CI = confidence interval; NE = not estimable - *
- Blinded Independent Central Review by RECIST v1.1
- †
- Stratified by osimertinib line of therapy (first-line or second-line), prior brain metastases (yes or no), and Asian race (yes or no).
- ‡
- Stratified Cox proportional hazards regression.
- §
- Stratified log-rank test.
- ¶
- Confirmed responses.
- #
- Stratified logistic regression analysis.
Progression-free survival (PFS)* Number of events 74 (56%) 171 (65%) Median, months (95% CI) 6.3 (5.6, 8.4) 4.2 (4.0, 4.4) HR (95% CI) †,‡; p-value †,§ 0.48 (0.36, 0.64); p<0.0001 Overall response rate*,¶ ORR, % (95% CI) 53% (44, 62) 29% (23, 35) p-value †,# p<0.0001 Complete response 0.8% 0% Partial response 52% 29% Duration of response*,¶ (DOR) Median (95% CI), months 6.9 (5.5, NE) 5.6 (4.2, 9.6) Figure 3: Kaplan-Meier curve of PFS in Previously Treated NSCLC Patients by BICR assessment- MARIPOSA-2
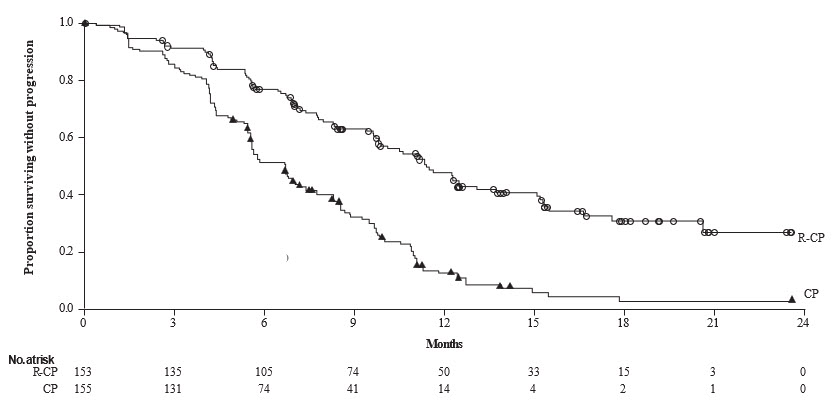
At the prespecified second interim analysis of OS, with 85% of the deaths needed for the final analysis, there was no statistically significant difference in OS. The median OS was 17.7 months (95% CI: 16.0, 22.4) in the ACP arm and 15.3 months (95% CI: 13.7, 16.8) in the CP arm, with a hazard ratio of 0.73 (95% CI: 0.54, 0.99).
Patients with asymptomatic or previously treated and stable intracranial metastases were eligible to be randomized in MARIPOSA-2. Baseline disease assessment, including brain magnetic resonance imaging (MRI) was performed at treatment initiation. All patients underwent serial brain MRI during the trial.
Pre-specified secondary analyses of intracranial ORR by BICR in the subset of 91 (23%) patients with baseline intracranial disease were performed. Data were only available for intracranial complete responses and not available for intracranial partial responses. Intracranial ORR was 20% (95% CI: 8, 39) in the 30 patients with baseline intracranial disease in the ACP arm and 7% (95% CI: 1.8, 16) in the 61 patients with baseline intracranial disease in the CP arm.
14.3 First Line Treatment of NSCLC with Exon 20 Insertion Mutations - PAPILLON
The efficacy of RYBREVANT was evaluated in PAPILLON (NCT04538664), in a randomized, open-label, multicenter study. Eligible patients were required to have previously untreated locally advanced or metastatic NSCLC with EGFR Exon 20 insertion mutations measurable disease per RECIST v1.1, Eastern Cooperative Oncology Group (ECOG) performance status (PS) ≤ 1, and adequate organ and bone marrow function. Patients with brain metastases at screening were eligible for participation once they were definitively treated, clinically stable, asymptomatic, and off corticosteroid treatment for at least 2 weeks prior to randomization. Patients with a medical history of interstitial lung disease or active ILD were excluded from the clinical study.
A total of 308 patients were randomized 1:1 to receive RYBREVANT in combination with carboplatin and pemetrexed (n=153) or carboplatin and pemetrexed (n=155). Patients received RYBREVANT intravenously at 1,400 mg (for patients < 80 kg) or 1,750 mg (for patients ≥ 80 kg) once weekly through 4 weeks, then every 3 weeks with a dose of 1,750 mg (for patients < 80 kg) or 2,100 mg (for patients ≥ 80 kg) starting at Week 7 until disease progression or unacceptable toxicity. Carboplatin was administered intravenously at area under the concentration-time curve 5 mg/mL per minute (AUC 5) once every 3 weeks, for up to 12 weeks. Pemetrexed was administered intravenously at 500 mg/m2 on once every 3 weeks until disease progression or unacceptable toxicity. Patients were stratified by Eastern Cooperative Oncology Group (ECOG) performance status (0 or 1) and prior brain metastases (yes or no).
The primary efficacy outcome measure was progression-free survival (PFS) as assessed by blinded independent central review (BICR). Additional efficacy outcome measures included overall response rate (ORR), duration of response (DOR) and overall survival (OS). Cross-over to single agent RYBREVANT was permitted for patients who had confirmed disease progression on carboplatin and pemetrexed.
The median age was 62 (range: 27 to 92) years, with 40% of the patients ≥ 65 years of age; 58% were female; 61% were Asian and 36% were White, 0.7% were Black or African American and race was not reported in 2.3% of patients; 93% were not Hispanic or Latino. Baseline ECOG performance status was 0 (35%) or 1 (65%); 58% were never smokers; 23% had history of brain metastasis and 84% had Stage IV cancer at initial diagnosis.
PAPILLON demonstrated a statistically significant improvement in progression free survival for patients randomized to RYBREVANT in combination with carboplatin and pemetrexed compared with carboplatin and pemetrexed.
Efficacy results are summarized in Table 21 and Figure 4.
Table 21: Efficacy Results in PAPILLON RYBREVANT+ carboplatin+ pemetrexed
(N=153)carboplatin+ pemetrexed
(N=155)CI = confidence interval Progression-free survival (PFS) Number of events (%) 84 (55) 132 (85) Median, months (95% CI) 11.4 (9.8, 13.7) 6.7 (5.6, 7.3) HR (95% CI) 0.40 (0.30, 0.53) p-value p<0.0001 Overall response rate (ORR)* ORR, % (95% CI) 67 (59, 75) 36 (29, 44) Complete response, % 4 1 Partial response, % 63 36 Duration of response (DOR)† Median (95% CI), months 10.1 (8.5, 13.9) 5.6 (4.4, 6.9) Figure 4: Kaplan-Meier Curve of PFS in Previously Untreated NSCLC Patients by BICR Assessment – PAPILLON Study

While OS results were immature at the current analysis, with 44% of pre-specified deaths for the final analysis reported, no trend towards a detriment was observed. Seventy-five (48%) of the treated patients crossed over from the carboplatin and pemetrexed arm after confirmation of disease progression to receive RYBREVANT as a single agent.
14.4 Previously Treated NSCLC with Exon 20 Insertion Mutations - CHRYSALIS
The efficacy of RYBREVANT was evaluated in patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations in a multicenter, open-label, multi-cohort clinical trial (CHRYSALIS, NCT02609776). The study included patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations whose disease had progressed on or after platinum-based chemotherapy. Patients with untreated brain metastases and patients with a history of ILD requiring treatment with prolonged steroids or other immunosuppressive agents within the last 2 years were not eligible for the study.
In the efficacy population, EGFR exon 20 insertion mutation status was determined by prospective local testing using tissue (94%) and/or plasma (6%) samples. Of the 81 patients with EGFR exon 20 insertion mutations identified by local testing, plasma samples from 78/81 (96%) patients were tested retrospectively using Guardant360 ®CDx, identifying 62/78 (79%) samples with an EGFR exon 20 insertion mutation; 16/78 (21%) samples did not have an EGFR exon 20 insertion mutation identified.
Patients received RYBREVANT at 1,050 mg (for patient baseline body weight < 80 kg) or 1,400 mg (for patient baseline body weight ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter until disease progression or unacceptable toxicity. The major efficacy outcome measure was overall response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1) as evaluated by Blinded Independent Central Review (BICR). An additional efficacy outcome measure was duration of response (DOR) by BICR.
The efficacy population included 81 patients with NSCLC with EGFR exon 20 insertion mutation with measurable disease who were previously treated with platinum-based chemotherapy. The median age was 62 (range: 42 to 84) years, 59% were female; 49% were Asian, 37% were White, 2.5% were Black; 74% had baseline body weight < 80 kg; 95% had adenocarcinoma; and 46% had received prior immunotherapy. The median number of prior therapies was 2 (range: 1 to 7). At baseline, 67% had Eastern Cooperative Oncology Group (ECOG) performance status of 1; 53% never smoked; all patients had metastatic disease; and 22% had previously treated brain metastases.
Efficacy results are summarized in Table 22.
Table 22: Efficacy Results for CHRYSALIS Prior Platinum-based Chemotherapy Treated
(N=81)Based on Kaplan-Meier estimates.
NE=Not Estimable; CI=confidence interval.Overall response rate(95% CI) 40% (29%, 51%) Complete response (CR) 3.7% Partial response (PR) 36% Duration of response (DOR) Median, months (95% CI), months 11.1 (6.9, NE) Patients with DOR ≥ 6 months 63% - 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Infusion-Related Reactions
Advise patients that RYBREVANT can cause infusion-related reactions (IRR), including anaphylaxis. Instruct patients to take glucocorticoids 48 hours prior to the first RYBREVANT infusion to reduce the risk of IRR [see Dosage and Administration (2.5)]. The majority of infusion-related reactions occurred with the first infusion. Advise patients to alert their healthcare provider immediately for any signs or symptoms of infusion-related reactions [see Warnings and Precautions (5.1)] .
Interstitial Lung Disease/Pneumonitis
Advise patients of the risks of interstitial lung disease (ILD)/pneumonitis. Advise patients to immediately contact their healthcare provider for new or worsening respiratory symptoms [see Warnings and Precautions (5.2)] .
Venous Thromboembolic Events with Concomitant Use with Lazertinib
When RYBREVANT is used in combination with lazertinib, advise patients of the risks of serious and life threatening venous thromboembolic (VTE) events, including deep venous thrombosis and pulmonary embolism. Advise patients that prophylactic anticoagulants are recommended to be used for the first four months of treatment. Advise patients to immediately contact their healthcare provider for signs and symptoms of venous thromboembolism [see Warnings and Precautions (5.3)] .
Dermatologic Adverse Reactions
Advise patients of the risk of dermatologic adverse reactions. Advise patients that prophylactic oral antibiotics are recommended starting on Day 1 for the first 12 weeks of treatment and, after completion of oral antibiotic treatment, topical antibiotic lotion to the scalp for the next 9 months of treatment. Advise patients to use a non-comedogenic skin moisturizer (ceramide-based or other formulations that provide long-lasting skin hydration and exclude drying components) on the face and whole body (except scalp) and 4% chlorhexidine solution to wash hands and feet, while on treatment. Advise patients to limit direct sun exposure during and for 2 months after treatment, to wear protective clothing, and to use broad-spectrum UVA/UVB sunscreen to reduce the risk and severity of dermatologic adverse reactions [see Warnings and Precautions (5.4)] .
Ocular Toxicity
Advise patients of the risk of ocular toxicity. Advise patients to contact their ophthalmologist if they develop eye symptoms and advise discontinuation of contact lenses until symptoms are evaluated [see Warnings and Precautions (5.5)] .
Paronychia/Nail Toxicity
Advise patients of the risk of paronychia. Advise patients to contact their healthcare provider for signs or symptoms of paronychia [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus, to use effective contraception during treatment with RYBREVANT and for 3 months after the last dose, and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with RYBREVANT and for 3 months after the last dose [see Use in Specific Populations (8.2)] .
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
RYBREVANT® (RYE–breh–vant)
(amivantamab-vmjw)
injection, for intravenous useThis Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 11/2025 What is RYBREVANT?
RYBREVANT is a prescription medicine used to treat adults with non-small cell lung cancer (NSCLC) that has spread to other parts of the body (metastatic) or cannot be removed by surgery, and has certain abnormal epidermal growth factor receptor (EGFR) gene(s):- in combination with lazertinib as a first-line treatment for non-small cell lung cancer (NSCLC)
- in combination with carboplatin and pemetrexed as a second-line treatment for NSCLC in patients whose disease has worsened on or after treatment with an EGFR tyrosine kinase inhibitor (TKI)
- in combination with carboplatin and pemetrexed as a first-line treatment for NSCLC
- alone for the treatment of NSCLC whose disease has worsened on or after platinum-based chemotherapy.
It is not known if RYBREVANT is safe and effective in children.Before you receive RYBREVANT, tell your healthcare provider about all of your medical conditions, including if you: - have a history of lung or breathing problems
- are pregnant or plan to become pregnant. RYBREVANT can harm your unborn baby.
Females who are able to become pregnant:- Your healthcare provider should do a pregnancy test before you start treatment with RYBREVANT.
- You should use effective birth control (contraception) during treatment and for 3 months after your last dose of RYBREVANT.
- Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with RYBREVANT.
- are breastfeeding or plan to breastfeed. It is not known if RYBREVANT passes into your breast milk. Do not breastfeed during treatment and for 3 months after your last dose of RYBREVANT.
How will I receive RYBREVANT? - RYBREVANT will be given to you by your healthcare provider by intravenous infusion into your vein.
- Your healthcare provider will decide the time between doses as well as how many treatments you will receive.
- Your healthcare provider will give you medicines before each dose of RYBREVANT to help reduce the risk of infusion-related reactions.
- Your healthcare provider may give you medicines in addition to RYBREVANT to reduce the risk and severity of skin and nail reactions.
- RYBREVANT may be given in combination with the medicines carboplatin and pemetrexed. If you have any questions about these medicines, ask your healthcare provider.
- If your treatment with RYBREVANT is given in combination with the medicine lazertinib, you should take your dose of lazertinib by mouth anytime before your infusion with RYBREVANT.
- If you miss any appointments, call your healthcare provider as soon as possible to reschedule your appointment.
RYBREVANT can cause skin reactions. You should limit your time in the sun during and for 2 months after your treatment with RYBREVANT. Wear protective clothing and use broad-spectrum UVA/UVB sunscreen during treatment with RYBREVANT. What are the possible side effects of RYBREVANT?
RYBREVANT may cause serious side effects, including:- infusion-related reactions. Infusion-related reactions are common but can be severe or serious and can include life-threatening (anaphylaxis) allergic reaction. Tell your healthcare provider right away if you get any of the following symptoms during your infusion of RYBREVANT:
- shortness of breath, difficulty breathing, or wheezing
- chest discomfort
- swelling of your eyes, lips, or tongue
- fever
- chills
- numbness of the tongue, lips, cheeks, or gums
- flushing
- skin rash, hives, or itching
- nausea or vomiting
- stomach cramps
- lightheadedness, dizziness, or fainting
- headache
- lung problems. RYBREVANT may cause lung problems that may lead to death. Symptoms may be similar to those symptoms from lung cancer. Tell your healthcare provider right away if you get any new or worsening lung symptoms, including shortness of breath, cough, or fever.
- blood clot problems. Blood clots are a serious, but common side effect of RYBREVANT, when given together with another drug called lazertinib, may cause blood clots in the veins of your legs (deep vein thrombosis) or lungs (pulmonary embolism) that may lead to death. Your healthcare provider will start you on medicine to reduce the risk of blood clots for the first 4 months of treatment. Tell your healthcare provider right away if you have any signs and symptoms of blood clots, including swelling, pain or tenderness in the leg, sudden unexplained chest pain, or shortness of breath.
- skin problems. RYBREVANT can cause severe rash; including blisters, peeling, skin pain and sores, redness, raised acne-like bumps, itching, and dry skin. Tell your healthcare provider right away if you get any skin reactions. Your healthcare provider may start you on an antibiotic for the first 3 months of treatment followed by an antibiotic lotion for your scalp for the next 9 months. During treatment with RYBREVANT, you should apply a non-comedogenic (does not clog pores) skin moisturizer (ceramide-based or other types of moisturizers that provide long-lasting skin hydration and does not include drying ingredients) on your face and whole body (except scalp) and wash your hands and feet every day with 4% chlorhexidine solution. Your healthcare provider may treat you with a medicine(s) or send you to see a skin specialist (dermatologist) if you get skin reactions during treatment with RYBREVANT. See "What should I avoid while receiving RYBREVANT?"
- eye problems. RYBREVANT may cause eye problems. Tell your healthcare provider right away if you get symptoms of eye problems which may include:
- eye pain
- inflammation of eye lids
- dry eyes
- eye redness
- blurred vision
- changes in vision
- itchy eyes
- excessive tearing
- sensitivity to light
Your healthcare provider may send you to see an eye specialist (ophthalmologist) if you get new or worsening eye problems during treatment with RYBREVANT. You should not use contact lenses until your eye symptoms are checked by a healthcare provider. The most common side effects of RYBREVANT when given in combination with lazertinib include: - rash
- infected skin around the nail
- muscle and joint pain
- sores in the mouth
- swelling of hands, ankles, feet, face, or all of your body
- unusual feeling in the skin (such as tingling or a crawling feeling)
- feeling very tired
- diarrhea
- constipation
- COVID-19
- dry skin
- bleeding
- decreased appetite
- itchy skin
- nausea
- changes in certain blood tests
The most common side effects of RYBREVANT when given in combination with carboplatin and pemetrexed include: - rash
- infected skin around the nail
- feeling very tired
- nausea
- sores in the mouth
- constipation
- swelling of hands, ankles, feet, face, or all of your body
- decreased appetite
- muscle and joint pain
- vomiting
- COVID-19
- changes in certain blood tests
The most common side effects of RYBREVANT when given alone: - rash
- infected skin around the nail
- muscle and joint pain
- shortness of breath
- nausea
- feeling very tired
- swelling of hands, ankles, feet, face, or all of your body
- sores in the mouth
- cough
- constipation
- vomiting
- changes in certain blood tests
Your healthcare provider may temporarily stop, decrease your dose, or completely stop your treatment with RYBREVANT if you have serious side effects.
These are not all of the possible side effects of RYBREVANT.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about safe and effective use of RYBREVANT
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your healthcare provider or pharmacist for information about RYBREVANT that is written for health professionals.What are the ingredients of RYBREVANT?
Active ingredient: amivantamab-vmjw
Inactive ingredients: EDTA disodium salt dihydrate, L-histidine, L-histidine hydrochloride monohydrate, L-methionine, polysorbate 80, sucrose, and water for injection.
Product of Ireland
Manufactured by: Janssen Biotech, Inc., Horsham, PA 19044, USA. U.S. License Number 1864
For patent information: www.janssenpatents.com
© Johnson & Johnson and its affiliates 2021-2025
For more information, call 1-800-526-7736 or go to www.RYBREVANT.com. - PRINCIPAL DISPLAY PANEL - 7 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
RYBREVANT
amivantamab injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:57894-501 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AMIVANTAMAB (UNII: 0JSR7Z0NB6) (AMIVANTAMAB - UNII:0JSR7Z0NB6) AMIVANTAMAB 350 mg Inactive Ingredients Ingredient Name Strength EDETATE DISODIUM (UNII: 7FLD91C86K) HISTIDINE (UNII: 4QD397987E) HISTIDINE HYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) METHIONINE (UNII: AE28F7PNPL) POLYSORBATE 20 (UNII: 7T1F30V5YH) SUCROSE (UNII: C151H8M554) WATER (UNII: 059QF0KO0R) Product Characteristics Color yellow (Colorlous to Pale Yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57894-501-01 1 in 1 CARTON 05/21/2021 1 NDC:57894-501-00 1 in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761210 05/21/2021 Labeler - Janssen Biotech, Inc. (099091753) Registrant - Janssen Pharmaceutical Sciences Unlimited Company (985639841) Establishment Name Address ID/FEI Business Operations Janssen Pharmaceutical Sciences Unlimited Company 985639841 API MANUFACTURE(57894-501) , ANALYSIS(57894-501) Establishment Name Address ID/FEI Business Operations Cilag AG 483237103 LABEL(57894-501) , MANUFACTURE(57894-501) , ANALYSIS(57894-501)
