Label: ZEMAIRA- alpha-1-proteinase inhibitor human kit
ZEMAIRA- .alpha.1-proteinase inhibitor human kit









-
NDC Code(s):
0053-7201-02,
0053-7202-02,
0053-7203-02,
0053-7211-01, view more0053-7212-01, 0053-7213-01, 0053-7653-12, 0053-7653-20, 0053-7653-80
- Packager: CSL Behring LLC
- Category: PLASMA DERIVATIVE
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated January 1, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ZEMAIRA safely and effectively. See full prescribing information for ZEMAIRA.
ZEMAIRA® (alpha1-proteinase inhibitor (human))
lyophilized powder for reconstitution for intravenous use
Initial U.S. Approval: 2003INDICATIONS AND USAGE
- ZEMAIRA is an alpha1-proteinase inhibitor (A1-PI) indicated for chronic augmentation and maintenance therapy in adults with A1-PI deficiency and clinical evidence of emphysema (1).
- The effect of augmentation therapy with ZEMAIRA or any A1-PI product on pulmonary exacerbations and on the progression of emphysema in A1-PI deficiency has not been demonstrated in randomized, controlled clinical studies (1).
- ZEMAIRA is not indicated as therapy for lung disease patients in whom severe A1-PI deficiency has not been established (1).
DOSAGE AND ADMINISTRATION
For intravenous use after reconstitution only (2).
- The recommended weekly dose of ZEMAIRA is 60 mg/kg body weight. Dose ranging studies using efficacy endpoints have not been performed with ZEMAIRA or any A1-PI product (2).
- Administer through a suitable 5 micron infusion filter (not supplied) at room temperature within 3 hours after reconstitution (2.2).
- Do not mix with other medicinal products. Administer through a separate dedicated infusion line (2.2).
- Administer at a rate of approximately 0.08 mL/kg/min as determined by the response and comfort of the patient (2.2).
- Monitor closely the infusion rate and the patient's clinical state, including vital signs, throughout the infusion. Slow or stop the infusion if adverse reactions occur. If symptoms subside promptly, the infusion may be resumed at a lower rate that is comfortable for the patient (2.2).
DOSAGE FORMS AND STRENGTHS
ZEMAIRA is supplied in a single-dose vial containing approximately 1000 mg, 4000 mg, or 5000 mg of functionally active A1-PI as a white to off-white lyophilized powder for reconstitution with 20 mL, 76 mL, or 95 mL of Sterile Water for Injection, USP. The amount of functional A1-PI is printed on the vial label and carton (3).
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Observe any signs of hypersensitivity such as tachycardia, hypotension, confusion, syncope, oxygen consumption decrease, and pharyngeal edema when administering ZEMAIRA to patients with known hypersensitivity to an A1-PI product (5.1).
- Patients with selective or severe IgA deficiency can develop antibodies to IgA and, therefore, have a greater risk of developing potentially severe hypersensitivity and anaphylactic reactions. If anaphylactic or severe anaphylactoid reactions occur, discontinue the infusion immediately (5.2).
- Because ZEMAIRA is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent (5.3).
ADVERSE REACTIONS
- Serious adverse reactions reported following administration of ZEMAIRA in pre-licensure clinical trials included one event each in separate subjects of bronchitis and dyspnea, and one event each in a single subject of chest pain, cerebral ischemia and convulsion.
- The most common adverse reactions occurring in at least 5% of subjects receiving ZEMAIRA in all pre-licensure clinical trials were headache, sinusitis, upper respiratory infection, bronchitis, asthenia, cough increased, fever, injection site hemorrhage, rhinitis, sore throat, and vasodilation (6).
To report SUSPECTED ADVERSE REACTIONS, contact CSL Behring Pharmacovigilance at 1-866-915-6958 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Preparation and Reconstitution
2.2 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity to Other A1-PI Products
5.2 Hypersensitivity to IgA
5.3 Transmissible Infectious Agents
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
ZEMAIRA is an alpha1-proteinase inhibitor (A1-PI) indicated for chronic augmentation and maintenance therapy in adults with A1-PI deficiency and clinical evidence of emphysema.
ZEMAIRA increases antigenic and functional (anti-neutrophil elastase capacity [ANEC]) serum levels and lung epithelial lining fluid (ELF) levels of A1-PI.
Clinical data demonstrating the long-term effects of chronic augmentation therapy of individuals with ZEMAIRA are not available.
The effect of augmentation therapy with ZEMAIRA or any A1-PI product on pulmonary exacerbations and on the progression of emphysema in A1-PI deficiency has not been demonstrated in randomized, controlled clinical studies.
ZEMAIRA is not indicated as therapy for lung disease patients in whom severe A1-PI deficiency has not been established.
-
2 DOSAGE AND ADMINISTRATION
For intravenous use after reconstitution only.
The recommended dose of ZEMAIRA is 60 mg/kg body weight administered once weekly. Dose ranging studies using efficacy endpoints have not been performed with ZEMAIRA or any A1-PI product.
2.1 Preparation and Reconstitution
- Check the expiration date on the vial label and carton. Do not use ZEMAIRA after the expiration date.
- Reconstitute prior to use according to the instructions provided below.
- Reconstitute ZEMAIRA using aseptic technique to maintain product sterility.
- Total reconstitution time for a 1g vial should be obtained within 5 minutes.
- Total reconstitution time for a 4g or 5g vial should be obtained within 10 minutes.
- Inspect the reconstituted solution prior to administration. The solution should be clear, colorless to slightly yellow, and free from visible particles.
- Reconstituted ZEMAIRA may be stored at room temperature. Do not freeze the reconstituted solution.
Follow the steps provided below for the preparation and reconstitution of ZEMAIRA:
- 1.
- Ensure that the ZEMAIRA vial and Sterile Water for Injection vial are at room temperature.
- 2.
- Remove the plastic flip-top cap from the Sterile Water for Injection vial.
- 3.
- Wipe the rubber stopper of the Sterile Water for Injection vial with antiseptic solution and allow it to dry.
- 4.
- Open the Mix2Vial® filter transfer set by peeling off the lid (Fig. 1). Do not remove the transfer set from the blister package.
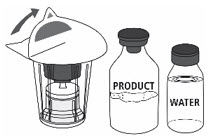
Fig. 1
- 5.
- Place the Sterile Water for Injection vial on an even, clean surface and hold the vial tight. Take the transfer set together with the blister package and vertically pierce the Sterile Water for Injection vial with the blue tip of the transfer set (Fig. 2).
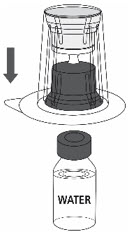
Fig. 2
- 6.
- Carefully remove the blister package from the transfer set by holding at the rim, and pulling vertically upwards. Make sure that you only pull away the blister package and not the transfer set (Fig. 3).
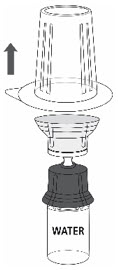
Fig. 3
- 7.
- Remove the plastic flip-top cap from the ZEMAIRA vial.
- 8.
- Wipe the rubber stopper of the ZEMAIRA vial with antiseptic solution and allow it to dry.
- 9.
- Place the ZEMAIRA vial on an even and firm surface. Invert the Sterile Water for Injection vial with the transfer set attached and vertically pierce the ZEMAIRA vial with the clear tip of the transfer set (Fig. 4). The Sterile Water for Injection will automatically flow into the ZEMAIRA vial.
Note: Ensure all water has transferred into the ZEMAIRA vial.
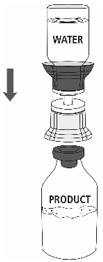
Fig. 4
- 10.
- Follow steps below to remove entire transfer set from ZEMAIRA vial:
- With one hand tightly grasp the ZEMAIRA vial as shown in Fig. 5.
- With the other hand tightly grasp Sterile Water for Injection vial and blue transfer set.
- Bend the entire transfer set to the side until it disconnects from the ZEMAIRA vial (Fig. 5).
- Discard the Sterile Water for Injection vial with the entire transfer set.

Fig. 5
- 11.
- Gently swirl the ZEMAIRA vial until the powder is completely dissolved (Fig. 6). DO NOT SHAKE. Take care not to touch the rubber vial stopper.

Fig. 6
If more than 1 vial of ZEMAIRA is needed to achieve the required dose, use aseptic technique to transfer the reconstituted solution from the vials into the administration container (e.g., empty intravenous bag or glass bottle).
2.2 Administration
For intravenous use only.
- Do not mix ZEMAIRA with other medicinal products; administer ZEMAIRA through a separate dedicated infusion line.
- Perform a visual inspection of the reconstituted solution. The solution should be clear, colorless to slightly yellow, and free from visible particles.
- Administer at room temperature within 3 hours after reconstitution.
- Filter the reconstituted solution during administration. To ensure proper filtration of ZEMAIRA, use an intravenous administration set with a suitable 5 micron infusion filter (not supplied).
- Administer ZEMAIRA intravenously at a rate of approximately 0.08 mL/kg/min as determined by the response and comfort of the patient. The recommended dosage of 60 mg/kg body weight will take approximately 15 minutes to infuse.
- Monitor closely the infusion rate and the patient's clinical state, including vital signs, throughout the infusion. Slow or stop the infusion if adverse reactions occur. If symptoms subside promptly, the infusion may be resumed at a lower rate that is comfortable for the patient.
- ZEMAIRA is for single dose only. Following administration, discard any unused solution and all administration equipment in an appropriate manner as per local requirements.
-
3 DOSAGE FORMS AND STRENGTHS
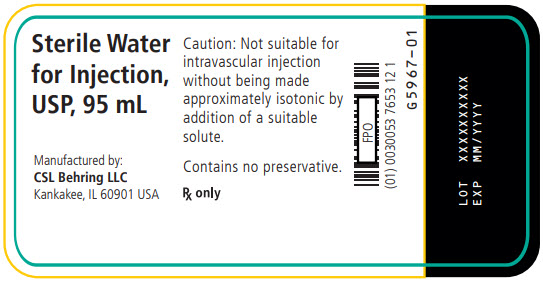
ZEMAIRA is supplied in a single-dose vial containing approximately 1000 mg, 4000 mg, or 5000 mg of functionally active A1-PI as a white to off-white lyophilized powder for reconstitution with 20 mL, 76 mL, or 95 mL of Sterile Water for Injection, USP. The amount of functional A1-PI is printed on the vial label and carton.
-
4 CONTRAINDICATIONS
- ZEMAIRA is contraindicated in patients with a history of anaphylaxis or severe systemic reactions to ZEMAIRA or A1-PI protein.
- ZEMAIRA is contraindicated in immunoglobulin A (IgA)-deficient patients with antibodies against IgA, due to the risk of severe hypersensitivity [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity to Other A1-PI Products
Observe any signs of hypersensitivity such as tachycardia, hypotension, confusion, syncope, oxygen consumption decrease, and pharyngeal edema when administering ZEMAIRA to patients with known hypersensitivity to an A1-PI product. If anaphylactic or severe anaphylactoid reactions occur, discontinue the infusion immediately. Have epinephrine and other appropriate supportive therapy available for the treatment of any acute anaphylactic or anaphylactoid reaction.
5.2 Hypersensitivity to IgA
ZEMAIRA may contain trace amounts of IgA. Patients with selective or severe IgA deficiency can develop antibodies to IgA and, therefore, have a greater risk of developing potentially severe hypersensitivity and anaphylactic reactions. If anaphylactic or severe anaphylactoid reactions occur, discontinue the infusion immediately. Have epinephrine and other appropriate supportive therapy available for the treatment of any acute anaphylactic or anaphylactoid reaction. ZEMAIRA is contraindicated in IgA-deficient patients with antibodies against IgA, due to the risk of severe hypersensitivity.
5.3 Transmissible Infectious Agents
Because ZEMAIRA is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. The risk of infectious agent transmission has been reduced by screening plasma donors for prior exposure to certain viruses, testing for the presence of certain current virus infections, and including virus inactivation/removal steps in the manufacturing process for ZEMAIRA [see Description (11)]. Despite these measures, ZEMAIRA, like other products made from human blood, may still potentially contain human pathogenic agents, including those not yet known or identified. Thus, the risk of transmission of infectious agents cannot be totally eliminated.
All infections thought by a physician to have been possibly transmitted by this product should be reported by the physician or other healthcare provider to the CSL Behring Pharmacovigilance Department at 1-866-915-6958 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
6 ADVERSE REACTIONS
Serious adverse reactions reported following administration of ZEMAIRA in pre-licensure clinical trials included one event each in separate subjects of bronchitis and dyspnea, and one event each in a single subject of chest pain, cerebral ischemia and convulsion.
The most common adverse reactions (ARs) occurring in at least 5% of subjects receiving ZEMAIRA in all pre-licensure clinical trials were headache, sinusitis, upper respiratory infection, bronchitis, asthenia, cough increased, fever, injection site hemorrhage, rhinitis, sore throat, and vasodilation.
Serious adverse reactions identified during postmarketing use were hypersensitivity reactions [see Warnings and Precautions (5.1)].
In post-licensure trials, the exposure adjusted incidence rate (EAIR) of serious exacerbations of chronic obstructive pulmonary disease (COPD) among subjects was higher during the RAPID Extension trial as compared to the rate observed during the preceding RAPID trial [see Adverse Reactions (6.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug product cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The following clinical trials were conducted with ZEMAIRA:
- Controlled, double-blind trial in 44 subjects, who received a weekly 60 mg/kg body weight dose of either ZEMAIRA (30 subjects) or Prolastin® (a commercially available Alpha1-Proteinase Inhibitor [Human] product) (14 subjects) for 10 weeks, followed by an open-label phase in which 43 subjects received ZEMAIRA weekly for 14 weeks;
- Open-label trial in 9 subjects who received a weekly 60 mg/kg body weight dose of ZEMAIRA for 26 weeks, followed by a 7-week to 22-week extension;
- Crossover, double-blind trial in 18 subjects who received a single 60 mg/kg dose of ZEMAIRA and a single 60 mg/kg dose of Prolastin;
- Open-label trial of 19 subjects who received a single 15 mg/kg (2 subjects), 30 mg/kg (5 subjects), 60 mg/kg (6 subjects), or 120 mg/kg (6 subjects) dose of ZEMAIRA; and
- Post-Licensure Randomized, Placebo-Controlled Trial of Augmentation Therapy in Alpha-1 Protease Inhibitor Deficiency (RAPID), in 180 subjects who received a weekly 60 mg/kg body weight dose of either ZEMAIRA (93 subjects) or placebo (87 subjects) for 24 months (referred to as years 1 and 2 in Table 3).
- Post-Licensure Open-label extension of the RAPID trial involving 140 subjects who had completed blinded treatment with ZEMAIRA or placebo for 24 months in the RAPID trial and who entered the extension trial and received open-label ZEMAIRA for up to an additional 24 months (referred to as years 3 and 4 in Table 3).
Table 1 summarizes the ARs, expressed as events per subject-year, and the corresponding number of ARs per infusion, expressed as % of all infusions, for each treatment in pre-licensure clinical trials of ZEMAIRA.
Table 1. Overall Adverse Reactions (ARs) and Serious ARs Number of Subjects*
(Events per Subject-Year†)Number of Infusions‡
(% of all Infusions)ZEMAIRA
(n=66, SY§=28.72)Prolastin
(n=32), SY§=3.83)ZEMAIRA
(n=1296)Prolastin
(n=160)- *
- Based on unique subjects. If a subject experienced more than one AR, the subject was only counted once.
- †
- The exposure adjusted event rate was based on total exposure time presented in subject-years and the total number of adverse reactions in the database.
- ‡
- If there were multiple occurrences of ARs following a single infusion, only one occurrence was counted.
- §
- SY=subject-year.
ARs (AEs assessed by investigator as at least possibly related or occurring during or within 72 hours after the end of the infusion or for which causality assessment was missing or indeterminate). 54 (5.6) 16 (3.8) 160 (12.3) 31 (19.4) Serious ARs (Serious AEs assessed by investigator as at least possibly related or occurring during or within 72 hours after the end of the infusion or for which causality assessment was missing or indeterminate). 4 (0.2) 1 (1.0) 6 (0.5) 1 (0.6) Table 2 summarizes the ARs occurring in 5% or more (>3) subjects, expressed as events per subject-year, and the corresponding number of ARs per infusion, expressed as % of all infusions, for each treatment in clinical trials of ZEMAIRA.
Table 2. Adverse Reactions Occurring in ≥5% of Subjects ARs (AEs assessed by investigator as at least possibly related or occurring during or within 72 hours after the end of the infusion or for which causality assessment was missing or indeterminate). Number of Subjects*
(Events per Subject-Year†)Number of Infusions‡
(% of all Infusions)ZEMAIRA
(n=66, SY§=28.72)Prolastin
(n=32, SY§=3.83)ZEMAIRA
(n=1296)Prolastin
(n=160)- *
- Based on unique subjects. If a subject experienced more than one AR of the same type, the subject was only counted once.
- †
- The exposure adjusted event rate was based on total exposure time presented in subject-years and the total number of adverse reactions in the database.
- ‡
- If more than one of the same type of an event occurred after an infusion, only one event was counted.
- §
- SY=subject-year.
Headache 13 (0.7) 5 (1.3) 19 (1.5) 5 (3.1) Sinusitis 10 (0.5) 1 (0.3) 13 (1.0) 1 (0.6) Upper Respiratory Infection 10 (0.4) 1 (0.3) 10 (0.8) 1 (0.6) Bronchitis 5 (0.2) 0 (0.0) 6 (0.5) 0 (0.0) Asthenia 5 (0.2) 2 (0.5) 5 (0.4) 2 (1.3) Cough Increased 5 (0.2) 1 (0.5) 5 (0.4) 2 (1.3) Fever 4 (0.1) 0 (0.0) 4 (0.3) 0 (0.0) Injection Site Hemorrhage 4 (0.1) 0 (0.0) 4 (0.3) 0 (0.0) Rhinitis 4 (0.1) 0 (0.0) 4 (0.3) 0 (0.0) Sore Throat 4 (0.1) 0 (0.0) 4 (0.3) 0 (0.0) Vasodilation 4 (0.1) 1 (0.3) 4 (0.3) 1 (0.6) Diffuse interstitial lung disease was noted on a routine chest x-ray of one subject at Week 24. Causality could not be determined.
Chronic Obstructive Pulmonary Disease (COPD) Exacerbations
In a retrospective analysis, during the 10-week blinded portion of the 24-week clinical trial, 6 subjects (20%) of the 30 treated with ZEMAIRA had a total of 7 exacerbations of their chronic obstructive pulmonary disease (COPD). Nine subjects (64%) of the 14 treated with Prolastin had a total of 11 exacerbations of their COPD. The observed difference between groups was 44% (95% confidence interval [CI] from 8% to 70%). Over the entire 24-week treatment period, of the 30 subjects in the ZEMAIRA treatment group, 7 subjects (23%) had a total of 11 exacerbations of their COPD.
In the RAPID study 25 serious exacerbations of COPD were reported in 15 ZEMAIRA subjects vs. 17 such events in 9 placebo subjects, corresponding to rates of 0.146 exacerbations per subject-year with ZEMAIRA and 0.115 exacerbations per subject-year with placebo, (ratio ZEMAIRA:Placebo [95% confidence interval]: 1.256 [0.457 - 3.454]).
Subjects who were randomized to ZEMAIRA in the 2-year RAPID trial who then entered and received open-label ZEMAIRA in the 2 year RAPID extension trial were in the "Early Start" group. Subjects who were randomized to Placebo in the 2-year RAPID trial who then entered and received open-label ZEMAIRA in the 2 year RAPID extension trial were in the "Delayed Start" group. During the RAPID Extension trial 37 serious exacerbations of COPD were reported in 19 subjects (25%) in the Early Start group, corresponding to rates of 0.25 exacerbations per subject-year. In comparison, 20 serious exacerbations were reported in 11 subjects (17%) in the Delayed Start group corresponding to rates of 0.16 exacerbations per subject-year (ratio Early: Delayed [95% confidence interval]: 1.58 [0.68 – 3.66], Table 3). Among the Early Start subjects who entered the RAPID extension trial (N = 76), the exposure adjusted incidence rate of serious exacerbations during the RAPID extension trial (years 3-4) was 0.25 compared to 0.12 for those subjects during the earlier RAPID trial (years 1-2), (ratio RAPID Extension:RAPID: 2.10 [95% confidence interval: 1.21 – 3.67]). Among the Delayed Start subjects who entered the RAPID extension trial (N = 64), the exposure adjusted incidence rate of serious exacerbations during the RAPID extension trial (years 3-4) was 0.16 compared to 0.10 for those subjects during the earlier RAPID trial (years 1-2), (ratio RAPID Extension:RAPID: 1.56 [95% confidence interval: 0.80 – 3.03]).
Table 3. Comparison of Exposure-Adjusted Incidence Rates (EAIR) for Serious COPD Exacerbations Occurring in the RAPID study between ZEMAIRA and Placebo subjects and in the RAPID Extension Studies between Early Start and Delayed Start subjects Serious COPD Exacerbations* Episode n % EAIR
(95% CI)Episode n % EAIR
95% CITreatment Ratio for EAIR
(95% CI)*N = total number of safety subjects, n = number of subjects within a category, % = (n/N)*100, CI = Confidence Interval.
Subject time at risk: ZEMAIRA = 171.14 years, Placebo = 147.75 years, Early Start Group = 146.46 years, Delay Start Group = 124.71 years.
EAIR = Exposure-Adjusted Incidence Rate (events/subject time at risk). The point estimates and confidence intervals for EAIR values were calculated using negative binomial models.- *
- Episode = Serious exacerbations of COPD identified by investigators as meeting the Anthonisen criteria1 plus Serious Adverse Event (SAE) terms COPD, Condition Aggravated, Bronchitis, Lower Respiratory Tract Infection, Pneumonia. Serious exacerbation events that overlap or occur within 1 day of one another were counted as single exacerbation episodes.
- †
- Early Start Group subjects were randomized to ZEMAIRA during the double-blind RAPID trial (years 1-2) and received open-label ZEMAIRA during the RAPID extension trial (years 3-4).
- ‡
- Delayed Start Group subjects were randomized to Placebo during the double-blind RAPID trial (years 1-2) and received open-label ZEMAIRA during the RAPID extension trial (years 3-4).
RAPID Study
(Years 1 – 2)ZEMAIRA (N = 93) Placebo (N = 87) ZEMAIRA: Placebo 25 15 16.1 0.15 (0.10-0.22) 17 9 10.3 0.12 (0.07-0.18) 1.26 (0.46 - 3.45) Extension Study
(Years 3-4)Early Start† (N = 76) Delayed Start‡ (N = 64) Early: Delayed 37 19 25.0 0.25 (0.18 – 0.35) 20 11 17.2 0.16 (0.10 – 0.25) 1.58 (0.68 – 3.66) In the 24-week double-blind trial, ZEMAIRA-treated subjects were tested for HAV, HBV, HCV, HIV, and parvovirus B19 (B19V), and no evidence of virus transmission was observed.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. No anti-A1PI antibodies have been detected in clinical trials of ZEMAIRA. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to ZEMAIRA with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience
Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
Table 4 lists the ARs that have been identified during postmarketing use of ZEMAIRA. This list does not include reactions already reported in clinical trials with ZEMAIRA [see Adverse Reactions (6.1)].
Table 4. ARs Reported During the Postmarketing Use of ZEMAIRA System Organ Class Preferred Term/Symptoms Blood and lymphatic system disorders Lymph node pain Gastrointestinal disorders Nausea General disorders and administration site conditions Chills, infusion site reactions, facial, periorbital, lip and extremity swelling, chest pain Nervous system disorders Hypoesthesia, paresthesia, dizziness Skin disorders Hyperhidrosis, pruritus, rash including exfoliative and generalized, urticaria Vascular disorders Flushing -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
No animal reproduction studies have been conducted with Zemaira and its safety for use in human pregnancy has not been established in controlled clinical trials. Since alpha1-proteinase inhibitor is an endogenous human protein, it is considered unlikely that Zemaira will cause harm to the fetus when given at recommended doses. However, Zemaira should be given with caution to pregnant women. In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the excretion of ZEMAIRA in human milk, the effect on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ZEMAIRA and any potential adverse effects on the breastfed infant from ZEMAIRA or from the underlying maternal condition.
-
11 DESCRIPTION
ZEMAIRA is a sterile, white to off-white, lyophilized preparation of purified alpha1-proteinase inhibitor (human) (A1-PI), also known as alpha1-antitrypsin, to be reconstituted and administered by the intravenous route. The specific activity of ZEMAIRA is ≥0.7 mg of functional A1-PI per milligram of total protein. The purity (total A1-PI/total protein) is ≥90% A1-PI. Each vial contains approximately 1000 mg, 4000 mg or 5000 mg of functionally active A1-PI. The measured amount per vial of functionally active A1-PI as determined by its capacity to neutralize human neutrophil elastase (NE) is printed on the vial label and carton. Following reconstitution with 20 mL, 76 mL or 95 mL of Sterile Water for Injection, USP, the ZEMAIRA solution contains 73 to 89 mM sodium, 30 to 39 mM chloride, 15 to 20 mM phosphate, and 121 to 168 mM mannitol. Hydrochloric acid and/or sodium hydroxide may have been added to adjust the pH. ZEMAIRA contains no preservative.
All plasma used in the manufacture of ZEMAIRA is obtained from US donors and is tested using serological assays for HBsAg and antibodies to HIV-1/2 and HCV. The plasma is tested with Nucleic Acid Testing (NAT) for HBV, HCV, HIV-1, and HAV, and found to be nonreactive (negative). The plasma is also tested by NAT for B19V. Only plasma that passed the virus screening is used for production. The limit for B19V in the fractionation pool is ≤104 International Units of B19V per mL.
ZEMAIRA is manufactured from large pools of human plasma by cold ethanol fractionation according to a modified Cohn process followed by additional purification steps. The manufacturing process includes two virus clearance steps: heat treatment at 60°C for 10 hours in an aqueous solution with stabilizers; and nanofiltration. These virus clearance steps have been validated in a series of in vitro experiments for their capacity to inactivate/remove both enveloped and non-enveloped viruses. Table 5 shows the virus clearance capacity of the ZEMAIRA manufacturing process, expressed as mean log10 reduction factor.
Table 5. Cumulative (Log10) Virus Inactivation/Removal in ZEMAIRA Manufacturing Step Virus Reduction Factor (Log10) Enveloped Viruses Non-Enveloped Viruses HIV-1 BVDV WNV PRV HAV CPV HIV, human immunodeficiency virus type 1, a model for HIV-1 and HIV-2.
BVDV, bovine viral diarrhea virus, a model for HCV.
WNV, West Nile virus.
PRV, pseudorabies virus, a non-specific model for large DNA viruses, e.g., herpes.
HAV, hepatitis A virus.
CPV, canine parvovirus, model for B19V.
na, not applicable.Heat treatment*† ≥6.8 ≥5.2 ≥8.3 4.4 ≥5.4 na Nanofiltration ≥5.5 ≥5.4 ≥8.4 ≥6.3 ≥5.3 ≥6.4 Cumulative Virus Reduction (log10) ≥12.3 ≥10.6 ≥16.7 ≥10.7 ≥10.7 ≥6.4 -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
A1-PI deficiency is a chronic, hereditary, autosomal, co-dominant disorder that is usually fatal in its severe form. Low blood levels of A1-PI (i.e., below 11 µM) are most commonly associated with progressive, severe emphysema that becomes clinically apparent by the third to fourth decade of life. In addition, PiSZ individuals, whose serum A1-PI levels range from approximately 9 to 23 µM, are considered to have a moderately increased risk for developing emphysema, regardless of whether their serum A1-PI levels are above or below 11 µM.2 Not all individuals with severe genetic variants of A1-PI deficiency have emphysema. Augmentation therapy with alpha1-proteinase inhibitor (human) is indicated only in patients with severe congenital A1-PI deficiency who have clinically evident emphysema. A registry study showed 54% of A1-PI deficient subjects had emphysema.3 Another registry study showed 72% of A1-PI deficient subjects had pulmonary symptoms.4 Smoking is an important risk factor for the development of emphysema in patients with A1-PI deficiency.
Approximately 100 genetic variants of A1-PI deficiency can be identified electrophoretically, only some of which are associated with the clinical disease.5,6 Ninety-five percent of clinically symptomatic A1-PI deficient individuals are of the severe PiZZ phenotype. Up to 39% of A1-PI deficient patients may have an asthmatic component to their lung disease, as evidenced by symptoms and/or bronchial hyperreactivity.3 Pulmonary infections, including pneumonia and acute bronchitis, are common in A1-PI deficient patients and contribute significantly to the morbidity of the disease.
Augmenting the levels of functional protease inhibitor by intravenous infusion is an approach to therapy for patients with A1-PI deficiency. However, the efficacy of augmentation therapy in affecting the progression of emphysema has not been demonstrated in randomized, controlled clinical studies. The intended theoretical goal is to provide protection to the lower respiratory tract by correcting the imbalance between NE and protease inhibitors. Whether augmentation therapy with ZEMAIRA or any A1-PI product actually protects the lower respiratory tract from progressive emphysematous changes has not been evaluated. Individuals with endogenous levels of A1-PI below 11 µM, in general, manifest a significantly increased risk for development of emphysema above the general population background risk.6,7,8,9 Although the maintenance of blood serum levels of A1-PI (antigenically measured) above 11 µM has been historically postulated to provide therapeutically relevant anti-neutrophil elastase protection10, this has not been proven. Individuals with severe A1-PI deficiency have been shown to have increased neutrophil and NE concentrations in lung epithelial lining fluid compared to normal PiMM individuals, and some PiSZ individuals with A1-PI above 11 µM have emphysema attributed to A1-PI deficiency.2 These observations underscore the uncertainty regarding the appropriate therapeutic target serum level of A1-PI during augmentation therapy.
Pulmonary disease, particularly emphysema, is the most frequent manifestation of A1-PI deficiency.6 The pathogenesis of emphysema is understood to evolve as described in the "protease-antiprotease imbalance" model. A1-PI is now understood to be the primary antiprotease in the lower respiratory tract, where it inhibits NE.11 Normal healthy individuals produce sufficient A1-PI to control the NE produced by activated neutrophils and are thus able to prevent inappropriate proteolysis of lung tissue by NE. Conditions that increase neutrophil accumulation and activation in the lung, such as respiratory infection and smoking, will in turn increase levels of NE. However, individuals who are severely deficient in endogenous A1-PI are unable to maintain an appropriate antiprotease defense and are thereby subject to more rapid proteolysis of the alveolar walls leading to chronic lung disease. ZEMAIRA serves as A1-PI augmentation therapy in this patient population, acting to increase and maintain serum levels and ELF levels of A1-PI.
12.2 Pharmacodynamics
Weekly repeated infusions of A1-PI at a dose of 60 mg/kg lead to serum A1-PI levels above the historical target threshold of 11 µM.
The clinical benefit of the increased blood levels of A1-PI at the recommended dose has not been established for any A1-PI product.
12.3 Pharmacokinetics
A double-blind, randomized, active-controlled, crossover pharmacokinetic study was conducted in 13 males and 5 females with A1-PI deficiency, ranging in age from 36 to 66 years. Nine subjects received a single 60 mg/kg dose of ZEMAIRA followed by Prolastin, and 9 subjects received Prolastin followed by a single 60 mg/kg dose of ZEMAIRA, with a wash-out period of 35 days between doses. A total of 13 post-infusion serum samples were taken at various time points up to Day 21. Table 6 shows the mean results for the ZEMAIRA pharmacokinetic parameters.
Table 6. Pharmacokinetic Parameters for Antigenic A1-PI in 18 Subjects Following a Single 60 mg/kg Dose of ZEMAIRA Pharmacokinetic Parameter Mean (SD)* - *
- n=18 subjects.
Area under the curve (AUC0-∞) 144 (±27) µM × day Maximum concentration (Cmax) 44.1 (±10.8) µM Terminal half-life (t1/2ß) 5.1 (±2.4) days Total clearance 603 (±129) mL/day Volume of distribution at steady state 3.8 (±1.3) L -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate carcinogenesis, mutagenesis, or impairment of fertility have not been conducted.
13.2 Animal Toxicology and/or Pharmacology
In a safety pharmacology study, dogs were administered a 60 or 240 mg/kg intravenous dose of ZEMAIRA. At the clinical dose of 60 mg/kg, no changes in cardiovascular and respiratory parameters or measured hematology, blood chemistry, or electrolyte parameters were attributed to the administration of ZEMAIRA. A minor transient decrease in femoral resistance and increase in blood flow were observed after administration of the 240 mg/kg dose.
In single-dose studies, mice and rats were administered a 0, 60, 240, or 600 mg/kg intravenous dose of ZEMAIRA and observed twice daily for 15 days. No signs of toxicity were observed up to 240 mg/kg. Transient signs of distress were observed in male mice and in male and female rats after administration of the highest dose (600 mg/kg).
In repeat-dose toxicity studies, rats and rabbits received 0, 60, or 240 mg/kg intravenous doses of ZEMAIRA once daily for 5 consecutive days. No treatment-related effects on clinical signs, body weight, hematology, coagulation, or urinalysis were observed in rats administered up to 240 mg/kg. No signs of toxicity were observed in rabbits administered 60 mg/kg. Changes in organ weights and minimal epidermal ulceration were observed in rabbits administered 240 mg/kg, but had no clinical effects.
The local tolerance of ZEMAIRA was evaluated in rabbits following intravenous, perivenous, and intraarterial administration. No treatment-related local adverse reactions were observed.
-
14 CLINICAL STUDIES
Clinical trials were conducted pre-licensure with ZEMAIRA in 89 subjects (59 males and 30 females). The subjects ranged in age from 29 to 68 years (median age 49 years). Ninety-seven percent of the treated subjects had the PiZZ phenotype of A1-PI deficiency, and 3% had the MMALTON phenotype. At screening, serum A1-PI levels were between 3.2 and 10.1 µM (mean of 5.6 µM). The objectives of the clinical trials were to demonstrate that ZEMAIRA augments and maintains serum levels of A1-PI above 11 µM (80 mg/dL) and increases A1-PI levels in ELF of the lower lung.
In a double-blind, controlled clinical trial to evaluate the safety and efficacy of ZEMAIRA, 44 subjects were randomized to receive 60 mg/kg of either ZEMAIRA or Prolastin once weekly for 10 weeks. After 10 weeks, subjects in both groups received ZEMAIRA for an additional 14 weeks. Subjects were followed for a total of 24 weeks to complete the safety evaluation [see Adverse Reactions (6.1)]. The mean trough serum A1-PI levels at steady state (Weeks 7-11) in the ZEMAIRA-treated subjects were statistically equivalent to those in the Prolastin-treated subjects within a range of ±3 µM. Both groups were maintained above 11 µM. The mean (range and standard deviation [SD]) of the steady state trough serum antigenic A1-PI level for ZEMAIRA-treated subjects was 17.7 µM (range 13.9 to 23.2, SD 2.5) and for Prolastin-treated subjects was 19.1 µM (range 14.7 to 23.1, SD 2.2). The difference between the ZEMAIRA and the Prolastin groups was not considered clinically significant and may be related to the higher specific activity of ZEMAIRA.
In a subgroup of subjects enrolled in the trial (10 ZEMAIRA-treated subjects and 5 Prolastin-treated subjects), bronchoalveolar lavage was performed at baseline and at Week 11. Four A1-PI related analytes in ELF were measured: antigenic A1-PI, A1-PI:NE complexes, free NE, and functional A1-PI (ANEC). A blinded retrospective analysis, which revised the prospectively established acceptance criteria showed that within each treatment group, ELF levels of antigenic A1-PI and A1-PI:NE complexes increased from baseline to Week 11 (Table 7). Free elastase was immeasurably low in all samples. The post-treatment ANEC values in ELF were not significantly different between the ZEMAIRA-treated and Prolastin-treated subjects (mean 1725 nM vs. 1418 nM). No conclusions can be drawn about changes of ANEC values in ELF during the trial period as baseline values in the ZEMAIRA-treated subjects were unexpectedly high. No A1-PI analytes showed any clinically significant differences between the ZEMAIRA and Prolastin treatment groups.
Table 7. Change in ELF From Baseline to Week 11 in a Subgroup Analysis Analyte Treatment Mean Change From Baseline 90% CI CI, confidence interval. A1-PI (nM) ZEMAIRA* 1358.3 822.6 to 1894.0 Prolastin† 949.9 460.0 to 1439.7 ANEC (nM) ZEMAIRA -588.1 -2032.3 to 856.1 Prolastin 497.5 -392.3 to 1387.2 A1-PI:NE Complexes (nM) ZEMAIRA 118.0 39.9 to 196.1 Prolastin 287.1 49.8 to 524.5 The clinical efficacy of ZEMAIRA or any A1-PI product in influencing the course of pulmonary emphysema or pulmonary exacerbations has not been demonstrated in adequately powered, randomized, controlled clinical trials.
-
15 REFERENCES
- Anthonisen NR, Connett, JE, Kiley, JP, et al. Effects of Smoking Intervention and the Use of an Inhaled Anticholinergic Bronchodilator and on the Rate of Decline of FEV1 – The Lung Study. JAMA. 1994;272(19):1497-1505.
- Turino GM, Barker AF, Brantly ML, et al. Clinical features of individuals with PI*SZ phenotype of α1-antitrypsin deficiency. Am J Respir Crit Care Med. 1996;154:1718-1725.
- Stoller JK, Brantly M, et al. Formation and current results of a patient-organized registry for α1-antitrypsin deficiency. Chest. 2000;118(3):843-848.
- McElvaney NG, Stoller JK, et al. Baseline characteristics of enrollees in the National Heart, Lung, and Blood Institute Registry of α1-Antitrypsin Deficiency. Chest. 1997;111:394-403.
- Crystal RG. α1-antitrypsin deficiency, emphysema, and liver disease; genetic basis and strategies for therapy. J Clin Invest. 1990;85:1343-1352.
- World Health Organization. Alpha-1-antitrypsin deficiency; Report of a WHO Meeting. Geneva. 18-20 March 1996.
- Eriksson S. Pulmonary emphysema and alpha1-antitrypsin deficiency. ACTA Med Scand. 1964;175(2):197-205.
- Eriksson S. Studies in α1-antitrypsin deficiency. ACTA Med Scan Suppl. 1965;432:1-85.
- Gadek JE, Crystal RG. α1-antitrypsin deficiency. In: Stanbury JB, Wyngaarden JB, Frederickson DS, et al., eds. The Metabolic Basis of Inherited Disease. 5th ed. New York, NY: McGraw-Hill; 1983:1450-1467.
- American Thoracic Society. Guidelines for the approach to the patient with severe hereditary alpha-1-antitrypsin deficiency. Am Rev Respir Dis. 1989;140:1494-1497.
- Gadek JE, Fells GA, Zimmerman RL, Rennard SI, Crystal RG. Antielastases of the human alveolar structures; implications for the protease-antiprotease theory of emphysema. J Clin Invest. 1981;68:889-898.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
ZEMAIRA is supplied in a single-dose vial containing the amount of functionally active A1-PI printed on the vial label and carton.
The product presentations include a package insert and the following components. Not made with natural rubber latex.
Table 8: How Supplied Presentation Kit NDC Number Components 1000 mg of functionally active A1-PI 0053-7201-02 - ZEMAIRA in a single-dose vial [NDC 0053-7211-01]
- 20 mL vial of Sterile Water for Injection, USP [NDC 0053-7653-20]
- One Mix2Vial filter transfer set for reconstitution
4000 mg of functionally active A1-P1 0053-7202-02 - ZEMAIRA in a single-dose vial [NDC 0053-7212-01]
- 76 mL vial of Sterile Water for Injection, USP [NDC 0053-7653-80]
- One Mix2Vial filter transfer set for reconstitution
5000 mg of functionally active A1-PI 0053-7203-02 - ZEMAIRA in a single-dose vial [NDC 0053-7213-01]
- 95 mL vial of Sterile Water for Injection, USP [NDC 0053-7653-12]
- One Mix2Vial filter transfer set for reconstitution
-
17 PATIENT COUNSELING INFORMATION
- Inform patients of the early signs of hypersensitivity reactions to ZEMAIRA (including hives, generalized urticaria, tightness of the chest, dyspnea, wheezing, faintness, hypotension, and anaphylaxis). Advise patients to discontinue use of ZEMAIRA and contact their physician and/or seek immediate emergency care, depending on the severity of the reaction, if these symptoms occur [see Warnings and Precautions (5.2)].
- Inform patients that because ZEMAIRA is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent [see Warnings and Precautions (5.3)].
- Inform patients that administration of ZEMAIRA has been demonstrated to raise the plasma level of A1-PI, but that the effect of this augmentation on the frequency of pulmonary exacerbations and on the rate of progression of emphysema has not been established by clinical trials.
- Dizziness may occur following the administration of ZEMAIRA; therefore, patients should rest for a while immediately following an infusion.
-
SPL UNCLASSIFIED SECTION
Manufactured by:
CSL Behring LLC
Kankakee, IL 60901 USA
US License No. 1767For patent information: http://www.cslbehring.com/products/patents
Prolastin is a registered trademark of Grifols Therapeutics Inc.
Mix2Vial® is a registered trademark of West Pharma. Services IL, Ltd., a subsidiary of West Pharmaceuticals Services, Inc.
-
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC 0053-7201-02
One single dose vial with diluent1000 mg Range
Alpha1-Proteinase Inhibitor
(Human)
Zemaira®For Intravenous Administration Only
Rx onlyThis package contains one vial of Zemaira®, one vial of Sterile Water for Injection, USP and
one Mix2Vial® filter transfer set for reconstitution.Storage: Zemaira® stored up to 25°C (77°F) is stable for the period indicated by the expiration date
on the label. Avoid freezing, which may damage the diluent vial. Discard any unused product and
all used disposable supplies.Manufactured by:
CSL Behring LLC
Kankakee, IL 60901 USA
US License No. 1767CSL Behring

- PRINCIPAL DISPLAY PANEL - 1000 mg Vial Label
- PRINCIPAL DISPLAY PANEL - 20 mL Vial Label
-
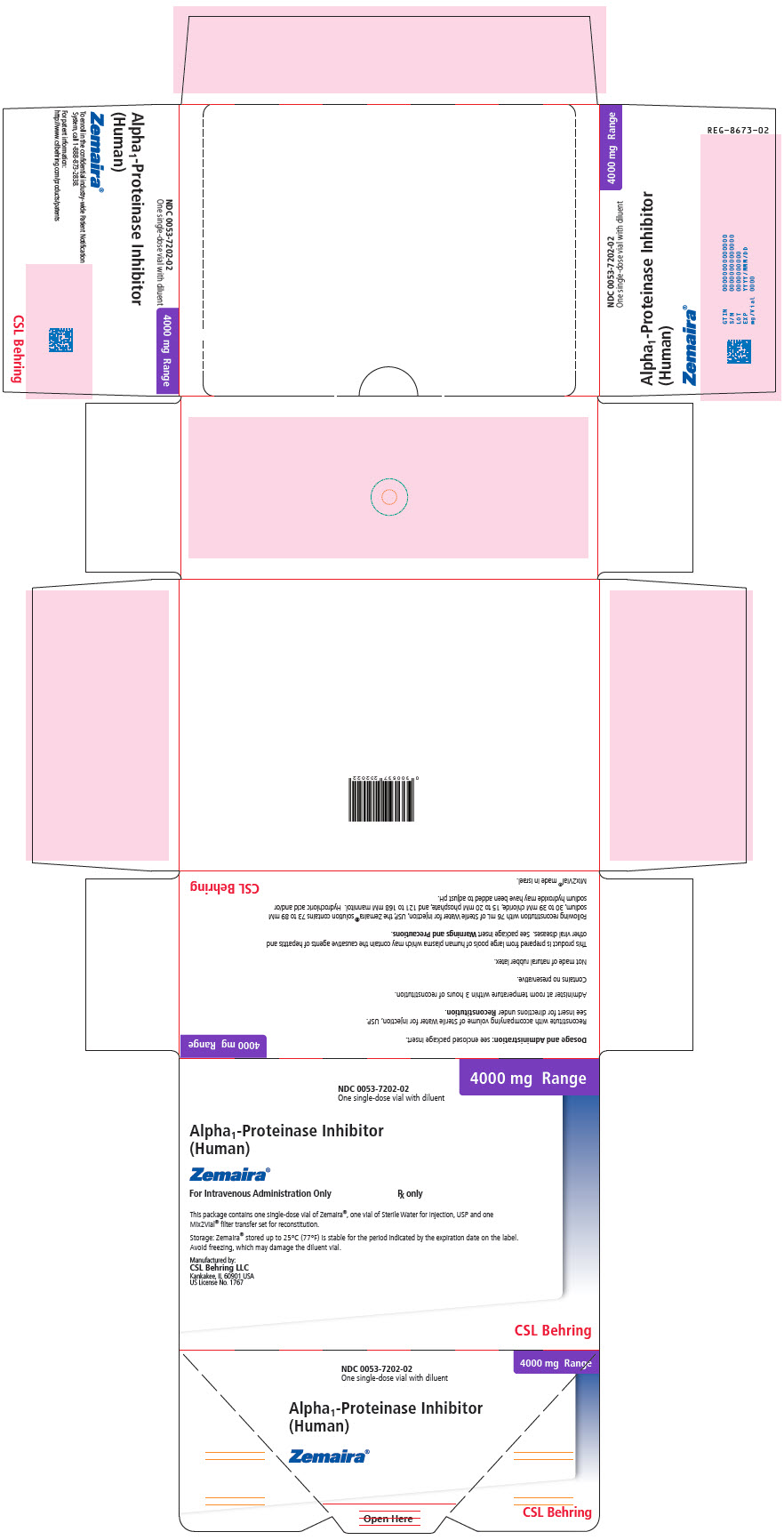
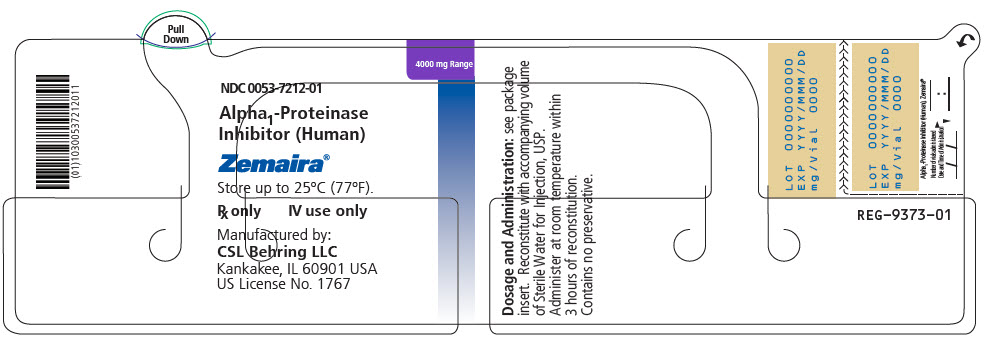
PRINCIPAL DISPLAY PANEL - 4000 mg Range - Kit Carton
NDC 0053-7202-02
One single-dose vial with diluent4000 mg Range
Alpha1-Proteinase Inhibitor
(Human)Zemaira®
For Intravenous Administration Only
Rx onlyThis package contains one single-dose vial of Zemaira®, one vial of Sterile Water for Injection, USP and one
Mix2Vial® filter transfer set for reconstitution.Storage: Zemaira® stored up to 25°C (77°F) is stable for the period indicated by the expiration date on the label.
Avoid freezing, which may damage the diluent vial.Manufactured by:
CSL Behring LLC
Kankakee, IL 60901 USA
US License No. 1767CSL Behring
- PRINCIPAL DISPLAY PANEL - 4000 mg Vial Label
- PRINCIPAL DISPLAY PANEL - 76 mL Vial Label
-
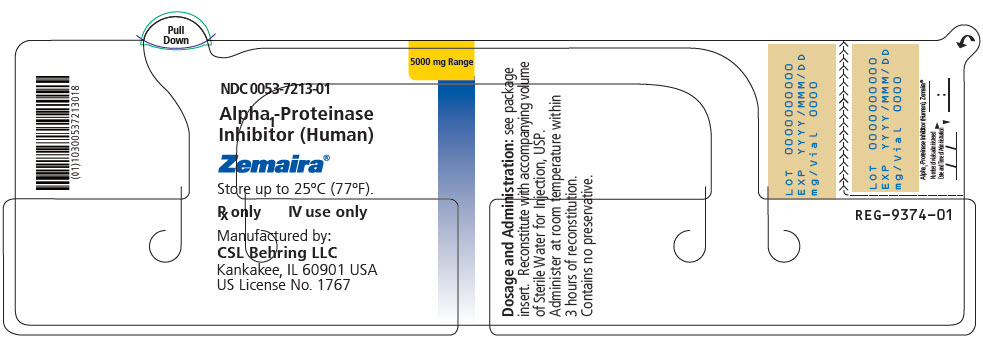
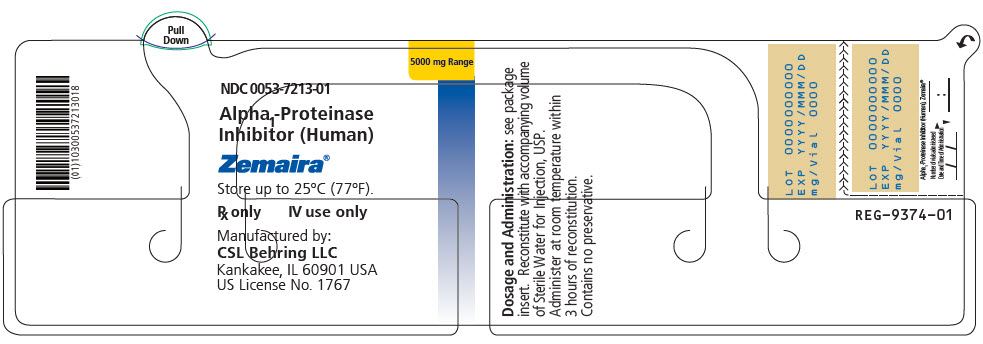
PRINCIPAL DISPLAY PANEL - 5000 mg Range - Kit Carton
NDC 0053-7203-02
One single dose vial with diluent5000 mg Range
Alpha1-Proteinase Inhibitor
(Human)Zemaira®
For Intravenous Administration Only
Rx onlyThis package contains one vial of Zemaira®, one vial of Sterile Water for Injection, USP and one
Mix2Vial® filter transfer set for reconstitution.Storage: Zemaira® stored up to 25°C (77°F) is stable for the period indicated by the expiration date on the label.
Avoid freezing, which may damage the diluent vial.Manufactured by:
CSL Behring LLC
Kankakee, IL 60901 USA
US License No. 1767CSL Behring
- PRINCIPAL DISPLAY PANEL - 5000 mg Vial Label
- PRINCIPAL DISPLAY PANEL - 95 mL Vial Label
-
INGREDIENTS AND APPEARANCE
ZEMAIRA
alpha-1-proteinase inhibitor human kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0053-7201 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7201-02 1 in 1 CARTON Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-DOSE 20 mL Part 2 1 VIAL, SINGLE-DOSE 20 mL Part 1 of 2 ALPHA-1-PROTEINASE INHIBITOR HUMAN
alpha-1-proteinase inhibitor human injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC:0053-7211 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength .ALPHA.1-PROTEINASE INHIBITOR HUMAN (UNII: F43I396OIS) (.ALPHA.1-PROTEINASE INHIBITOR HUMAN - UNII:F43I396OIS) .ALPHA.1-PROTEINASE INHIBITOR HUMAN 1000 mg in 20 mL Inactive Ingredients Ingredient Name Strength Sodium Chloride (UNII: 451W47IQ8X) 41 mg in 20 mL Sodium Phosphate (UNII: SE337SVY37) 47 mg in 20 mL Mannitol (UNII: 3OWL53L36A) 525 mg in 20 mL hydrochloric acid (UNII: QTT17582CB) sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7211-01 20 mL in 1 VIAL, SINGLE-DOSE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 07/08/2003 Part 2 of 2 DILUENT
water injectionProduct Information Item Code (Source) NDC:0053-7653 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength Water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7653-20 20 mL in 1 VIAL, SINGLE-DOSE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 07/08/2003 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 07/08/2003 ZEMAIRA
.alpha.1-proteinase inhibitor human kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0053-7202 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7202-02 1 in 1 CARTON Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 76 mL Part 2 1 VIAL, SINGLE-USE 76 mL Part 1 of 2 .ALPHA.1-PROTEINASE INHIBITOR HUMAN
.alpha.1-proteinase inhibitor human injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC:0053-7212 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength .ALPHA.1-PROTEINASE INHIBITOR HUMAN (UNII: F43I396OIS) (.ALPHA.1-PROTEINASE INHIBITOR HUMAN - UNII:F43I396OIS) .ALPHA.1-PROTEINASE INHIBITOR HUMAN 4000 mg in 76 mL Inactive Ingredients Ingredient Name Strength Sodium Chloride (UNII: 451W47IQ8X) 163 mg in 76 mL Sodium Phosphate (UNII: SE337SVY37) 188 mg in 76 mL Mannitol (UNII: 3OWL53L36A) 2100 mg in 76 mL hydrochloric acid (UNII: QTT17582CB) sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7212-01 76 mL in 1 VIAL, SINGLE-USE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 09/30/2023 Part 2 of 2 DILUENT
water injectionProduct Information Item Code (Source) NDC:0053-7653 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength Water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7653-80 76 mL in 1 VIAL, SINGLE-USE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 04/24/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 04/24/2019 ZEMAIRA
.alpha.1-proteinase inhibitor human kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0053-7203 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7203-02 1 in 1 CARTON Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 95 mL Part 2 1 VIAL, SINGLE-USE 95 mL Part 1 of 2 .ALPHA.1-PROTEINASE INHIBITOR HUMAN
.alpha.1-proteinase inhibitor human injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC:0053-7213 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength .ALPHA.1-PROTEINASE INHIBITOR HUMAN (UNII: F43I396OIS) (.ALPHA.1-PROTEINASE INHIBITOR HUMAN - UNII:F43I396OIS) .ALPHA.1-PROTEINASE INHIBITOR HUMAN 5000 mg in 95 mL Inactive Ingredients Ingredient Name Strength Sodium Chloride (UNII: 451W47IQ8X) 204 mg in 95 mL Sodium Phosphate (UNII: SE337SVY37) 235 mg in 95 mL Mannitol (UNII: 3OWL53L36A) 2625 mg in 95 mL hydrochloric acid (UNII: QTT17582CB) sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7213-01 95 mL in 1 VIAL, SINGLE-USE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 09/30/2023 Part 2 of 2 DILUENT
water injectionProduct Information Item Code (Source) NDC:0053-7653 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength Water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0053-7653-12 95 mL in 1 VIAL, SINGLE-USE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 04/24/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125078 04/24/2019 Labeler - CSL Behring LLC (058268293) Establishment Name Address ID/FEI Business Operations CSL Behring LLC 058268293 MANUFACTURE(0053-7201, 0053-7202, 0053-7203)