Label: VIRACEPT- nelfinavir mesylate tablet, film coated


- NDC Code(s): 63010-010-30, 63010-027-70
- Packager: AGOURON
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated March 22, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VIRACEPT safely and effectively. See full prescribing information for VIRACEPT.
VIRACEPT® (nelfinavir mesylate) Tablets, for oral use
VIRACEPT® (nelfinavir mesylate) Oral Powder, for oral use
Initial U.S. Approval: 1997INDICATIONS AND USAGE
VIRACEPT is a protease inhibitor indicated for the treatment of HIV-1 infection in combination with other antiretroviral agents. (1)
DOSAGE AND ADMINISTRATION
- •
- See full prescribing information for administration instructions (2)
- •
- Adults and adolescents 13 years and older (tablets): 1250 mg twice daily or 750 mg three times daily with a meal (2.1)
- •
- Children 2 to less than 13 years (oral powder or 250 mg tablets): 45 to 55 mg/kg twice daily or 25 to 35 mg/kg three times daily with a meal. Refer to Tables 1 and 2 of the full prescribing information for specific dosing guidelines based on age and body weight (2.2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- •
- Coadministration with drugs that are highly dependent on CYP3A for clearance and which elevated concentrations are associated with serious and/or life-threatening events (4)
WARNINGS AND PRECAUTIONS
ALERT: Find out about medicines that should not be taken with VIRACEPT.
- •
- The concomitant use of VIRACEPT and certain other drugs may result in known or potentially significant drug interactions. Consult the full prescribing information prior to and during treatment for potential drug interactions (5.1, 7.3)
- •
- Hepatic impairment: should not be used in patients with either moderate or severe hepatic impairment (2.4, 5.2)
- •
- Phenylketonuria: the oral powder contains 11.2 mg phenylalanine per gram of powder (5.3)
- •
- Diabetes mellitus/hyperglycemia: new onset or exacerbation of pre-existing diabetes mellitus and hyperglycemia reported with protease inhibitors. In some cases after treatment discontinuation, hyperglycemia persisted (5.4)
- •
- Hemophilia: increased bleeding, including spontaneous skin hematomas and hemarthrosis reported with protease inhibitors. In more than half of the cases, protease inhibitors was continued or reintroduced (5.5)
- •
- Fat redistribution: observed with antiretroviral therapy (5.6)
- •
- Immune reconstitution syndrome: reported with combination antiretroviral therapy, including VIRACEPT. Patients may develop an inflammatory response to indolent or residual opportunistic infections (5.7)
ADVERSE REACTIONS
- •
- Most common adverse reactions (≥2%) of moderate or severe intensity in adults and adolescents (13 years and older) are diarrhea, nausea, rash, and flatulence (6.1)
- •
- Most common adverse reactions in pediatric patients (2 to less than 13 years) are diarrhea, leukopenia/neutropenia, rash, anorexia, and abdominal pain. (6.2)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- •
- Coadministration of VIRACEPT with other drugs (CYP3A substrates) can alter the concentration of these other drugs, and other drugs (inhibitors and/or inducers of CYP3A or CYP2C19) may alter the concentrations of nelfinavir. The potential drug-drug concentrations must be considered prior to and during therapy (4, 7, 12.3)
- •
- VIRACEPT should be given with food one hour after or more than 2 hours before didanosine (7)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Adults and Adolescents (13 years and older)
2.2 Pediatric Patients (2 to less than 13 years)
2.3 Method of Administration
2.4 Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Serious Adverse Reactions Due to Drug Interactions
5.2 Hepatic Impairment
5.3 Phenylketonurics
5.4 Diabetes Mellitus/Hyperglycemia
5.5 Hemophilia
5.6 Fat Redistribution
5.7 Immune Reconstitution Syndrome
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience: Adults and Adolescents (13 years and older)
6.2 Clinical Trials Experience: Pediatrics (2 to less than 13 years of age)
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Potential for VIRACEPT to Affect Other Drugs
7.2 Potential for Other Drugs to Affect VIRACEPT
7.3 Established and Other Potentially Significant Drug Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Studies in Antiretroviral Treatment Naïve Adult Patients
14.2 Studies in Antiretroviral Treatment Experienced Adult Patients
14.3 Studies in Pediatric Patients
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Adults and Adolescents (13 years and older)
The recommended dose is 1250 mg (five 250 mg tablets or two 625 mg tablets) twice daily or 750 mg (three 250 mg tablets) three times daily. VIRACEPT should be taken with a meal. Patients unable to swallow the 250 or 625 mg tablets may dissolve the tablets in a small amount of water [see Dosage and Administration (2.3)].
2.2 Pediatric Patients (2 to less than 13 years)
In children 2 years of age and older, the recommended oral dose of VIRACEPT Oral Powder or 250 mg tablets is 45 to 55 mg/kg twice daily or 25 to 35 mg/kg three times daily. All doses should be taken with a meal. Doses higher than the adult maximum dose of 2500 mg per day have not been studied in children.
For children unable to swallow tablets, VIRACEPT 250 mg tablet(s) may be dissolved in a small amount of water or, VIRACEPT Oral Powder may be administered [see Dosage and Administration (2.3)].
The healthcare provider should assess appropriate formulation and dosage for each patient. Tables 1 and 2 provide dosing guidelines for VIRACEPT tablets and powder based on age and body weight.
Table 1: Dosing Table for Children 2 to less than 13 years of age (tablets) Body weight Twice daily (BID)
45 – 55 mg/kg
≥2 yearsThree times daily (TID)
25 – 35 mg/kg
≥2 yearsNumber of tablets
(250 mg)Number of tablets
(250 mg)Kg 10 – 12
2
1
13 – 18
3
2
19 – 20
4
2
≥21
4 – 5*
3†
Table 2: Dosing Table for Children 2 to less than 13 years of age (powder) Body weight Twice daily (BID)
45 – 55 mg/kgThree times daily (TID)
25 – 35 mg/kgKg
Scoops of powder
(50 mg/1 g)Teaspoons* of powder
Scoops of powder
(50 mg/1 g)Teaspoons* of powder
9.0 to <10.5
10
2½
6
1½
10.5 to <12
11
2¾
7
1¾
12 to <14
13
3¼
8
2
14 to <16
15
3¾
9
2¼
16 to <18
Not recommended†
Not recommended†
10
2½
18 to <23
Not recommended†
Not recommended†
12
3
≥23
Not recommended†
Not recommended†
15
3¾
2.3 Method of Administration
For Patients Unable to Swallow Viracept Tablets
- •
- Place VIRACEPT tablet(s) in small amount of water.
- •
- Once dissolved, mix the cloudy liquid well, and consume it immediately.
- •
- The glass should be rinsed with water and the rinse swallowed to ensure the entire dose is consumed.
Viracept Oral Powder
- •
- Mix VIRACEPT Oral Powder with a small amount of water, milk, formula, soy formula, soy milk, or dietary supplements
- •
- Once mixed, the entire contents must be consumed in order to obtain the full dose.
- •
- If the mixture is not consumed immediately, it must be stored under refrigeration, but storage must not exceed 6 hours.
- •
- Acidic food or juice (e.g., orange juice, apple juice, or apple sauce) are not recommended for mixing VIRACEPT Oral Powder because the combination may result in a bitter taste.
- •
- VIRACEPT Oral Powder should not be reconstituted with water in its original container.
2.4 Hepatic Impairment
VIRACEPT can be used in patients with mild hepatic impairment without any dose adjustment. VIRACEPT should not be used in patients with either moderate or severe hepatic impairment [see Warnings and Precautions (5.2), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
VIRACEPT 250 mg Tablet: Light-blue, capsule-shaped tablets with a clear film coating engraved with "VIRACEPT" on one side and "250 mg" on the other.
VIRACEPT 625 mg Tablet: White oval tablet with a clear film coating engraved with "V" on one side and "625" on the other.
VIRACEPT Oral Powder: Off-white powder containing 50 mg (as nelfinavir-free base) in each level scoopful (1 gram).
-
4 CONTRAINDICATIONS
Coadministration of VIRACEPT is contraindicated with drugs that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events. These drugs and other contraindicated drugs (which may lead to reduced efficacy of nelfinavir) are listed in Table 3 [also see Drug Interactions (7), Table 6].
Table 3: Drugs That Are Contraindicated With VIRACEPT Drug Class Drugs Within Class That Are Contraindicated With VIRACEPT Clinical Comment - *
- See Drug Interactions, Table 6 for coadministration of sildenafil and tadalafil when dosed for erectile dysfunction.
Alpha 1-adrenoreceptor antagonist
Alfuzosin
Potentially increased alfuzosin concentrations can result in hypotension.
Antiarrhythmics
Amiodarone, quinidine
Potential for serious and/or life-threatening cardiac arrhythmia.
Antimycobacterial Agents
Rifampin
Plasma concentrations of nelfinavir can be reduced by concomitant use of rifampin. This may lead to loss of therapeutic effect and possible development of resistance to VIRACEPT or other coadministered antiretroviral agents.
Antipsychotics
Lurasidone
PimozidePotential for serious and/or life-threatening reactions.
Potential for serious and/or life threatening reactions such as cardiac arrhythmias.Ergot Derivatives
Dihydroergotamine, ergotamine, methylergonovine
Potential for serious and/or life threatening reactions such as ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues.
GI Motility Agent
Cisapride
Potential for serious and/or life threatening reactions such as cardiac arrhythmias.
Herbal products
St. John's wort (Hypericum perforatum)
Plasma concentrations of nelfinavir can be reduced by concomitant use of the herbal preparation St. John's wort. This may lead to loss of therapeutic effect and possible development of resistance to VIRACEPT or other coadministered antiretroviral agents.
HMG-CoA Reductase Inhibitors
Lovastatin, Simvastatin
Potential for serious reactions such as myopathy including rhabdomyolysis.
PDE5 Inhibitors
Sildenafil (Revatio®) [for treatment of pulmonary arterial hypertension]*
A safe and effective dose has not been established when used with nelfinavir. There is increased potential for sildenafil-associated adverse events (which include visual disturbances, hypotension, prolonged erection, and syncope).
Sedative/Hypnotics
Triazolam, oral midazolam
Potential for serious and/or life threatening reactions such as prolonged or increased sedation or respiratory depression.
-
5 WARNINGS AND PRECAUTIONS
ALERT: Find out about medicines that should not be taken with VIRACEPT. This statement is included on the product's bottle label.
5.1 Risk of Serious Adverse Reactions Due to Drug Interactions
Initiation of VIRACEPT, a CYP3A inhibitor, in patients receiving medications metabolized by CYP3A or initiation of medications metabolized by CYP3A in patients already receiving VIRACEPT, may increase plasma concentrations of medications metabolized by CYP3A. Initiation of medications that inhibit or induce CYP3A may increase or decrease concentrations of VIRACEPT, respectively. These interactions may lead to:
- •
- Clinically significant adverse reactions, potentially leading to severe, life threatening, or fatal events from greater exposures of concomitant medications.
- •
- Clinically significant adverse reactions from greater exposures of VIRACEPT.
- •
- Loss of therapeutic effect of VIRACEPT and possible development of resistance.
See Table 6 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations [see Drug Interactions (7)]. Consider the potential for drug interactions prior to and during VIRACEPT therapy; review concomitant medications during VIRACEPT therapy; and monitor for the adverse reactions associated with the concomitant medications [see Contraindications (4) and Drug Interactions (7)].
5.2 Hepatic Impairment
VIRACEPT should not be used in patients with either moderate or severe hepatic impairment (Child-Pugh B or C, score greater than or equal to 7) [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
5.3 Phenylketonurics
Viracept Oral Powder contains phenylalanine, a component of aspartame. Each gram of VIRACEPT powder contains 11.2 mg phenylalanine. Phenylalanine can be harmful to patients with phenylketonuria.
5.4 Diabetes Mellitus/Hyperglycemia
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus and hyperglycemia have been reported during post-marketing surveillance in HIV-infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established.
5.5 Hemophilia
There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis, in patients with hemophilia type A and B treated with protease inhibitors. In some patients, additional factor VIII was given. In more than half of the reported cases, treatment with protease inhibitors was continued or reintroduced. A causal relationship has not been established.
5.6 Fat Redistribution
Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement ("buffalo hump"), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
5.7 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including VIRACEPT. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections [such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jiroveci pneumonia (PCP), or tuberculosis], which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
-
6 ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience: Adults and Adolescents (13 years and older)
The safety of VIRACEPT was studied in over 5000 patients who received drug either alone or in combination with nucleoside analogues. The majority of adverse events were of mild intensity. The most frequently reported adverse event among patients receiving VIRACEPT was diarrhea, which was generally of mild to moderate intensity.
Drug-related clinical adverse experiences of moderate or severe intensity in ≥2% of patients treated with VIRACEPT coadministered with d4T and 3TC (Study 542) for up to 48 weeks, or with ZDV plus 3TC (Study 511) for up to 24 weeks are presented in Table 4.
Table 4: Percentage of Patients with Treatment-Emergent* Adverse Events of Moderate or Severe Intensity Reported in ≥ 2% of Adult and Adolescent Patients Study 511 Study 542 24 weeks 48 weeks
Adverse EventsPlacebo
+ ZDV/3TC
(n=101)500 mg TID VIRACEPT +
ZDV/3TC
(n=97)750 mg TID VIRACEPT + ZDV/3TC
(n=100)1250 mg BID VIRACEPT + d4T/3TC
(n=344)750 mg TID VIRACEPT + d4T/3TC
(n=210)- *
- Includes those adverse events at least possibly, probably or definitely related to study drug or of unknown relationship and excludes concurrent HIV conditions
Digestive System
Diarrhea
3%
14%
20%
20%
15%
Nausea
4%
3%
7%
3%
3%
Flatulence
0
5%
2%
1%
1%
Skin/Appendages
Rash
1%
1%
3%
2%
1%
Adverse events occurring in less than 2% of patients receiving VIRACEPT in all phase 2 and 3 clinical trials and considered at least possibly related or of unknown relationship to treatment and of at least moderate severity are listed below.
Body as a Whole: abdominal pain, accidental injury, allergic reaction, asthenia, back pain, fever, headache, malaise, pain, and redistribution/accumulation of body fat [see Warnings and Precautions (5.7)].
Digestive System: anorexia, dyspepsia, epigastric pain, gastrointestinal bleeding, hepatitis, mouth ulceration, pancreatitis, and vomiting.
Hemic/Lymphatic System: anemia, leukopenia, and thrombocytopenia.
Metabolic/Nutritional System: increases in alkaline phosphatase, amylase, creatine phosphokinase, lactic dehydrogenase, SGOT, SGPT, and gamma-glutamyl transpeptidase; hyperlipemia, hyperuricemia, hyperglycemia, hypoglycemia, dehydration, and liver function tests abnormal.
Musculoskeletal System: arthralgia, arthritis, cramps, myalgia, myasthenia, and myopathy.
Nervous System: anxiety, depression, dizziness, emotional lability, hyperkinesia, insomnia, migraine, paresthesia, seizures, sleep disorder, somnolence, and suicide ideation.
Respiratory System: dyspnea, pharyngitis, rhinitis, and sinusitis.
Skin/Appendages: dermatitis, folliculitis, fungal dermatitis, maculopapular rash, pruritus, sweating, and urticaria.
Special Senses: acute iritis and eye disorder.
Urogenital System: kidney calculus, sexual dysfunction, and urine abnormality.
Laboratory Abnormalities
The percentage of patients with marked laboratory abnormalities in Studies 542 and 511 are presented in Table 5. Marked laboratory abnormalities are defined as a Grade 3 or 4 abnormality in a patient with a normal baseline value, or a Grade 4 abnormality in a patient with a Grade 1 abnormality at baseline.
Table 5: Percentage of Patients by Treatment Group with Marked Laboratory Abnormalities* in >2% of Patients Study 511 Study 542 Placebo + ZDV/3TC
(n=101)500 mg TID VIRACEPT +
ZDV/3TC
(n=97)750 mg TID VIRACEPT + ZDV/3TC
(n=100)1250 mg BID VIRACEPT + d4T/3TC
(n=344)750 mg TID VIRACEPT + d4T/3TC
(n=210)- *
- Marked laboratory abnormalities are defined as a shift from Grade 0 at baseline to at least Grade 3 or from Grade 1 to Grade 4
Hematology
Hemoglobin
6%
3%
2%
0
0
Neutrophils
4%
3%
5%
2%
1%
Lymphocytes
1%
6%
1%
1%
0
Chemistry
ALT (SGPT)
6%
1%
1%
2%
1%
AST (SGOT)
4%
1%
0
2%
1%
Creatine Kinase
7%
2%
2%
NA
NA
6.2 Clinical Trials Experience: Pediatrics (2 to less than 13 years of age)
VIRACEPT has been studied in approximately 400 pediatric patients in clinical trials from birth to 13 years of age. The adverse event profile seen during pediatric clinical trials was similar to that for adults.
The most commonly reported drug-related, treatment-emergent adverse events reported in the pediatric studies included: diarrhea, leukopenia/neutropenia, rash, anorexia, and abdominal pain. Diarrhea, regardless of assigned relationship to study drug, was reported in 39% to 47% of pediatric patients receiving VIRACEPT in 2 of the larger treatment trials. Leukopenia/neutropenia was the laboratory abnormality most commonly reported as a significant event across the pediatric studies.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of VIRACEPT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a Whole: hypersensitivity reactions (including bronchospasm, moderate to severe rash, fever, and edema).
Cardiovascular System: QTc prolongation, torsades de pointes.
Digestive System: jaundice.
Metabolic/Nutritional System: bilirubinemia, metabolic acidosis.
-
7 DRUG INTERACTIONS
7.1 Potential for VIRACEPT to Affect Other Drugs
Nelfinavir is an inhibitor of CYP3A. Coadministration of VIRACEPT and drugs primarily metabolized by CYP3A (e.g., dihydropyridine calcium channel blockers, HMG-CoA reductase inhibitors, immunosuppressants, and PDE5 inhibitors) may result in increased plasma concentrations of such drugs that could increase or prolong both its therapeutic and adverse effects (see Tables 3 and 6).
7.2 Potential for Other Drugs to Affect VIRACEPT
Nelfinavir is metabolized by CYP3A and CYP2C19. Coadministration of VIRACEPT and drugs that induce CYP3A or CYP2C19, such as rifampin, may decrease nelfinavir plasma concentrations and reduce its therapeutic effect. Coadministration of VIRACEPT and drugs that inhibit CYP3A or CYP2C19 may increase nelfinavir plasma concentrations.
7.3 Established and Other Potentially Significant Drug Interactions
Table 6 provides the effect on concentrations of VIRACEPT or concomitant drug as a result of coadministration with VIRACEPT. These recommendations are based on either drug interaction studies or predicted interactions due to the expected magnitude of interaction and potential for serious adverse events or loss of efficacy.
Table 6: Established and Other Potentially Significant Drug Interactions: Alteration in Dose or Regimen May Be Recommended Based on Drug Interaction Studies [see Clinical Pharmacology (12.3) (Tables 12 and 13) for magnitude of interaction]
Concomitant Drug Class:
Drug NameEffect on
ConcentrationClinical Comment HIV Antiviral Agents: Reverse Transcriptase Inhibitors Delavirdine
↑ nelfinavir (Cmin)
↓ delavirdineConcentrations of nelfinavir were increased while concentrations of delavirdine were decreased when the two agents were coadministered. Appropriate doses of the combination, with respect to safety and efficacy, have not been established.
Nevirapine
↓ nelfinavir (Cmin)
Concentrations of nelfinavir were decreased when coadministered with nevirapine. An appropriate dose of nelfinavir with respect to safety and efficacy has not been established.
Didanosine
↔ nelfinavir
There was no change in nelfinavir concentration when coadministered with didanosine. However, it is recommended that didanosine be administered on an empty stomach; therefore, didanosine should be given one hour before or two hours after VIRACEPT (given with food).
HIV Antiviral Agents: Protease Inhibitors
Indinavir
↑ nelfinavir
↑ indinavirConcentrations of both indinavir and nelfinavir were increased when the two agents were coadministered. Appropriate doses for these combinations, with respect to safety and efficacy, have not been established.
Ritonavir
↑ nelfinavir
↔ ritonavirConcentrations of nelfinavir were increased when coadministered with ritonavir. An appropriate dose of nelfinavir for this combination, with respect to safety and efficacy, has not been established.
Saquinavir
↑ nelfinavir
↑ saquinavirConcentrations of both saquinavir and nelfinavir were increased when the two agents were coadministered. Appropriate doses for these combinations, with respect to safety and efficacy, have not been established.
ANTICOAGULANT
Warfarin
Warfarin
Coadministration of warfarin and VIRACEPT may affect concentrations of warfarin. It is recommended that the INR (international normalized ratio) be monitored carefully during treatment with VIRACEPT, especially when commencing therapy.
ANTICONVULSANTS
Carbamazepine Phenobarbital
Phenytoin
↓ nelfinavir
↓ phenytoinConcentrations of nelfinavir may be decreased. VIRACEPT may not be effective due to decreased nelfinavir plasma concentrations in patients taking these agents concomitantly.
Phenytoin plasma/serum concentrations should be monitored; phenytoin dose may require adjustment to compensate for altered phenytoin concentration.ANTIDEPRESSANT
Trazodone
↑ trazodone
Concomitant use of trazodone and VIRACEPT may increase plasma concentrations of trazodone. Adverse events of nausea, dizziness, hypotension and syncope have been observed following coadministration of trazodone and ritonavir. If trazodone is used with a CYP3A4 inhibitor such as VIRACEPT, the combination should be used with caution and a lower dose of trazodone should be considered.
ANTIGOUT
Colchicine
↑ colchicines
Patients with renal or hepatic impairment should not be given colchicine with VIRACEPT due to the risk of colchicine toxicity.
Treatment of gout flares –
co- administration of colchicine in patients on VIRACEPT:
0.6 mg (1 tablet) × 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Dose to be repeated no earlier than 3 days.
Prophylaxis of gout-flares –
coadministration of colchicine in patients on VIRACEPT:
If the original colchicine regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg once a day.
If the original colchicine regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once every other day.
Treatment of familial Mediterranean fever (FMF)– coadministration of colchicine in patients on VIRACEPT:
Maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day).ANTIMYCOBACTERIAL
Rifabutin
↑ rifabutin
↓ nelfinavir
(750 mg TID)
↔ nelfinavir
(1250 mg BID)It is recommended that the dose of rifabutin be reduced to one-half the usual dose when administered with VIRACEPT; 1250 mg BID is the preferred dose of VIRACEPT when coadministered with rifabutin.
ENDOTHELIN RECEPTOR ANTAGONIST
Bosentan
↑ bosentan
Concentrations of bosentan may be increased when coadministered with VIRACEPT. Coadministration of bosentan in patients on VIRACEPT or coadministration of VIRACEPT in patients on bosentan:
Start at or adjust bosentan to 62.5 mg once daily or every other day based upon individual tolerability.HMG-CoA REDUCTASE INHIBITORS
Atorvastatin
Rosuvastatin↑ atorvastatin
↑ rosuvastatinTitrate atorvastatin dose carefully and use the lowest necessary dose; do not exceed atorvastatin 40 mg/day.
IMMUNOSUPPRESSANTS
Cyclosporine
Tacrolimus
Sirolimus↑ immuno-suppressants
↑ nelfinavirConcentrations of these immunosuppressants and nelfinavir may be increased by coadministration of these agents with nelfinavir.
INHALED BETA AGONIST
Salmeterol
↑ salmeterol
Concurrent administration of salmeterol with VIRACEPT is not recommended. The combination may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations and sinus tachycardia.
INHALED/NASAL STEROID
Fluticasone
↑ fluticasone
Concomitant use of fluticasone propionate and VIRACEPT may increase plasma concentrations of fluticasone propionate. Use with caution. Consider alternatives to fluticasone propionate, particularly for long-term use.
MACROLIDE ANTIBIOTIC
Azithromycin
↑ azithromycin
Dose adjustment of azithromycin is not recommended, but close monitoring for known side effects such as liver enzyme abnormalities and hearing impairment is warranted.
NARCOTIC ANALGESIC
Methadone
↓ methadone
Concentrations of methadone were decreased when coadministered with VIRACEPT. Dosage of methadone may need to be increased when coadministered with VIRACEPT.
HORMONAL CONTRACEPTIVES
Ethinyl estradiol
Norethindrone↓ ethinyl estradiol
↓ norethindroneConcentrations of ethinyl estradiol and norethindrone were decreased when coadministered with VIRACEPT. Alternative or additional contraceptive measures should be used when oral contraceptives containing ethinyl estradiol or norethindrone and VIRACEPT are coadministered.
PDE5 INHIBITORS
Sildenafil
Vardenafil
Tadalafil↑ PDE5 Inhibitors
Concomitant use of PDE5 inhibitors and VIRACEPT should be undertaken with caution.
May result in an increase in PDE5 inhibitor-associated adverse events, including hypotension, syncope, visual disturbances, and priapism.
Use of PDE5 inhibitors for pulmonary arterial hypertension (PAH):
• Use of sildenafil (REVATIO) is contraindicated when used for the treatment of pulmonary arterial hypertension (PAH) [see Contraindications (4)].
• The following dose adjustments are recommended for use of tadalafil (ADCIRCA™) with VIRACEPT:
Coadministration of ADCIRCA in patients on VIRACEPT or coadministration of VIRACEPT in patients on ADCIRCA:
Start at or adjust ADCIRCA to 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability.
Use of PDE5 inhibitors for erectile dysfunction:
Sildenafil at a single dose not exceeding 25 mg in 48 hours, vardenafil at a single dose not exceeding 2.5 mg in 24 hours, or tadalafil at a single dose not exceeding 10 mg dose in 72 hours, is recommended. Use with increased monitoring for adverse events.PROTON PUMP INHIBITORS
Omeprazole
↓ nelfinavir
Omeprazole decreases the plasma concentrations of nelfinavir. Concomitant use of proton pump inhibitors and VIRACEPT may lead to a loss of virologic response and development of resistance.
ANTIPSYCHOTICS
Quetiapine
↑ quetiapine
Initiation of VIRACEPT in patients taking quetiapine:
Consider alternative antiretroviral therapy to avoid increases in quetiapine drug exposures. If coadministration is necessary, reduce the quetiapine dose to 1/6 of the current dose and monitor for quetiapine-associated adverse reactions. Refer to the quetiapine prescribing information for recommendations on adverse reaction monitoring.
Initiation of quetiapine in patients taking VIRACEPT:
Refer to the quetiapine prescribing information for initial dosing and titration of quetiapine. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to VIRACEPT during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Published reports of hepatic adverse events ranging from elevated liver enzymes to hepatic failure in pregnant patients exposed to nelfinavir have been reported (see Clinical Considerations). Due to VIRACEPT's overall adverse event profile, including hepatic adverse events, and literature reports of decreased exposures in second and third trimesters, consider alternative antiretroviral drugs during pregnancy.
Available data from the APR suggests a statistically significant increase in overall risks of major birth defects with first trimester exposure with nelfinavir (3.9%) when compared with the background rate of 2.7% in one U.S. reference population (the Metropolitan Atlanta Congenital Defects Program [MACDP]), but the risk is similar to the background rate of 4.2% reported in another U.S. reference population (the Texas Birth Defects Registry [TBDR]). No pattern of defects was identified by the APR. The clinical relevance of this statistical finding is uncertain (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2 to 4% and 15 to 20% respectively.
In animal reproductive studies, no effects on embryo-fetal development were observed when nelfinavir was administered orally to pregnant rats and rabbits during organogenesis at systemic exposures similar to or less than human exposure (based on AUC) at the maximum recommended human dose (MRHD), respectively (see Data).
Maternal Adverse Reactions
There have been reports of hepatic adverse events ranging from elevated liver enzymes to hepatic failure in pregnant patients receiving nelfinavir as part of combination treatment of HIV-1 infection. Consider alternative antiretroviral drugs during pregnancy. If VIRACEPT is used during pregnancy, clinical monitoring is recommended [see Warnings and Precautions (5.2) and Use in Specific Populations (8.6)].
Human Data
Based on prospective reports to the APR of over 1,200 live births following first trimester and over 2,700 live births following second and third trimester exposure to nelfinavir-containing regimens, there was an increase in overall rates of major birth defects (n=47) with nelfinavir when compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of birth defects in live births was 3.9% (95% CI: 2.9% to 5.1%) with first trimester exposure to nelfinavir-containing regimens and 3.1% (95% CI: 2.5% to 3.9%) with the second/third trimester exposure to nelfinavir-containing regimens. The rate for nelfinavir-containing regimens is greater than the overall rates of defects in the MACDP (2.7%) but less than rates of defects in the TBDR (4.2%). Methodological limitations of the APR include the use of MACDP as the external comparator group. Limitations of using an external comparator include differences in methodology and populations, as well as confounding due to the underlying disease; these limitations preclude an accurate comparison of outcomes. The MACDP population is not disease specific, evaluates women and infants from a limited geographic area, and does not include outcomes for births that occurred at <20 weeks gestation.
Animal Data
Nelfinavir was administered orally to pregnant rats and rabbits during organogenesis, from gestation day 6 to 17 (rats) and 7 to 20 (rabbits). There were no effects on fetal development or maternal toxicity when nelfinavir was administered to pregnant rats at systemic exposures (AUC) similar to human exposure. Administration of nelfinavir to pregnant rabbits resulted in no fetal development effects up to a dose at which a slight decrease in maternal body weight was observed; however, even at the highest dose evaluated, systemic exposure in rabbits was significantly lower than human exposure. Additional studies in rats indicated that exposure to nelfinavir in females from mid-pregnancy through lactation (gestation day 6 to lactation day 20) had no effect on the survival, growth, and development of the offspring. Subsequent reproductive performance of these offspring was also not affected by maternal exposure to nelfinavir.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-1 infected patients in the United States not breastfeed their infants to avoid risking potential transmission of HIV-1 infection. Based on limited published data, nelfinavir is present in low levels in human milk, and adverse effects in infants exposed to nelfinavir have been reported. Because of the potential for (1) HIV-1 transmission (in HIV-negative infants), (2) developing viral resistance (in HIV-positive infants), and (3) the potential for serious adverse reactions in breastfed infants similar to those seen in adults, instruct mothers not to breastfeed if they are receiving VIRACEPT.
8.3 Females and Males of Reproductive Potential
Contraception
Use of VIRACEPT may reduce the efficacy of estrogen-based oral contraceptives. Advise patients to use alternative or additional contraceptive measures when oral contraceptives containing ethinyl estradiol or norethindrone and VIRACEPT are coadministered [see Drug Interactions (7.3)].
8.4 Pediatric Use
The safety, tolerability, pharmacokinetic profile and efficacy of VIRACEPT were evaluated in HIV infected pediatric patients from 2 to 13 years of age in multicenter clinical trials, Study 556 and PACTG 337 [see Clinical Pharmacology (12.3) and Clinical Studies (14.3)]. In patients less than 2 years of age, VIRACEPT was found to be safe at the doses studied, but a reliably effective dose could not be established [see Dosage and Administration (2.2), Adverse Reactions (6.2), and Clinical Pharmacology (12.3)]. The pharmacokinetic profile, safety and antiviral activity of VIRACEPT in adolescent patients 13 years and older is supported by data from the adult clinical trials where some trials allowed enrolment of subjects 13 years and older. Thus, the data for adolescents and adults were analyzed collectively [see Adverse Reactions (6.1)].
8.5 Geriatric Use
Clinical studies of VIRACEPT did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Hepatic Impairment
VIRACEPT should not be used in patients with either moderate or severe hepatic impairment (Child-Pugh B or C, score greater than or equal to 7) [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. No dose adjustment of VIRACEPT is necessary for patients with mild hepatic impairment (Child-Pugh A, score 5–6).
-
10 OVERDOSAGE
Human experience of acute overdose with VIRACEPT is limited. There is no specific antidote for overdose with VIRACEPT. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage. Administration of activated charcoal may also be used to aid removal of unabsorbed drug. Since nelfinavir is highly protein bound, dialysis is unlikely to significantly remove drug from blood.
-
11 DESCRIPTION
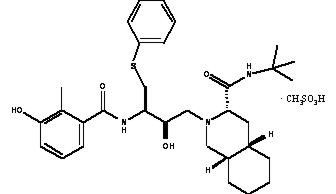
VIRACEPT® (nelfinavir mesylate) is an inhibitor of the human immunodeficiency virus type 1 (HIV-1) protease. VIRACEPT Tablets are available for oral administration as a light-blue, capsule-shaped tablet with a clear film coating in 250 mg strength (as nelfinavir-free base) and as a white oval tablet with a clear film coating in 625 mg strength (as nelfinavir-free base). Each tablet contains the following common inactive ingredients: calcium silicate, crospovidone, magnesium stearate, hypromellose, and triacetin. In addition, the 250 mg tablet contains FD&C blue #2 powder and the 625 mg tablet contains colloidal silicon dioxide. VIRACEPT Oral Powder is available for oral administration in a 50 mg/g strength (as nelfinavir-free base) in bottles. The oral powder also contains the following inactive ingredients: microcrystalline cellulose, maltodextrin, dibasic potassium phosphate, crospovidone, hypromellose, aspartame, sucrose palmitate, and natural and artificial flavor. The chemical name for nelfinavir mesylate is [3S-[2(2S*, 3S*), 3α,4aβ,8aβ]]-N-(1,1-dimethylethyl)decahydro-2-[2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-(phenylthio)butyl]-3-isoquinoline carboxamide mono-methanesulfonate (salt) and the molecular weight is 663.90 (567.79 as the free base). Nelfinavir mesylate has the following structural formula:
Nelfinavir mesylate is a white to off-white amorphous powder, slightly soluble in water at pH ≤4 and freely soluble in methanol, ethanol, 2-propanol and propylene glycol.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Nelfinavir is an inhibitor of the HIV-1 protease [see Microbiology (12.4)].
12.2 Pharmacodynamics
Effects on Electrocardiogram
The effect of Viracept at the recommended dose of 1250 mg twice daily on the QTcF interval administered with a low fat meal (20% fat) was evaluated in a randomized, placebo and active (moxifloxacin 400 mg once daily) controlled, crossover study in 66 healthy subjects. The maximum mean time-matched (95% upper confidence bound) differences in QTcF interval from placebo after baseline-correction was below 10 milliseconds, the threshold of clinical concern. This finding was unchanged when a single supratherapeutic dose of Viracept 3125 mg was administered following a 3-day administration of Viracept 1250 mg twice daily. The exposure at 3125 mg was 1.4-fold that at 1250 mg. The dose of 3125 mg in this study did not achieve the anticipated exposures in patients taking a high fat meal (50% fat) or with concomitant administration of drugs that could increase nelfinavir exposure [see Pharmacokinetics (12.3)].
No subject in any group had an increase in QTcF of ≥60 milliseconds from baseline. No subject experienced an interval exceeding the potentially clinically relevant threshold of 500 milliseconds.
12.3 Pharmacokinetics
The pharmacokinetic properties of nelfinavir were evaluated in healthy volunteers and HIV-infected patients; no substantial differences were observed between the two groups.
Absorption
Pharmacokinetic parameters of nelfinavir (area under the plasma concentration-time curve during a 24-hour period at steady-state [AUC24], peak plasma concentrations [Cmax], morning and evening trough concentrations [Ctrough]) from a pharmacokinetic study in HIV-positive patients after multiple dosing with 1250 mg (five 250 mg tablets) twice daily (BID) for 28 days (10 patients) and 750 mg (three 250 mg tablets) three times daily (TID) for 28 days (11 patients) are summarized in Table 7.
Table 7: Summary of a Pharmacokinetic Study in HIV-positive Patients With Multiple Dosing of 1250 mg (Five 250 mg Tablets) BID for 28 Days and 750 mg (Three 250 mg Tablets) TID for 28 Days Regimen AUC24
mg∙h/LCmax
mg/LCtrough
Morning
mg/LCtrough
Afternoon or Evening
mg/LData are mean ± SD 1250 mg BID
52.8±15.7
4.0±0.8
2.2±1.3
0.7±0.4
750 mg TID
43.6±17.8
3.0±1.6
1.4±0.6
1.0±0.5
The difference between morning and afternoon or evening trough concentrations for the TID and BID regimens was also observed in healthy volunteers who were dosed at precisely 8- or 12-hour intervals.
In healthy volunteers receiving a single 1250 mg dose, the 625 mg tablet was not bioequivalent to the 250 mg tablet formulation. Under fasted conditions (n=27), the AUC and Cmax were 34% and 24% higher, respectively, for the 625 mg tablets. In a relative bioavailability assessment under fed conditions (n=28), the AUC was 24% higher for the 625 mg tablet; the Cmax was comparable for both formulations. In HIV-1 infected subjects (N=21) receiving multiple doses of 1250 mg BID under fed conditions, the 625 mg formulation was bioequivalent to the 250 mg formulation based on similarity in steady state exposure (Cmax and AUC).
Table 8 shows the summary of the steady state pharmacokinetic parameters (mean ± SD) of nelfinavir after multiple dose administration of 1250 mg BID (2 × 625 mg tablets) to HIV-infected patients (N=21) for 14 days.
Table 8: Summary of the Steady State Pharmacokinetic Parameters (Mean ± SD) of Nelfinavir After Multiple Dose Administration of 1250 mg BID (2 × 625 mg Tablets) to HIV-infected Patients (N=21) for 14 Days. Regimen AUC12
mg∙h/LCmax
mg/LCmin
mg/LAUC12: Steady state AUC Cmax: Maximum plasma concentration at steady state Cmin: Minimum plasma concentration at steady state 1250 mg BID
35.3 (16.4)
4.7 (1.9)
1.5 (1.0)
In healthy volunteers receiving a single 750 mg dose under fed conditions, nelfinavir concentrations were similar following administration of the 250 mg tablet and oral powder.
Effect of Food on Oral Absorption
Food increases nelfinavir exposure and decreases nelfinavir pharmacokinetic variability relative to the fasted state. In one study, healthy volunteers received a single dose of 1250 mg of VIRACEPT 250 mg tablets (5 tablets) under fasted or fed conditions (three different meals). In a second study, healthy volunteers received single doses of 1250 mg VIRACEPT (5 × 250 mg tablets) under fasted or fed conditions (two different fat content meals). The results from the two studies are summarized in Table 9 and Table 10, respectively.
Table 9: Increase in AUC, Cmax and Tmax for Nelfinavir in Fed State Relative to Fasted State Following 1250 mg VIRACEPT (5 × 250 mg Tablets) Number of Kcal % Fat Number of subjects AUC fold increase Cmax fold increase Increase in Tmax (hr) 125
20
n=21
2.2
2.0
1.00
500
20
n=22
3.1
2.3
2.00
1000
50
n=23
5.2
3.3
2.00
Table 10: Increase in Nelfinavir AUC, Cmax and Tmax in Fed Low Fat (20%) versus High Fat (50%) State Relative to Fasted State Following 1250 mg VIRACEPT (5 × 250 mg Tablets) Number of Kcal % Fat Number of subjects AUC fold increase Cmax fold increase Increase in Tmax (hr) 500
20
n=22
3.1
2.5
1.8
500
50
n=22
5.1
3.8
2.1
Nelfinavir exposure can be increased by increasing the calorie or fat content in meals taken with VIRACEPT.
A food effect study has not been conducted with the 625 mg tablet. However, based on a cross-study comparison (n=26 fed vs. n=26 fasted) following single dose administration of nelfinavir 1250 mg, the magnitude of the food effect for the 625 mg nelfinavir tablet appears comparable to that of the 250 mg tablets. VIRACEPT should be taken with a meal.
Distribution
The apparent volume of distribution following oral administration of nelfinavir was 2–7 L/kg. Nelfinavir in serum is extensively protein-bound (>98%).
Metabolism
Unchanged nelfinavir comprised 82–86% of the total plasma radioactivity after a single oral 750 mg dose of 14C-nelfinavir. In vitro, multiple cytochrome P-450 enzymes including CYP3A and CYP2C19 are responsible for metabolism of nelfinavir. One major and several minor oxidative metabolites were found in plasma. The major oxidative metabolite has in vitro antiviral activity comparable to the parent drug.
Elimination
The terminal half-life in plasma was typically 3.5 to 5 hours. The majority (87%) of an oral 750 mg dose containing 14C-nelfinavir was recovered in the feces; fecal radioactivity consisted of numerous oxidative metabolites (78%) and unchanged nelfinavir (22%). Only 1–2% of the dose was recovered in urine, of which unchanged nelfinavir was the major component.
Specific Populations
Hepatic Impairment
The steady-state pharmacokinetics of nelfinavir (1250 mg BID for 2 weeks) was studied in HIV-seronegative subjects with mild (Child-Pugh Class A; n=6) or moderate (Child-Pugh Class B; n=6) hepatic impairment. When compared with subjects with normal hepatic function, the Cmax and AUC of nelfinavir were not significantly different in subjects with mild hepatic impairment but were increased by 22% and 62%, respectively, in subjects with moderate hepatic impairment. The steady-state pharmacokinetics of nelfinavir has not been studied in HIV-seronegative subjects with severe hepatic impairment.
The steady-state pharmacokinetics of nelfinavir has not been studied in HIV-positive patients with any degree of hepatic impairment.
Renal Impairment
The pharmacokinetics of nelfinavir have not been studied in patients with renal impairment.
Gender and Race
No significant pharmacokinetic differences have been detected between males and females. Pharmacokinetic differences due to race have not been evaluated.
Pediatrics
The pharmacokinetics of nelfinavir have been investigated in 5 studies in pediatric patients from birth to 13 years of age either receiving VIRACEPT three times or twice daily. The dosing regimens and associated AUC24 values are summarized in Table 11.
Table 11: Summary of Steady-state AUC24 of Nelfinavir in Pediatric Studies Ctrough values are not presented in the table because they are not available for all studies Protocol number
Dosing regimen*
N†
Age
AUC24 (mg∙hr/L)
arithmetic mean ± SDAG1343-524
20 (19–28) mg/kg TID
14
2–13 years
56.1±29.8
PACTG-725
55 (48–60) mg/kg BID
6
3–11 years
101.8±56.1
PENTA 7
40 (34–43) mg/kg TID
4
2–9 months
33.8±8.9
PENTA 7
75 (55–83) mg/kg BID
12
2–9 months
37.2±19.2
PACTG-353
40 (14–56) mg/kg BID
10
6 weeks
44.1±27.4
1 week
45.8±32.1
Pharmacokinetic data are also available for 86 patients (age 2 to 12 years) who received VIRACEPT 25–35 mg/kg TID in Study AG1343-556. The pharmacokinetic data from Study AG1343-556 were more variable than data from other studies conducted in the pediatric population; the 95% confidence interval for AUC24 was 9 to 121 mg∙hr/L.
Overall, use of VIRACEPT in the pediatric population is associated with highly variable drug exposure. The high variability may be due to inconsistent food intake in pediatric patients [see Dosage and Administration (2.2)].
Geriatric Patients
The pharmacokinetics of nelfinavir have not been studied in patients over 65 years of age.
Drug Interactions
CYP3A and CYP2C19 appear to be the predominant enzymes that metabolize nelfinavir in humans. The potential ability of nelfinavir to inhibit the major human cytochrome P450 enzymes (CYP3A, CYP2C19, CYP2D6, CYP2C9, CYP1A2 and CYP2E1) has been investigated in vitro. Only CYP3A was inhibited at concentrations in the therapeutic range. Specific drug interaction studies were performed with nelfinavir and a number of drugs. Table 12 summarizes the effects of nelfinavir on the geometric mean AUC, Cmax and Cmin of coadministered drugs. Table 13 shows the effects of coadministered drugs on the geometric mean AUC, Cmax and Cmin of nelfinavir.
Table 12: Drug Interactions: Changes in Pharmacokinetic Parameters for Coadministered Drug in the Presence of VIRACEPT % Change of Coadministered Drug Pharmacokinetic Parameters* (90% CI) Coadministered Drug Nelfinavir Dose N AUC Cmax Cmin NA: Not relevant for single-dose treatment; ND: Cannot be determined - *
- ↑ Indicates increase; ↓ Indicates decrease; ↔ Indicates no change (geometric mean exposure increased, or decreased <10%)
- †
- Using the soft-gelatin capsule formulation of saquinavir 1200 mg
- ‡
- Rifabutin 150 mg qd changes are relative to Rifabutin 300 mg qd × 8 days without coadministration with nelfinavir
- §
- Comparable changes in rifabutin concentrations were observed with VIRACEPT 1250 mg q12h × 7 days
- ¶
- Changes are reported for total plasma methadone; changes for the individual R-enantiomer and S-enantiomer were similar
- #
- Phenytoin exposure measures are reported for total phenytoin exposure. The effect of nelfinavir on unbound phenytoin was similar
HIV-Protease Inhibitors
Indinavir 800 mg Single Dose
750 mg q8h × 7 days
6
↑51%
(↑29–↑77%)↓10%
(↓28–↑13%)NA
Ritonavir 500 mg Single Dose
750 mg q8h × 5 doses
10
↔
↔
NA
Saquinavir 1200 mg Single Dose†
750 mg TID × 4 days
14
↑392%
(↑291–↑521%)↑179%
(↑117–↑259%)NA
Amprenavir 800 mg TID × 14 days
750 mg TID × 14 days
6
↔
↓14%
(↓38–↑20%)↑189%
(↑52–↑448%)Nucleoside Reverse Transcriptase Inhibitors
Lamivudine 150 mg Single Dose
750 mg q8h × 7–10 days
11
↑10%
(↑2–↑18%)↑31%
(↑9–↑56%)NA
Zidovudine 200 mg Single Dose
750 mg q8h × 7–10 days
11
↓35%
(↓29–↓40%)↓31%
(↓13–↓46%)NA
Non-nucleoside Reverse Transcriptase Inhibitors
Efavirenz 600 mg qd × 7 days
750 mg q8h × 7 days
10
↓12%
(↓31–↑12%)↓12%
(↓29–↑8%)↓22%
(↓54–↑32%)Delavirdine 400 mg q8h × 14 days
750 mg q8h × 7 days
7
↓31%
(↓57–↑10%)↓27%
(↓49–↑4%)↓33%
(↓70–↑49%)Anti-infective Agents
Rifabutin 150 mg qd × 8 days‡
750 mg q8h × 7–8 days§
12
↑83%
(↑72–↑96%)↑19%
(↑11–↑28%)↑177%
(↑144–↑215%)Rifabutin 300 mg qd × 8 days
750 mg q8h × 7–8 days
10
↑207%
(↑161–↑263%)↑146%
(↑118–↑178%)↑305%
(↑245–↑375%)Azithromycin 1200 mg Single Dose
750 mg TID × 11 days
12
↑112%
(↑80–↑150%)↑136%
(↑77–↑215%)NA
HMG-CoA Reductase Inhibitors
Atorvastatin 10 mg qd × 28 days
1250 mg BID × 14 days
15
↑74%
(↑41–↑116%)↑122%
(↑68–↑193%)↑39%
(↓21–↑145%)Simvastatin 20 mg qd × 28 days
1250 mg BID × 14 days
16
↑505%
(↑393–↑643%)↑517%
(↑367–↑715%)ND
Other Agents
Ethinyl estradiol 35 µg qd × 15 days
750 mg q8h × 7 days
12
↓47%
(↓42–↓52%)↓28%
(↓16–↓37%)↓62%
(↓57–↓67%)Norethindrone 0.4 mg qd × 15 days
750 mg q8h × 7 days
12
↓18%
(↓13–↓23%)↔
↓46%
(↓38–↓53%)Methadone 80 mg ± 21 mg qd¶ >1 month
1250 mg BID × 8 days
13
↓47%
(↓42–↓51%)↓46%
(↓42–↓49%)↓53%
(↓49–↓57%)Phenytoin 300 mg qd × 14 days#
1250 mg BID × 7 days
12
↓29%
(↓17–↓39%)↓21%
(↓12–↓29%)↓39%
(↓27–↓49%)Table 13: Drug Interactions: Changes in Pharmacokinetic Parameters for Nelfinavir in the Presence of the Coadministered Drug % Change of Nelfinavir Pharmacokinetic Parameters* (90% CI) Coadministered Drug Nelfinavir Dose N AUC Cmax Cmin NA: Not relevant for single-dose treatment HIV-Protease Inhibitors
Indinavir 800 mg q8h × 7 days
750 mg Single Dose
6
↑83%
(↑42–↑137%)↑31%
(↑16–↑48%)NA
Ritonavir 500 mg q12h × 3 doses
750 mg Single Dose
10
↑152%
(↑96–↑224%)↑44%
(↑28–↑63%)NA
Saquinavir 1200 mg TID × 4 days†
750 mg Single Dose
14
↑18%
(↑7–↑30%)↔
NA
Nucleoside Reverse Transcriptase Inhibitors
Didanosine 200 mg Single Dose
750 mg Single Dose
9
↔
↔
NA
Zidovudine 200 mg + Lamivudine 150 mg Single Dose
750 mg q8h × 7–10 days
11
↔
↔
↔
Non-nucleoside Reverse Transcriptase Inhibitors
Efavirenz 600 mg qd × 7 days
750 mg q8h × 7 days
7
↑20%
(↑8–↑34%)↑21%
(↑10–↑33%)↔
Nevirapine 200 mg qd × 14 days followed by 200 mg BID × 14 days
750 mg TID × 36 days
23
↔
↔
↓32%
(↓50–↑5%)Delavirdine 400 mg q8h × 7 days
750 mg q8h × 14 days
12
↑107%
(↑83–↑135%)↑88%
(↑66–↑113%)↑136%
(↑103–↑175%)Anti-infective Agents
Ketoconazole 400 mg qd × 7 days
500 mg q8h × 5–6 days
12
↑35%
(↑24–↑46%)↑25%
(↑11–↑40%)↑14%
(↓23–↑69%)Rifabutin 150 mg qd × 8 days
750 mg q8h × 7–8 days
11
↓23%
(↓14–↓31%)↓18%
(↓8–↓27%)↓25%
(↓8–↓39%)1250 mg q12h × 7–8 days
11
↔
↔
↓15%
(↓43–↑27%)Rifabutin 300 mg qd × 8 days
750 mg q8h × 7–8 days
10
↓32%
(↓15–↓46%)↓24%
(↓10–↓36%)↓53%
(↓15–↓73%)Rifampin 600 mg qd × 7 days
750 mg q8h × 5–6 days
12
↓83%
(↓79–↓86%)↓76%
(↓69–↓82%)↓92%
(↓86–↓95%)Azithromycin 1200 mg Single Dose
750 mg tid × 9 days
12
↓15%
(↓7–↓22%)↓10%
(↓19–↑1%)↓29%
(↓19–↓38%)Other Agents
Phenytoin 300 mg qd × 7 days
1250 mg BID × 14 days
15
↔
↔
↓18%
(↓45–↑23%)Omeprazole 40 mg qd × 4 days administered 30 minutes before nelfinavir
1250 mg BID × 4 days
19
↓36%
(↓20–↓49%)↓37%
(↓23–↓49%)↓39%
(↓15–↓57%)12.4 Microbiology
Mechanism of Action
Nelfinavir is an inhibitor of the HIV-1 protease. Inhibition of the viral protease prevents cleavage of the gag and gag-pol polyprotein resulting in the production of immature, non-infectious virus.
Antiviral Activity in Cell Culture
The antiviral activity of nelfinavir has been demonstrated in both acute and/or chronic HIV infections in lymphoblastoid cell lines, peripheral blood lymphocytes, and monocytes/macrophages. Nelfinavir was found to be active against several laboratory strains and clinical isolates of HIV-1, and the HIV-2 strain ROD. The EC95 (95% effective concentration) of nelfinavir ranged from 7 to 196 nM. Drug combination studies with other HIV-1 protease inhibitors showed nelfinavir had antagonistic interactions with indinavir, additive interactions with ritonavir or saquinavir, and synergistic interactions with amprenavir and lopinavir. Minimal to no cellular cytotoxicity was observed with any of these protease inhibitors alone or in combination with nelfinavir. In combination with reverse transcriptase inhibitors, nelfinavir demonstrated additive (didanosine or stavudine) to synergistic (abacavir, delavirdine, efavirenz, emtricitabine, lamivudine, nevirapine, tenofovir, zalcitabine, or zidovudine) antiviral activity without enhanced cytotoxicity. Nelfinavir's anti-HIV activity was not antagonized by the anti-HCV drug ribavirin.
Resistance
HIV-1 isolates with reduced susceptibility to nelfinavir have been selected in cell culture. HIV-1 isolates from selected patients treated with nelfinavir alone or in combination with reverse transcriptase inhibitors were monitored for phenotypic (n=19) and genotypic (n=195, 157 of which were evaluable) changes in clinical trials over a period of 2 to 82 weeks. One or more viral protease mutations at amino acid positions 30, 35, 36, 46, 71, 77, and 88 were detected in the HIV-1 of >10% of patients with evaluable isolates. The overall incidence of the D30N substitution in the viral protease of evaluable isolates (n=157) from patients receiving nelfinavir monotherapy or nelfinavir in combination with zidovudine and lamivudine or stavudine was 54.8%. The overall incidence of other substitutions associated with primary protease inhibitor resistance was 9.6% for the L90M substitution, whereas substitutions at 48, 82, or 84 were not observed. Of the 19 clinical isolates for which both phenotypic and genotypic analyses were performed, 9 showed reduced susceptibility (5- to 93-fold) to nelfinavir in cell culture. All 9 isolates possessed one or more mutations in the viral protease gene. Amino acid position 30 appeared to be the most frequent mutation site.
Cross-resistance
Non-clinical Studies: Patient-derived recombinant HIV-1 isolates containing the D30N substitution (n=4) and demonstrating high-level (>10-fold) nelfinavir-resistance remained susceptible (<2.5-fold resistance) to amprenavir, indinavir, lopinavir, and saquinavir in cell culture. Patient-derived recombinant HIV-1 isolates containing the L90M substitution (n=8) demonstrated moderate to high-level resistance to nelfinavir and had varying levels of susceptibility to amprenavir, indinavir, lopinavir, and saquinavir in cell culture. Most patient-derived recombinant isolates with phenotypic and genotypic evidence of reduced susceptibility (>2.5-fold) to amprenavir, indinavir, lopinavir, and/or saquinavir demonstrated high-level cross-resistance to nelfinavir. Amino acid substitutions associated with resistance to other protease inhibitors (e.g., G48V, V82A/F/T, I84V, L90M) appeared to confer high-level cross-resistance to nelfinavir. Following ritonavir therapy 6 of 7 clinical isolates with decreased ritonavir susceptibility (8- to 113-fold) compared to baseline also exhibited decreased susceptibility to nelfinavir (5- to 40-fold). Cross-resistance between nelfinavir and reverse transcriptase inhibitors is unlikely because different enzyme targets are involved. Clinical isolates (n=5) with decreased susceptibility to lamivudine, nevirapine, or zidovudine remain fully susceptible to nelfinavir.
Clinical Studies: There have been no controlled or comparative studies evaluating the virologic response to subsequent protease inhibitor-containing regimens in subjects who have demonstrated loss of virologic response to a nelfinavir-containing regimen. However, virologic response was evaluated in a single-arm prospective study of 26 subjects with extensive prior antiretroviral experience with reverse transcriptase inhibitors (mean 2.9) who had received nelfinavir for a mean duration of 59.7 weeks and were switched to a ritonavir (400 mg BID)/saquinavir hard-gel (400 mg BID)-containing regimen after a prolonged period of nelfinavir failure (median 48 weeks). Sequence analysis of HIV-1 isolates prior to switch demonstrated a D30N or an L90M substitution in 18 and 6 subjects, respectively. Subjects remained on therapy for a mean of 48 weeks (range 40 to 56 weeks) where 17 (65%) and 13 (50%) of the 26 subjects were treatment responders with HIV-1 RNA below the assay limit of detection (<500 HIV-1 RNA copies/mL, Chiron bDNA) at 24 and 48 weeks, respectively.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies in mice and rats were conducted with nelfinavir at oral doses up to 1000 mg/kg/day. No evidence of a tumorigenic effect was noted in mice at systemic exposures (Cmax) up to 9-fold those measured in humans at the recommended therapeutic dose (750 mg TID or 1250 mg BID). In rats, thyroid follicular cell adenomas and carcinomas were increased in males at 300 mg/kg/day and higher and in females at 1000 mg/kg/day. Systemic exposures (Cmax) at 300 and 1000 mg/kg/day were 1- to 3-fold, respectively, those measured in humans at the recommended therapeutic dose. Repeated administration of nelfinavir to rats produced effects consistent with hepatic microsomal enzyme induction and increased thyroid hormone deposition; these effects predispose rats, but not humans, to thyroid follicular cell neoplasms. Nelfinavir showed no evidence of mutagenic or clastogenic activity in a battery of in vitro and in vivo genetic toxicology assays. These studies included bacterial mutation assays in S. typhimurium and E. coli, a mouse lymphoma tyrosine kinase assay, a chromosomal aberration assay in human lymphocytes, and an in vivo mouse bone marrow micronucleus assay.
Nelfinavir produced no effects on either male or female mating and fertility or embryo survival in rats at systemic exposures comparable to the human therapeutic exposure.
-
14 CLINICAL STUDIES
Description of Clinical Studies
The efficacy of VIRACEPT is based on analyses of multiple clinical studies in HIV-1 infected antiretroviral treatment-naïve and experienced adult patients. In the adult clinical studies described below, efficacy was evaluated by the percent of patients with plasma HIV RNA <400 copies/mL (Studies 511 and 542), <500 copies/mL (Study ACTG 364), or <50 copies/mL (Study Avanti 3). In the analysis presented in each figure, patients who terminated the study early for any reason, switched therapy due to inadequate efficacy or who had a missing HIV-RNA measurement that was either preceded or followed by a measurement above the limit of assay quantification were considered to have HIV-RNA above 400 copies/mL, above 500 copies/mL, or above 50 copies/mL at subsequent time points, depending on the study's definition of virologic failure.
14.1 Studies in Antiretroviral Treatment Naïve Adult Patients
Study 511: VIRACEPT + zidovudine + lamivudine versus zidovudine + lamivudine
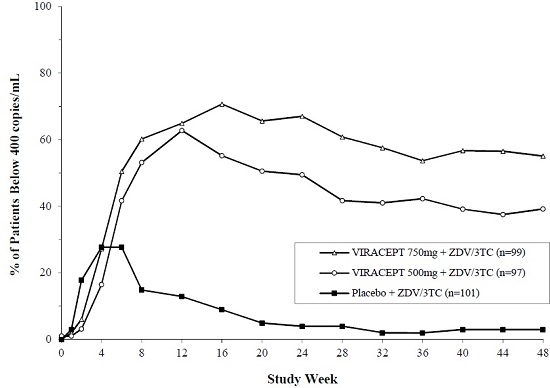
Study 511 is a double-blind, randomized, placebo-controlled trial comparing treatment with zidovudine (ZDV; 200 mg TID) and lamivudine (3TC; 150 mg BID) plus 2 doses of VIRACEPT (750 mg and 500 mg TID) to zidovudine (200 mg TID) and lamivudine (150 mg BID) alone in 297 antiretroviral naïve HIV-1 infected patients. The median age was 35 years [range 21 to 63]; 89% were male and 78% were Caucasian. Mean baseline CD4 cell count was 288 cells/mm3 and mean baseline plasma HIV RNA was 5.21 log10 copies/mL (160,394 copies/mL). The proportion of patients with plasma HIV RNA <400 copies/mL at Week 48 was 86%, as summarized in Figure 1. The mean change in CD4 cell count at Week 48 was 207.6 cells/mm3.
Figure 1
Study 511: Percentage of Patients With HIV RNA Below 400 Copies/mLStudy 542: VIRACEPT BID + stavudine + lamivudine compared to VIRACEPT TID + stavudine + lamivudine
Study 542 is a, randomized, open-label trial comparing the HIV RNA suppression achieved by VIRACEPT 1250 mg BID versus VIRACEPT 750 mg TID in patients also receiving stavudine (d4T; 30–40 mg BID) and lamivudine (3TC; 150 mg BID). Patients had a median age of 36 years (range 18 to 83), were 84% male, and were 91% Caucasian. Patients had received less than 6 months of therapy with nucleoside transcriptase inhibitors and were naïve to protease inhibitors. Mean baseline CD4 cell count was 296 cells/mm3 and mean baseline plasma HIV RNA was 5.0 log10 copies/mL (100,706 copies/mL).
Results showed that there was no significant difference in mean CD4 cell count among treatment groups; the mean increases from baseline for the BID and TID arms were 150 cells/mm3 at 24 weeks and approximately 200 cells/mm3 at 48 weeks.
The percent of patients with HIV RNA <400 copies/mL and the outcomes of patients through 48 weeks of treatment are summarized in Table 14.
Table 14: Outcomes of Randomized Treatment Through 48 Weeks Outcome VIRACEPT 1250 mg BID Regimen VIRACEPT 750 mg TID Regimen - *
- Twelve patients in the BID arm and fourteen patients in the TID arm had not yet reached 48 weeks of therapy.
- †
- These rates only reflect dose-limiting toxicities that were counted as the initial reason for treatment failure in the analysis [see Adverse Reactions (6)].
- ‡
- Consent withdrawn, lost to follow-up, intercurrent illness, noncompliance or missing data; all assumed as failures.
Number of patients evaluable*
323
192
HIV-1 RNA <400 copies/mL
198 (61%)
111 (58%)
HIV-1 RNA ≥400 copies/mL
46 (14%)
22 (11%)
Discontinued due to VIRACEPT toxicity†
9 (3%)
2 (1%)
Discontinued due to other antiretroviral agents' toxicity†
3 (1%)
3 (2%)
Others‡
67 (21%)
54 (28%)
Study Avanti 3: VIRACEPT TID + zidovudine + lamivudine compared to zidovudine + lamivudine
Study Avanti 3 was a placebo-controlled, randomized, double-blind study designed to evaluate the safety and efficacy of VIRACEPT (750 mg TID) in combination with zidovudine (ZDV; 300 mg BID) and lamivudine (3TC; 150 mg BID) (n=53) versus placebo in combination with ZDV and 3TC (n=52) administered to antiretroviral-naïve patients with HIV infection and a CD4 cell count between 150 and 500 cells/μL. Patients had a mean age of 35 (range 22–59), were 89% male, and 88% Caucasian. Mean baseline CD4 cell count was 304 cells/mm3 and mean baseline plasma HIV RNA was 4.8 log10 copies/mL (57,887 copies/mL). The percent of patients with plasma HIV RNA <50 copies/mL at 52 weeks was 54% for the (VIRACEPT + ZDV + 3TC)-treatment group and 13% for the (ZDV + 3TC)-treatment group.
14.2 Studies in Antiretroviral Treatment Experienced Adult Patients
Study ACTG 364: VIRACEPT TID + 2NRTIs compared to efavirenz + 2NRTIs compared to VIRACEPT + efavirenz + 2NRTIs
Study ACTG 364 was a randomized, double-blind study that evaluated the combination of VIRACEPT 750 mg TID and/or efavirenz 600 mg QD with 2 NRTIs (either didanosine [ddI] + d4T, ddI + 3TC, or d4T + 3TC) in patients with prolonged prior nucleoside exposure who had completed 2 previous ACTG studies. Patients had a mean age of 41 years (range 18 to 75), were 88% male, and were 74% Caucasian. Mean baseline CD4 cell count was 389 cells/mm3 and mean baseline plasma HIV RNA was 3.9 log10 copies/mL (7,954 copies/mL).
The percent of patients with plasma HIV RNA <500 copies/mL at 48 weeks was 42%, 62%, and 72% for the VIRACEPT (n=66), EFV (n=65), and VIRACEPT + EFV (n=64) treatment groups, respectively.
14.3 Studies in Pediatric Patients
The pharmacokinetic profile, safety and antiviral activity of VIRACEPT in pediatric patients 2 years of age up to 13 years were evaluated in 2 randomized studies.
Study 556 was a randomized, double-blind, placebo-controlled trial with VIRACEPT or placebo coadministered with ZDV and ddI in 141 HIV-positive children who had received minimal antiretroviral therapy. The mean age of the children was 3.9 years. Ninety four (67%) children were between 2–12 years, and 47 (33%) were < 2 years of age. The mean baseline HIV RNA value was 5.0 log for all patients and the mean CD4 cell count was 886 cells/mm3 for all patients. The efficacy of VIRACEPT measured by HIV RNA <400 at 48 weeks in children ≥2 years of age was 26% compared to 2% of placebo patients (p=0.0008). In the children < 2 years of age, only 1 of 27 and 2 of 20 maintained an undetectable HIV RNA level at 48 weeks for placebo and VIRACEPT patients, respectively.
PACTG 377 was an open-label study that randomized 181 HIV treatment-experienced pediatric patients to receive: d4T+NVP+RTV, d4T+3TC+NFV, or d4T+3TC+NVP+NFV with NFV given on a TID schedule. The median age was 5.9 years and 46% were male. At baseline the median HIV RNA was 4.4 log and median CD4 cell count was 690 cells/mm3. Substudy PACTG 725 evaluated d4T+3TC+NFV with NFV given on a BID schedule. The proportion of patients with detectable viral load at baseline achieving HIV RNA <400 copies/mL at 48 weeks was: 41% for d4T+NVP+RTV, 42% for d4T+3TC+NFV, 30% for d4T+NVP+NFV, and 52% for d4T+3TC+NVP+NFV. No significant clinical differences were identified between patients receiving VIRACEPT in BID or TID schedules.
VIRACEPT has been evaluated in 2 studies of young infants. The PENTA 7 study was an open-label study to evaluate the toxicity, tolerability, pharmacokinetics, and activity of NFV+d4T+ddI in 20 HIV-infected infants less than 12 weeks of age. PACTG 353 evaluated the pharmacokinetics and safety of VIRACEPT in infants born to HIV-infected women receiving NFV as part of combination therapy during pregnancy.
The following issues should be considered when initiating VIRACEPT in pediatric patients:
- •
- In pediatric patients ≥2 years of age receiving VIRACEPT as part of triple combination antiretroviral therapy in randomized studies, the proportion of patients achieving a HIV RNA level <400 copies/mL through 48 weeks ranged from 26% to 42%.
- •
- Response rates in children <2 years of age appeared to be poorer than those in patients ≥2 years of age in some studies.
- •
- Highly variable drug exposure remains a significant problem in the use of VIRACEPT in pediatric patients. Unpredictable drug exposure may be exacerbated in pediatric patients because of increased clearance compared to adults and difficulties with compliance and adequate food intake with dosing. Pharmacokinetic results from the pediatric studies are reported in Table 11 [see Clinical Pharmacology (12.3)].
The pharmacokinetic profile, safety and antiviral activity of VIRACEPT in adolescent patients 13 years and older is supported by data from the adult clinical trials where some trials allowed enrolment of subjects 13 years and older. Thus, the data for adolescents and adults were analyzed collectively.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
VIRACEPT (nelfinavir mesylate) 250 mg: Light-blue, capsule-shaped tablets with a clear film coating engraved with "VIRACEPT" on one side and "250 mg" on the other.
- Bottles of 300 (250 mg) tablets – NDC 63010-010-30
VIRACEPT (nelfinavir mesylate) 625 mg: White oval tablet with a clear film coating engraved with "V" on one side and "625" on the other.
- Bottles of 120 (625 mg) tablets – NDC 63010-027-70
VIRACEPT (nelfinavir mesylate) Oral Powder is available as a 50 mg/g off-white powder containing 50 mg (as nelfinavir free base) in each level scoopful (1 gram).
- Multiple use bottles of 144 grams of powder with scoop …….NDC 63010-011-90
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information)
A statement to patients and healthcare providers is included on the product's bottle label: ALERT: Find out about medicines that should NOT be taken with VIRACEPT.
Instruction for Use
For optimal absorption, patients should be advised to take VIRACEPT with food.
Patients should be informed that VIRACEPT Tablets are film-coated and that this film-coating is intended to make the tablets easier to swallow.
If a dose of VIRACEPT is missed, patients should take the dose as soon as possible and then return to their normal schedule. However, if a dose is skipped, the patient should not double the next dose.
Adult or pediatric patients unable to swallow the tablets may dissolve the tablets in a small amount of water:
- •
- Place VIRACEPT tablet(s) in small amount of water
- •
- Once dissolved, mix the cloudy liquid well, and consume it immediately.
- •
- The glass should be rinsed with water and the rinse swallowed to ensure the entire dose is consumed
Pediatric patients unable to swallow tablets can also use the powder formulation:
- •
- Mix VIRACEPT Oral Powder with a small amount of water, milk, formula, soy formula, soy milk, or dietary supplements
- •
- Once mixed, the entire contents must be consumed in order to obtain the full dose.
- •
- If the mixture is not consumed immediately, it must be stored under refrigeration, but storage must not exceed 6 hours.
- •
- Acidic food or juice (e.g., orange juice, apple juice, or apple sauce) are not recommended for mixing VIRACEPT Oral Powder because the combination may result in a bitter taste.
- •
- VIRACEPT Oral Powder should not be reconstituted with water in its original container.
Drug Interactions
VIRACEPT may interact with some drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription, non-prescription medication or herbal products, including St. John's wort [see Contraindications (4), Drug Interactions (7.3), and Use in Specific Populations (8.3)].
Hepatic Impairment
Patients should be informed that VIRACEPT should not be used if there is moderate or severe hepatic impairment.
Phenylketonuria
Physicians should alert patients with phenylketonuria that VIRACEPT Oral Powder contains phenylalanine
Fat Redistribution
Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy, including PREZISTA/ritonavir, and that the cause and long-term health effects of these conditions are not known at this time
The most frequent adverse event associated with VIRACEPT is diarrhea, which can usually be controlled with non-prescription drugs, such as loperamide, which slow gastrointestinal motility.
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to VIRACEPT during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Instruct patients with HIV-1 infection not to breastfeed because HIV-1 can be passed to the infant in the breast milk [see Use in Specific Populations (8.2)].
Females and Males of Reproductive Potential
Advise females of reproductive potential that VIRACEPT may reduce the effectiveness of estrogen-based contraceptives, and to use alternative non-hormonal contraception [see Drug Interactions (7.3), Use in Specific Populations (8.3)].
-
SPL UNCLASSIFIED SECTION
VIRACEPT is a registered trademark of Agouron Pharmaceuticals, LLC
REVATIO is a registered trademark of Pfizer, Inc.
ADCIRCA is a trademark of Eli Lilly and Company
This product's labeling may have been updated. For the most recent prescribing information, please visit www.pfizer.com.
LAB-0174-29.0
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
VIRACEPT (VI-ra-cept)
(nelfinavir mesylate)
TabletsVIRACEPT (VI-ra-cept)
(nelfinavir mesylate)
Oral PowderImportant: VIRACEPT can interact with other medicines and cause serious side effects. It is important to know the medicines that should not be taken with VIRACEPT. See the section "Do not take VIRACEPT if you:"
What is VIRACEPT?
VIRACEPT is a prescription HIV-1 medicine used with other HIV-1 medicines to treat human immunodeficiency virus (HIV-1) infection. HIV-1 is the virus that causes Acquired Immune Deficiency Syndrome (AIDS).
It is not known if VIRACEPT is safe and effective in children less than 2 years of age.
- •
- are taking any of the following medicines. VIRACEPT may cause serious life-threatening side effects or death when used with these medicines:
- o
- alfuzosin
- o
- amiodarone
- o
- cisapride
- o
- ergot-containing medicines:
- ▪
- dihydroergotamine
- ▪
- ergotamine
- ▪
- methylergonovine
- o
- lovastatin
- o
- lurasidone
- o
- midazolam, when taken by mouth for sedation
- o
- pimozide
- o
- quinidine
- o
- rifampin
- o
- sildenafil (Revatio®), when used for the treatment of pulmonary arterial hypertension (PAH)
- o
- simvastatin
- o
- St. John's wort (Hypericum perforatum)
- o
- triazolam
Before taking VIRACEPT, tell your healthcare provider about all of your medical conditions, including if you:
- •
- have liver problems
- •
- have kidney problems
- •
- have phenylketonuria. VIRACEPT Oral Powder contains aspartame.
- •
- have high blood sugar (diabetes)
- •
- have hemophilia
- •
- are pregnant or plan to become pregnant. VIRACEPT may harm your unborn baby.
- o
- Tell your healthcare provider right away if you become pregnant during treatment with VIRACEPT.
- o
- Hormonal forms of birth control, such as injections, vaginal rings or implants, contraceptive patches, and some birth control pills may not work during treatment with VIRACEPT. Talk to your healthcare provider about forms of birth control that may be used during treatment with VIRACEPT.
- o
- Pregnancy Registry. There is a pregnancy registry for women who take VIRACEPT during pregnancy. The purpose of the registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry.
- •
- are breastfeeding or plan to breastfeed. Do not breastfeed if you take VIRACEPT.
- o
- You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.
- o
- VIRACEPT can pass into your breast milk.
- o
- Talk to your healthcare provider about the best way to feed your baby.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Some medicines interact with VIRACEPT. Keep a list of your medicines to show your healthcare provider and pharmacist.
- •
- You can ask your healthcare provider or pharmacist for a list of medicines that interact with VIRACEPT.
- •
-
Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take VIRACEPT with other medicines.
- o
- Especially tell your healthcare provider if you take medicines that contain didanosine. Take VIRACEPT with food one hour after or more than two hours before you take didanosine.
How should I take VIRACEPT?
- •
- You should stay under a healthcare provider's care during treatment with VIRACEPT.
- •
- Take VIRACEPT every day exactly as prescribed by your healthcare provider.
- •
- Do not change your dose of VIRACEPT or stop treatment without talking to your healthcare provider first.
- •
- Take VIRACEPT with a meal.
- •
- VIRACEPT is usually taken 2 or 3 times each day. Your healthcare provider will tell you how much VIRACEPT to take and when to take it.
- •
- If your child is taking VIRACEPT, your child's healthcare provider will decide the right dose based on your child's weight.
- •
- VIRACEPT Tablets are film-coated to help make the tablets easier to swallow.
- •
- If you or your child are unable to swallow the tablets:
- o
- You may dissolve the tablets in a small amount of water. Once the tablets are dissolved, the liquid will be cloudy.
- o
- Mix the cloudy liquid well, and then drink it right away.
- o
- Rinse the glass with water and drink the water to ensure that you take the full dose of VIRACEPT.
- •
- If you miss a dose of VIRACEPT, take it as soon as possible. If you skip a dose, do not double the next dose.
- •
- If you take too much VIRACEPT, call your healthcare provider or go to the nearest hospital emergency room right away.
- •
- Do not run out of VIRACEPT. Get your VIRACEPT refilled from your healthcare provider or pharmacy before you run out.
How should VIRACEPT Oral Powder be prepared?
- •
- Your healthcare provider should tell you how much VIRACEPT Oral Powder to use.
- •
- VIRACEPT Oral Powder comes with a scoop for measuring. Ask your healthcare provider or pharmacist for help measuring the correct amount of VIRACEPT Oral Powder.
- •
- Do not mix VIRACEPT Oral Powder in the container that it comes in. Measure the correct amount of VIRACEPT Oral Powder into a cup.
- •
- You may mix VIRACEPT Oral Powder with a small amount of water, milk, formula, soy formula, soy milk, or liquid dietary supplements.
- •
- You should not mix VIRACEPT Oral Powder with acidic food or juice, such as orange juice, apple juice, or applesauce because the mixture may taste bitter.
- •
- Your child should drink all of the mixture to be sure that the entire dose of VIRACEPT is taken.
- •
- If your child does not take the dose right away, store in the refrigerator until you are ready to use it. Do not store a mixed dose of VIRACEPT Oral Powder for more than 6 hours.
What are the possible side effects of VIRACEPT?
VIRACEPT can cause serious side effects, including:
- •
- Diabetes and high blood sugar (hyperglycemia). Some people who take protease inhibitors including VIRACEPT can get high blood sugar, develop diabetes, or your diabetes can get worse. Some people have had to start taking medicine to treat diabetes or have had to change their diabetes medicine. Tell your healthcare provider if you notice an increase in thirst or urinate often while taking VIRACEPT.
- •
- Increased bleeding in people with hemophilia. Some people with hemophilia have increased bleeding with protease inhibitors including VIRACEPT.
- •
- Changes in body fat. These changes can happen in people taking HIV-1 medicines. The changes may include an increased amount of fat in the upper back and neck ("buffalo hump"), breast, and around the middle of your body (trunk). Loss of fat from the legs, arms, and face may also happen. The exact cause and long-term health effects of these conditions are not known.
- •
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Call your healthcare provider right away if you start having new symptoms after starting your HIV-1 medicine.
The most common side effects of VIRACEPT in adults and adolescents (13 years of age and older) include:
- •
- diarrhea
- •
- nausea
- •
- rash
- •
- gas
The most common side effects of VIRACEPT in children (2 years of age to less than 13 years of age) include:
- •
- diarrhea
- •
- low white blood cell count (leukopenia and neutropenia)
- •
- rash
- •
- loss of appetite
- •
- stomach-area (abdominal) pain
These are not all the possible side effects of VIRACEPT.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VIRACEPT?
- •
- Store VIRACEPT Tablets and Oral Powder at room temperature, between 59ºF to 86ºF (15ºC to 30ºC).
- •
- Store VIRACEPT in the original container.
- •
- Keep the container closed tightly.
Keep VIRACEPT and all medicines out of the reach of children.
General information about the safe and effective use of VIRACEPT.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VIRACEPT for a condition for which it was not prescribed. Do not give VIRACEPT to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about VIRACEPT that is written for health professionals.
What are the ingredients in VIRACEPT?
Active ingredient: nelfinavir mesylate
Tablet inactive ingredients: calcium silicate, crospovidone, magnesium stearate, hypromellose, and triacetin. In addition, the 250 mg tablet contains FD&C blue #2 powder and the 625 mg tablet contains colloidal silicon dioxide.
Oral powder inactive ingredients: microcrystalline cellulose, maltodextrin, dibasic potassium phosphate, crospovidone, hypromellose, aspartame, sucrose palmitate, and natural and artificial flavor.

VIRACEPT is a registered trademark of Agouron Pharmaceuticals, LLC
For more information, call 1-800-438-1985.
Trademarks are the property of their respective owners.
LAB-0346-15.0This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 10/2023
-
PRINCIPAL DISPLAY PANEL - 250 mg Tablet Bottle Label
NDC 63010-010-30
300 TabletsVIRACEPT®
(nelfinavir mesylate)
TabletsEach tablet contains nelfinavir mesylate
equivalent to 250 mg of nelfinavir (free base).ALERT: Find out about
medicines that should NOT
be taken with VIRACEPT.250mg
Viracept
Note to Pharmacist: Do not
cover ALERT box with
pharmacy label.Agouron®
Pharmaceuticals, LLC
A Pfizer CompanyRx only
300 Tablets
- PRINCIPAL DISPLAY PANEL - 625 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
VIRACEPT
nelfinavir mesylate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63010-010 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NELFINAVIR MESYLATE (UNII: 98D603VP8V) (NELFINAVIR - UNII:HO3OGH5D7I) NELFINAVIR 250 mg Inactive Ingredients Ingredient Name Strength CALCIUM SILICATE (UNII: S4255P4G5M) CROSPOVIDONE (120 .MU.M) (UNII: 68401960MK) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TRIACETIN (UNII: XHX3C3X673) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color BLUE (light blue) Score no score Shape OVAL (capsule-shaped) Size 19mm Flavor Imprint Code VIRACEPT;250;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63010-010-30 300 in 1 BOTTLE; Type 0: Not a Combination Product 03/14/1997 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020779 03/14/1997 VIRACEPT
nelfinavir mesylate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63010-027 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NELFINAVIR MESYLATE (UNII: 98D603VP8V) (NELFINAVIR - UNII:HO3OGH5D7I) NELFINAVIR 625 mg Inactive Ingredients Ingredient Name Strength CALCIUM SILICATE (UNII: S4255P4G5M) CROSPOVIDONE (120 .MU.M) (UNII: 68401960MK) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TRIACETIN (UNII: XHX3C3X673) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Product Characteristics Color WHITE Score no score Shape OVAL Size 26mm Flavor Imprint Code V;625 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63010-027-70 120 in 1 BOTTLE; Type 0: Not a Combination Product 04/30/2003 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021503 04/30/2003 Labeler - AGOURON (145772760) Registrant - Pfizer Inc (113480771) Establishment Name Address ID/FEI Business Operations Pharmacia & Upjohn Company LLC 618054084 ANALYSIS(63010-010, 63010-027) , API MANUFACTURE(63010-010, 63010-027) , PACK(63010-010, 63010-027) Establishment Name Address ID/FEI Business Operations Pfizer Manufacturing Deutschland GmbH 341970073 ANALYSIS(63010-010, 63010-027) , MANUFACTURE(63010-010, 63010-027) , PACK(63010-010, 63010-027) , LABEL(63010-010, 63010-027)