Label: PRAVASTATIN SODIUM tablet




-
NDC Code(s):
68462-195-05,
68462-195-90,
68462-196-05,
68462-196-90, view more68462-197-05, 68462-197-90, 68462-198-05, 68462-198-90
- Packager: Glenmark Pharmaceuticals Inc., USA
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated July 8, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PRAVASTATIN SODIUM TABLETS safely and effectively. See full prescribing information for PRAVASTATIN SODIUM TABLETS.
PRAVASTATIN SODIUM tablets, for oral use
Initial U.S. Approval: 1991RECENT MAJOR CHANGES
INDICATIONS AND USAGE
Pravastatin sodium tablets are an HMG-CoA reductase inhibitor (statin) indicated (1):
- •
- To reduce the risk of myocardial infarction, myocardial revascularization procedures, and cardiovascular mortality in adults with elevated low-density lipoprotein cholesterol (LDL-C) without clinically evident coronary heart disease (CHD).
- •
- To reduce the risk of coronary death, myocardial infarction, myocardial revascularization procedures, stroke or transient ischemic attack, and slow the progression of coronary atherosclerosis in adults with clinically evident CHD.
- •
- As an adjunct to diet to reduce LDL-C in adults with primary hyperlipidemia.
- •
- As an adjunct to diet to reduce LDL-C in pediatric patients ages 8 years and older with heterozygous familial hypercholesterolemia (HeFH).
- •
- As an adjunct to diet for the treatment of adults with:
- •
- Primary dysbetalipoproteinemia.
- •
- Hypertriglyceridemia.
DOSAGE AND ADMINISTRATION
- •
- Take orally once daily at any time of the day, with or without food (2.1).
- •
- For patients that require a high-intensity statin or are unable to achieve their LDL-C goal receiving pravastatin sodium tablets 80 mg daily, prescribe alternative LDL-C-lowering treatment (2.1).
- •
- Assess LDL-C when clinically appropriate, as early as 4 weeks after initiating pravastatin sodium, and adjust the dosage if necessary (2.1).
- •
- Adults: recommended starting dosage is pravastatin sodium tablets 40 mg to 80 mg once daily. (2.2)
- •
- Pediatric Patients (2.3):
- •
- aged 8 to 13 years, the recommended dosage is 20 mg once daily.
- •
- aged 14 to 18 years, the recommended starting dosage is 40 mg once daily.
- •
- Severe renal impairment: recommended starting dosage is pravastatin sodium 10 mg once daily. Recommended maximum pravastatin sodium tablets dosage is 40 mg once daily. (2.4)
- •
- See full prescribing information for dosage modifications due to drug interactions (2.5,7).
DOSAGE FORMS AND STRENGTHS
- •
- Tablets: 10 mg, 20 mg, 40 mg, 80 mg of pravastatin sodium, USP. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- •
- Myopathy and Rhabdomyolysis: Risk factors include age 65 years or greater, uncontrolled hypothyroidism, renal impairment, concomitant use with certain other drugs, and higher pravastatin sodium dosage. Discontinue pravastatin sodium if markedly elevated CK levels occur or myopathy is diagnosed or suspected. Temporarily discontinue pravastatin sodium in patients experiencing an acute or serious condition at high risk of developing renal failure secondary to rhabdomyolysis. Inform patients of the risk of myopathy and rhabdomyolysis when starting or increasing pravastatin sodium dosage. Instruct patients to promptly report unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever. (5.1, 7.1, 8.5, 8.6)
- •
- Immune-Mediated Necrotizing Myopathy (IMNM): Rare reports of IMNM, an autoimmune myopathy, have been reported. Discontinue pravastatin sodium if IMNM is suspected (5.2).
- •
- Hepatic Dysfunction: Increases in serum transaminases have occurred, some persistent. Rare reports of fatal and non-fatal hepatic failure have occurred. Consider testing liver enzymes before initiating therapy and as clinically indicated thereafter. If serious hepatic injury with clinical symptoms and/or hyperbilirubinemia or jaundice occurs, promptly discontinue (5.3).
ADVERSE REACTIONS
In short-term clinical trials, the most commonly reported adverse reactions (≥2% and greater than placebo) were: musculoskeletal pain, nausea/vomiting, upper respiratory infection, diarrhea, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Glenmark Pharmaceuticals Inc., USA at 1 (888) 721-7115 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- •
- See full prescribing information for details regarding concomitant use of pravastatin sodium with other drugs that increase the risk of myopathy and rhabdomyolysis. (2.5, 7.1)
- •
- Bile Acid Sequestrants: in patients taking a bile acid sequestrant, administer pravastatin sodium at least 1 hour before or at least 4 hours after the bile acid sequestrant (7.2)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
Important Dosage and Administration Information
2.2 Recommended Dosage in Adult Patients
Recommended Dosage in Pediatric Patients 8 Years of Age and Older with HeFH
2.4 Recommended Dosage in Patients with Renal Impairment
2.5 Dosage and Administration Modifications Due to Drug Interactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myopathy and Rhabdomyolysis
5.2 Immune-Mediated Necrotizing Myopathy
5.3 Hepatic Dysfunction
5.4 Increases in HbA1c and Fasting Serum Glucose Levels
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drug Interactions that Increase the Risk of Myopathy and Rhabdomyolysis with Pravastatin Sodium
7.2 Drug Interactions that Decrease the Efficacy of Pravastatin Sodium
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
- Pravastatin sodium tablets are indicated:
- 1.
- To reduce the risk of myocardial infarction, myocardial revascularization procedures, and cardiovascular mortality in adults with elevated low-density lipoprotein cholesterol (LDL-C) without clinically evident coronary heart disease (CHD).
- 2.
- To reduce the risk of coronary death, myocardial infarction, myocardial revascularization procedures, stroke or transient ischemic attack, and slow the progression of coronary atherosclerosis in adults with clinically evident CHD.
- 3.
- As an adjunct to diet to reduce LDL-C in adults with primary hyperlipidemia.
- 4.
- As an adjunct to diet to reduce LDL-C in pediatric patients ages 8 years and older with heterozygous familial hypercholesterolemia (HeFH).
- 5.
- As an adjunct to diet for the treatment of adults with:
- •
- Primary dysbetalipoproteinemia.
- •
- Hypertriglyceridemia.
-
2 DOSAGE AND ADMINISTRATION
Important Dosage and Administration Information
- •
- Take pravastatin sodium tablets orally once daily as a single dose at any time of the day, with or without food.
- •
- For patients that require a high-intensity statin or are unable to achieve their LDL-C goal receiving pravastatin sodium tablets 80 mg daily, prescribe alternative LDL-C-lowering treatment.
- •
- Assess LDL-C when clinically appropriate, as early as 4 weeks after initiating pravastatin sodium tablets, and adjust the dosage if necessary.
2.2 Recommended Dosage in Adult Patients
The recommended starting dosage is pravastatin sodium tablets 40 mg to 80 mg once daily.
Recommended Dosage in Pediatric Patients 8 Years of Age and Older with HeFH
- •
- In pediatric patients aged 8 to 13 years, the recommended dosage is pravastatin sodium tablets 20 mg once daily.
- •
- In pediatric patients aged 14 to 18 years, the recommended starting dosage is pravastatin sodium tablets 40 mg once daily.
2.4 Recommended Dosage in Patients with Renal Impairment
- •
- In patients with severe renal impairment, the recommended starting dosage is pravastatin sodium 10 mg once daily. The maximum recommended dosage of pravastatin sodium tablets in patients with severe renal impairment is 40 mg once daily [see Clinical Pharmacology (12.3)].
- •
- The recommended dosage of pravastatin sodium tablets for patients with mild or moderate renal impairment is the same as patients with normal renal function.
2.5 Dosage and Administration Modifications Due to Drug Interactions
- •
- In patients taking a bile acid sequestrant, administer pravastatin sodium tablets at least 1 hour before or 4 hours after the bile acid sequestrant [See Drug Interactions (7.2)].
- •
-
Concomitant use of pravastatin sodium tablets with the following drugs requires dosage modifications of pravastatin sodium tablets [see Warnings and Precautions (5.1) and Drug Interactions (7.1)]:
- •
- Cyclosporine
- In patients taking cyclosporine, the recommended starting dosage is pravastatin sodium 10 mg once daily. The maximum recommended dosage of pravastatin sodium tablets in patients taking cyclosporine is 20 mg once daily.
- •
- Clarithromycin and Erythromycin
- The maximum recommended dosage is pravastatin sodium tablets 40 mg once daily.
-
3 DOSAGE FORMS AND STRENGTHS
Pravastatin Sodium Tablets, USP are supplied as:
- •
- 10 mg of pravastatin sodium:Yellow colored, circular shaped, flat faced tablets with “G5” debossed on one side and “10” debossed on the other side.
- •
- 20 mg of pravastatin sodium:Yellow, rounded-rectangular, biconvex tablets with “G5” debossed on one side and “20” debossed on the other side.
- •
- 40 mg of pravastatin sodium:Green, rounded-rectangular, biconvex tablets with “G5” debossed on one side and “40” debossed on the other side.
- •
- 80 mgof pravastatin sodium: Yellow, oval, biconvex tablets with “G5” debossed on one side and “80” debossed on the other side.
-
4 CONTRAINDICATIONS
Acute liver failure or decompensated cirrhosis [see Warnings and Precautions (5.3)].
Hypersensitivity to any pravastatin or any excipients in pravastatin sodium tablets.
-
5 WARNINGS AND PRECAUTIONS
5.1 Myopathy and Rhabdomyolysis
- Pravastatin sodium may cause myopathy and rhabdomyolysis. Acute kidney injury secondary to myoglobinuria and rare fatalities have occurred as a result of rhabdomyolysis in patients treated with statins, including pravastatin sodium. Myopathy, defined as muscle aching or muscle weakness in conjunction with increases in creatine phosphokinase (CK) to greater than 10 times the upper limit of normal (ULN), occurred <0.1% in pravastatin sodium-treated patients in clinical trials.
- Risk Factors for Myopathy
- Risk factors for myopathy include age 65 years or greater, uncontrolled hypothyroidism, renal impairment, concomitant use with certain other drugs (including other lipid-lowering therapies), and higher pravastatin sodium dosage [see Drug Interactions (7.1)].
- Steps to Prevent or Reduce the Risk of Myopathy and Rhabdomyolysis
- Pravastatin sodium is not recommended in patients taking gemfibrozil [see Drug Interactions (7)]. There are pravastatin sodium dosage restrictions for patients taking cyclosporin and select macrolide antibiotics [see Dosage and Administration (2.5)]. The following drugs when used concomitantly with pravastatin sodium may also increase the risk of myopathy and rhabdomyolysis: niacin, fibrates, and colchicine [see Drug Interactions (7)].
- Discontinue pravastatin sodium if markedly elevated CK levels occur or myopathy is diagnosed or suspected. Muscle symptoms and CK increases may resolve if pravastatin sodium is discontinued. Temporarily discontinue pravastatin sodium in patients experiencing an acute or serious condition at high risk of developing renal failure secondary to rhabdomyolysis, e.g., sepsis; shock; severe hypovolemia; major surgery; trauma; severe metabolic, endocrine, or electrolyte disorders; or uncontrolled epilepsy.
- Inform patients of the risk of myopathy and rhabdomyolysis when starting or increasing the pravastatin sodium dosage. Instruct patients to promptly report any unexplained muscle pain, tenderness or weakness, particularly if accompanied by malaise or fever.
5.2 Immune-Mediated Necrotizing Myopathy
There have been rare reports of immune-mediated necrotizing myopathy (IMNM), an autoimmune myopathy, associated with statin use, including reports of recurrence when the same or a different statin was administered. IMNM is characterized by proximal muscle weakness and elevated serum creatine kinase that persist despite discontinuation of statin treatment; positive anti-HMG CoA reductase antibody; muscle biopsy showing necrotizing myopathy; and improvement with immunosuppressive agents. Additional neuromuscular and serologic testing may be necessary. Treatment with immunosuppressive agents may be required. Discontinue pravastatin sodium if IMNM is suspected.
5.3 Hepatic Dysfunction
- Increases in serum transaminases have been reported with use of pravastatin sodium [see Adverse Reactions (6.1)]. In most cases, these changes appeared soon after initiation, were transient, were not accompanied by symptoms, and resolved or improved on continued therapy or after a brief interruption in therapy. Persistent increases to more than three times the ULN in serum transaminases have occurred in approximately 1% of patients receiving either pravastatin sodium or placebo in clinical studies. Marked persistent increases of hepatic transaminases have also occurred with pravastatin sodium. There have been rare postmarketing reports of fatal and non-fatal hepatic failure in patients taking statins, including pravastatin sodium.
- Patients who consume substantial quantities of alcohol and/or have a history of liver disease may be at increased risk for hepatic injury.
- Consider liver enzyme testing before pravastatin sodium initiation and when clinically indicated thereafter. pravastatin sodium is contraindicated in patients with acute liver failure or decompensated cirrhosis [see Contraindications (4)]. If serious hepatic injury with clinical symptoms and/or hyperbilirubinemia or jaundice occurs, promptly discontinue pravastatin sodium.
-
6 ADVERSE REACTIONS
The following important adverse reactions are described below and elsewhere in the labeling:
- •
- Myopathy and Rhabdomyolysis [see Warnings and Precautions (5.1)]
- •
- Immune-Mediated Necrotizing Myopathy [see Warnings and Precautions (5.2)]
- •
- Hepatic Dysfunction [see Warnings and Precautions (5.3)]
- •
- Increases in HbA1c and Fasting Serum Glucose Levels [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
- Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
- In pravastatin sodium placebo-controlled clinical trials, 1313 patients (age range 20 to 76 years, 32% women, 93.5% White, 5% Black, 0.9% Hispanic, 0.4% Asian, 0.2% Other) with a median treatment duration of 14 weeks, 3.3% of patients on pravastatin sodium and 1.2% patients on placebo discontinued due to adverse reactions (regardless of causality). The most common adverse reactions that led to treatment discontinuation and occurred at an incidence greater than placebo were: hepatic transaminase elevations, nausea, anxiety/depression, and dizziness.
Adverse reactions (regardless of causality) reported in ≥2% of pravastatin sodium-treated patients in placebo-controlled trials of up to 8 months duration are identified in Table 1:
Table 1: Adverse Reactions in ≥2% of Patients Treated with Pravastatin (Any Dose) and at an Incidence Greater Than Placebo in Short-Term Placebo-Controlled Trials % Placebo
N=411
% Any Dose
N=902
Nausea/Vomiting
7.1
7.4
Diarrhea
5.6
6.7
Headache
4.6
6.3
Upper Respiratory Infection
5.8
5.9
Angina Pectoris
3.4
4.5
Rash
1.4
4.5
CPK Increased
3.6
4.1
Dizziness
3.4
3.5
ALT Increased
1.2
2.9
Chest Pain
1.9
2.7
Cough
1.7
2.5
Myalgia
1.2
2.3
Influenza
0.7
2
g-GT Increased
1.2
2
Adverse Reactions (regardless of causality)
The safety and tolerability of pravastatin sodium at a dose of 80 mg in 2 controlled trials with a mean exposure of 8.6 months was similar to that of pravastatin sodium at lower doses except that 4 out of 464 patients taking 80 mg of pravastatin had a single elevation of CK >10 times ULN compared to 0 out of 115 patients taking 40 mg of pravastatin.
In pravastatin sodium placebo-controlled clinical trials, 21,483 patients (age range 24 to 75 years, 10.3% women, 52.3% White, 0.8% Black, 0.5% Hispanic, 0.1% Asian, 0.1% Other, 46.1% not recorded) had a median treatment duration of 261 weeks.
Adverse reactions (regardless of causality) were pooled from 7 double-blind, placebo-controlled trials (West of Scotland Coronary Prevention Study [WOS]; Cholesterol and Recurrent Events study [CARE]; Long-term Intervention with Pravastatin in Ischemic Disease study [LIPID]; Pravastatin Limitation of Atherosclerosis in the Coronary Arteries study [PLAC I]; Pravastatin, Lipids and Atherosclerosis in the Carotids study [PLAC II]; Regression Growth Evaluation Statin Study [REGRESS]; and Kuopio Atherosclerosis Prevention Study [KAPS]) involving a total of 10,764 patients treated with pravastatin sodium 40 mg and 10,719 patients treated with placebo. Patients were exposed to pravastatin sodium for a mean of 4 to 5.1 years in WOS, CARE, and LIPID and 1.9 to 2.9 years in PLAC I, PLAC II, KAPS, and REGRESS. Adverse reactions (regardless of causality) occurring in ≥5% of patients treated with pravastatin sodium in these studies are identified in Table 2.
Table 2: Adverse Reactions in ≥5% of Patients Treated with Pravastatin 40 mg and at an Incidence Greater than Placebo in Long-Term Placebo-Controlled Trials Placebo
(N=10,719)
% of patients
Pravastatin sodium
(N=10,764)
% of patients
Musculoskeletal Pain
24.4
24.9
Upper Respiratory Tract Infection
20.2
21.2
Musculoskeletal Traumatism
9.6
10.2
Chest Pain
9.8
10
Influenza
9
9.2
Fatigue
7.8
8.4
Cough
7.4
8.2
Dizziness
6.6
7.3
Rash (including dermatitis)
7.1
7.2
Sinus Abnormality
6.7
7
Muscle Cramp
4.6
5.1
Adverse Reactions (regardless of causality)
No new adverse reactions were identified in a study of pediatric patients with HeFH.
Laboratory Abnormalities
Increases in ALT, AST values and CK have been observed.
Transient, asymptomatic eosinophilia has been reported. Eosinophil counts usually returned to normal despite continued therapy. Anemia, thrombocytopenia, and leukopenia have been reported with statins.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of pravastatin sodium. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Musculoskeletal: myopathy, rhabdomyolysis, tendon disorder, polymyositis, immune-mediated necrotizing myopathy associated with statin use.
Nervous System: dysfunction of certain cranial nerves (including alteration of taste, impairment of extraocular movement, facial paresis), peripheral nerve palsy. Rare postmarketing reports of cognitive impairment (e.g., memory loss, forgetfulness, amnesia, memory impairment, confusion) associated with statin use. Cognitive impairment was generally nonserious, and reversible upon statin discontinuation, with variable times to symptom onset (1 day to years) and symptom resolution (median of 3 weeks).
Hypersensitivity: anaphylaxis, angioedema, lupus erythematosus-like syndrome, polymyalgia rheumatica, dermatomyositis, vasculitis, purpura, hemolytic anemia, positive ANA, ESR increase, arthritis, arthralgia, asthenia, photosensitivity, chills, malaise, toxic epidermal necrolysis, erythema multiforme (including Stevens-Johnson syndrome).
Gastrointestinal: abdominal pain, constipation, pancreatitis, hepatitis (including chronic active hepatitis), cholestatic jaundice, fatty change in liver, cirrhosis, fulminant hepatic necrosis, hepatoma, fatal and non-fatal hepatic failure.
Dermatologic: a variety of skin changes (e.g., nodules, discoloration, dryness of mucous membranes, changes to hair/nails), lichen planus.
Renal: urinary abnormality (including dysuria, frequency, nocturia).
Respiratory: dyspnea, interstitial lung disease.
Psychiatric: nightmare.
Reproductive: gynecomastia.
Laboratory Abnormalities: liver function test abnormalities, thyroid function abnormalities.
-
7 DRUG INTERACTIONS
7.1 Drug Interactions that Increase the Risk of Myopathy and Rhabdomyolysis with Pravastatin Sodium
Pravastatin sodium is a substrate of the transport protein OATP1B1. Pravastatin sodium plasma levels can be significantly increased with concomitant administration of inhibitors of OATP1B1. Table 3 includes a list of drugs that increase the risk of myopathy and rhabdomyolysis when used concomitantly with pravastatin sodium and instructions for preventing or managing them [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
Table 3: Drug Interactions that Increase the Risk of Myopathy and Rhabdomyolysis with Pravastatin Sodium
Gemfibrozil
Clinical Impact:
- There is an increased risk of myopathy/rhabdomyolysis when pravastatin Sodium is administered with gemfibrozil
Intervention:
- Avoid concomitant use of gemfibrozil with pravastatin sodium.
Cyclosporine
Clinical Impact:
- The risk of myopathy and rhabdomyolysis is increased with concomitant use of cyclosporine with pravastatin sodium.
Intervention:
- Initiate with a dosage of pravastatin sodium 10 mg once daily. Do not exceed pravastatin sodium 20 mg once daily [see Dosage and Administration (2.5)].
Select Macrolide Antibiotics
Clinical Impact:
- The risk of myopathy and rhabdomyolysis is increased by concomitant use of clarithromycin or erythromycin with pravastatin sodium. Other macrolides (e.g., azithromycin) have the potential to increase pravastatin sodium exposures and increase the risk of myopathy and rhabdomyolysis when used concomintantly.
Intervention:
- For patients taking erythromycin or clarithromycin, do not exceed 40 mg pravastatin sodium once daily [see Dosage and Administration (2.5)].
Niacin
Clinical Impact:
- Cases of myopathy and rhabdomyolysis have been observed with concomitant use of niacin with pravastatin sodium.
Intervention:
- Consider if the benefit of using niacin concomitantly with pravastatin sodium outweighs the increased risk of myopathy and rhabdomyolysis. If concomitant use is decided, monitor patients for signs and symptoms of myopathy, particularly during initiation of therapy and during upward dose titration of either drug.
Fibrates (other than Gemfibrozil)
Clinical Impact:
- Fibrates may cause myopathy when given alone. The risk of myopathy and rhabdomyolysis is increased with concomitant use of fibrates with pravastatin sodium.
Intervention:
- Consider if the benefit of using fibrates concomitantly with pravastatin sodium outweighs the increased risk of myopathy and rhabdomyolysis. If concomitant use is decided, monitor patients for signs and symptoms of myopathy, particularly during initiation of therapy and during upward dose titration of either drug.
Colchicine
Clinical Impact:
- Cases of myopathy and rhabdomyolysis have been reported with concomitant use of colchicine with pravastatin sodium.
Intervention:
- Consider if the benefit of using colchicine concomitantly with pravastatin sodium outweighs the increased risk of myopathy and rhabdomyolysis. If concomitant use is decided, monitor patients for signs and symptoms of myopathy, particularly during initiation of therapy and during upward dose titration of either drug.
7.2 Drug Interactions that Decrease the Efficacy of Pravastatin Sodium
Table 4 presents drug interactions that may decrease the efficacy of pravastatin sodium and instructions for preventing or managing them.
Table 4: Drug Interactions that Decrease the Efficacy of Pravastatin Sodium
Bile Acid Sequestrants
Clinical Impact:
Concomitant cholestyramine or colestipol administration decreased the mean exposure of pravastatin approximately 51% and 47%, respectively [see Clinical Pharmacology (12.3)].
Intervention:
In patients taking a bile acid sequestrant, administer pravastatin sodium at least 1 hour before or at least 4 hours after the bile acid sequestrant [see Dosage and Administration (2.5)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Discontinue pravastatin sodium when pregnancy is recognized. Alternatively, consider the ongoing therapeutic needs of the individual patient. pravastatin sodium decreases synthesis of cholesterol and possibly other biologically active substances derived from cholesterol; therefore, pravastatin sodium may cause fetal harm when administered to pregnant patients based on the mechanism of action [see Clinical Pharmacology (12.1)]. In addition, treatment of hyperlipidemia is not generally necessary during pregnancy. Atherosclerosis is a chronic process and the discontinuation of lipid- lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hyperlipidemia for most patients. Available data from case series and prospective and retrospective observational cohort studies over decades of use with statins in pregnant women have not identified a drug-associated risk of major congenital malformations. Published data from prospective and retrospective observational cohort studies with pravastatin sodium use in pregnant women are insufficient to determine if there is a drug-associated risk of miscarriage (see Data). In animal reproduction studies, no evidence of fetal malformations was seen in pregnant rats or rabbits orally administered pravastatin during the period of organogenesis at doses that resulted in 10 times and 120 times, respectively, the human exposure at the maximum recommended human dose (MRHD) of 80 mg/day, based on body surface area (mg/m2). An imbalance in some fetal skeletal variations, increased offspring mortality, and developmental delays occurred when pregnant rats were exposed to 10 times to 12 times the MRHD during organogenesis to parturition (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
A Medicaid cohort linkage study of 1152 statin-exposed pregnant women compared to 886,996 controls did not find a significant teratogenic effect from maternal use of statins in the first trimester of pregnancy, after adjusting for potential cofounders – including maternal age, diabetes mellitus, hypertension, obesity, and alcohol and tobacco use – using propensity score-based methods. The relative risk of congenital malformations between the group with statin use and the group with no statin use in the first trimester was 1.07 (95% confidence interval 0.85 to 1.37) after controlling for confounders, particularly pre-existing diabetes mellitus. There were also no statistically significant increases in any of the organ-specific malformations assessed after accounting for cofounders. In the majority of pregnancies, statin treatment was initiated prior to pregnancy and was discontinued at some point in the first trimester when pregnancy was identified. Study limitations include reliance on physician coding to define the presence of a malformation, lack of control for certain confounders such as body mass index, use of prescription dispensing as verification for the use of a statin, and lack of information on non-live births.
Animal Data
Embryofetal and neonatal mortality was observed in rats given pravastatin during the period of organogenesis or during organogenesis continuing through weaning. In pregnant rats given oral gavage doses of 4, 20, 100, 500, and 1000 mg/kg/day from gestation days 7 through 17 (organogenesis) increased mortality of offspring and increased cervical rib skeletal anomalies were observed at ≥100 mg/kg/day systemic exposure, 10 times the human exposure at 80 mg/day MRHD based on body surface area (mg/m2).
In other studies, no teratogenic effects were observed when pravastatin was dosed orally during organogenesis in rabbits (gestation days 6 through 18) up to 50 mg/kg/day or in rats (gestation days 7 through 17) up to 1000 mg/kg/day. Exposures were 10 times (rabbit) or 120 times (rat) the human exposure at 80 mg/day MRHD based on body surface area (mg/m2).
In pregnant rats given oral gavage doses of 10, 100, and 1000 mg/kg/day from gestation day 17 through lactation day 21 (weaning), developmental delays were observed at ≥100 mg/kg/day systemic exposure, corresponding to 12 times the human exposure at 80 mg/day MRHD, based on body surface area (mg/m2).
In pregnant rats, pravastatin crosses the placenta and is found in fetal tissue at 30% of the maternal plasma levels following administration of a single dose of 20 mg/day orally on gestation day 18, which corresponds to exposure 2 times the MRHD of 80 mg daily based on body surface area (mg/m2).
8.2 Lactation
Risk Summary
- Based on one lactation study in published literature, pravastatin is present in human milk. There is no available information on the effects of the drug on the breastfed infant or the effects of the drug on milk production. Statins, including pravastatin sodium, decrease cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol and may cause harm to the breastfed infant.
Because of the potential for serious adverse reactions in a breastfed infant, based on the mechanism of action, advise patients that breastfeeding is not recommended during treatment with pravastatin sodium [see Use in Specific Populations (8.1), Clinical Pharmacology (12.1)].
8.4 Pediatric Use
The safety and effectiveness of pravastatin sodium as an adjunct to diet to reduce LDL-C have been established in pediatric patients 8 years of age and older with HeFH. Use of pravastatin sodium for this indication is based on a double-blind, placebo-controlled clinical study in 214 pediatric patients (100 males and 114 females) 8 years of age and older with HeFH. Doses greater than 40 mg daily have not been studied in this population.
The safety and effectiveness of pravastatin sodium have not been established in pediatric patients younger than 10 years of age with HeFH or in pediatric patients with other types of hyperlipidemia (other than HeFH).
8.5 Geriatric Use
In clinical studies, 4,797 (36.4%) pravastatin sodium-treated patients were aged 65 and older and 110 (0.8%) were aged 75 and older. No significant differences in efficacy or safety were observed between geriatric patients and younger patients.
Mean pravastatin AUCs are 25% to 50% higher in elderly subjects than in healthy young subjects, but mean maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), and half-life (t½) values are similar in both age groups and substantial accumulation of pravastatin would not be expected in the elderly [see Clinical Pharmacology (12.3)].
- Advanced age (≥65 years) is a risk factor for pravastatin sodium-associated myopathy and rhabdomyolysis. Dose selection for an elderly patient should be cautious, recognizing the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy and the higher risk of myopathy. Monitor geriatric patients receiving pravastatin sodium for the increased risk of myopathy [see Warnings and Precautions (5.1)].
8.6 Renal Impairment
Renal impairment is a risk factor for myopathy and rhabdomyolysis. Monitor all patients with renal impairment for development of myopathy.
In patients with severe renal impairment, the recommended starting dose is pravastatin sodium 10 mg once daily. The maximum recommended dosage in patients with severe renal impairment is pravastatin sodium 40 mg once daily. The recommended dosage for patients with mild or moderate renal impairment is the same as patients with normal renal function. [see Dosage and Administration (2.4), Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Pravastatin sodium shows a large inter-subject variability in pharmacokinetics in patients with liver cirrhosis [Clinical Pharmacology (12.3)]. Pravastatin sodium is contraindicated in patients with acute liver failure or decompensated cirrhosis [see Contraindications (4), Warnings and Precautions (5.3)].
- 10 OVERDOSAGE
-
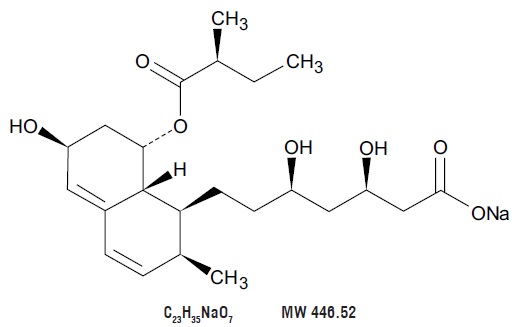
11 DESCRIPTION
Pravastatin Sodium Tablets, USP is a statin, an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase.
Pravastatin sodium, USP is designated chemically as 1-Naphthaleneheptanoic acid, 1,2,6,7,8,8a-hexahydro-β,δ,6-trihydroxy-2-methyl-8-(2-methyl-1-oxobutoxy)-, monosodium salt, [1S-[1α(βS*,δS*),2α,6α,8β(R*),8aα]]-.
Structural formula:
Pravastatin sodium, USP is an odorless, white to off-white, fine or crystalline powder. It is a relatively polar hydrophilic compound with a partition coefficient (octanol/water) of 0.59 at a pH of 7.0. It is soluble in methanol and water (>300 mg/mL), slightly soluble in isopropanol, and practically insoluble in acetone, acetonitrile, chloroform, and ether.
Pravastatin Sodium Tablets, USP for oral use contain 10 mg, 20 mg, 40 mg, and 80 mg pravastatin sodium, which is equivalent to 9.48 mg, 18.97 mg, 37.94 mg and 75.88 mg of pravastatin, respectively. Inactive ingredients include: colloidal silicon dioxide, crospovidone, hydroxypropyl methylcellulose, magnesium stearate, mannitol, meglumine, microcrystalline cellulose and starch. The 10 mg, 20 mg and 80 mg tablets also contain D&C yellow no. 10 aluminum lake and the 40 mg tablet also contains D&C yellow no. 10 aluminum lake and FD&C blue no. 1 aluminum lake.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pravastatin is a reversible inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts HMG-CoA to mevalonate, a precursor of cholesterol.
12.2 Pharmacodynamics
Inhibition of HMG-CoA reductase by pravastatin accelerates the expression of LDL-receptors, followed by the uptake of LDL-C from blood to the liver, leading to a decrease in plasma LDL-C and total cholesterol. Sustained inhibition of cholesterol synthesis in the liver also decreases levels of very-low-density lipoproteins. The maximum LDL-C reduction of pravastatin sodium is usually achieved by 4 weeks and is maintained after that.
12.3 Pharmacokinetics
Absorption
Pravastatin sodium is administered orally in the active form. Peak plasma pravastatin concentrations occurred 1 to 1.5 hours upon oral administration. Based on urinary recovery of total radiolabeled drug, the average oral absorption of pravastatin is 34% and absolute bioavailability is 17%. While the presence of food in the gastrointestinal tract reduces area under the concentration-time curve (AUC) and Cmax by 31% and 49%, respectively, the lipid-lowering effects of the drug are similar whether taken with or 1 hour prior to meals.
Pravastatin plasma concentrations, including AUC, Cmax, and steady-state minimum (Cmin), are directly proportional to administered dose. Systemic bioavailability of pravastatin administered following a bedtime dose was decreased 60% compared to that following an AM dose. Despite this decrease in systemic bioavailability, the efficacy of pravastatin administered once daily in the evening, although not statistically significant, was marginally more effective than that after a morning dose.
The coefficient of variation (CV), based on between-subject variability, was 50% to 60% for AUC. The geometric means of pravastatin Cmax and AUC following a 20 mg dose in the fasted state were 26.5 ng/mL and 59.8 ng*hr/mL, respectively.
Steady-state AUCs, Cmax, and Cmin plasma concentrations showed no evidence of pravastatin accumulation following once or twice daily administration of pravastatin sodium tablets.
Distribution
Approximately 50% of the circulating drug is bound to plasma proteins.
Elimination
Metabolism
The major biotransformation pathways for pravastatin are: (a) isomerization to 6-epi pravastatin and the 3α-hydroxyisomer of pravastatin (SQ 31,906) and (b) enzymatic ring hydroxylation to SQ 31,945. The 3α-hydroxyisomeric metabolite (SQ 31,906) has 1/10 to 1/40 the HMG-CoA reductase inhibitory activity of the parent compound. Pravastatin undergoes extensive first-pass extraction in the liver (extraction ratio 0.66).
Excretion
Approximately 20% of a radiolabeled oral dose is excreted in urine and 70% in the feces. After intravenous administration of radiolabeled pravastatin to normal volunteers, approximately 47% of total body clearance was via renal excretion and 53% by non-renal routes (i.e., biliary excretion and biotransformation).
Following single dose oral administration of 14C-pravastatin, the radioactive elimination t½ for pravastatin is 1.8 hours in humans and the elimination half-life (t½) for total radioactivity (pravastatin plus metabolites) is 77 hours.
Specific Populations
Renal Impairment
A single 20 mg oral dose of pravastatin was administered to 24 patients with varying degrees of renal impairment (as determined by creatinine clearance). No effect was observed on the pharmacokinetics of pravastatin or its 3α-hydroxy isomeric metabolite (SQ 31,906). Compared to healthy subjects with normal renal function, patients with severe renal impairment had 69% and 37% higher mean AUC and Cmax values, respectively, and a 0.61 hour shorter t½ for the inactive enzymatic ring hydroxylation metabolite (SQ 31,945) [see Use in Specific Populations (8.6)].
Hepatic Impairment
In a study comparing the kinetics of pravastatin in patients with biopsy confirmed cirrhosis (N=7) and normal subjects (N=7), the mean AUC varied 18-fold in cirrhotic patients and 5-fold in healthy subjects. Similarly, the peak pravastatin values varied 47-fold for cirrhotic patients compared to 6-fold for healthy subjects [see Use in Specific Populations (8.6)].
Geriatric
In a single oral dose study using pravastatin 20 mg, the mean AUC for pravastatin was approximately 27% greater and the mean cumulative urinary excretion (CUE) approximately 19% lower in elderly men (65 to 75 years old) compared with younger men (19 to 31 years old). In a similar study conducted in women, the mean AUC for pravastatin was approximately 46% higher and the mean CUE approximately 18% lower in elderly women (65 to 78 years old) compared with younger women (18 to 38 years old). In both studies, Cmax, Tmax, and t½ values were similar in older and younger subjects [seeUse in Specific Populations (8.5)].
Pediatric
After 2 weeks of once-daily 20 mg oral pravastatin administration, the geometric means of AUC were 80.7 (CV 44%) and 44.8 (CV 89%) ng*hr/mL for pediatric patients 8 to 11 years (N=14) and 12 to 16 years (N=10), respectively. The corresponding values for Cmax were 42.4 (CV 54%) and 18.6 ng/mL (CV 100%) for pediatric patients 8 to 11 years and 12 to 16 years, respectively. No conclusion can be made based on these findings due to the small number of samples and large variability [seeUse in Specific Populations (8.4)].
Drug-Drug Interactions
Table 5: Effect of Coadministered Drugs on the Pharmacokinetics of Pravastatin Pravastatin Coadministered Drug and Dosing Regimen Dose (mg) Change in AUC Change in Cmax BID = twice daily; OD = once daily; QID = four times daily Cyclosporine 5 mg/kg single dose
40 mg single dose
↑282%
↑327%
Clarithromycin 500 mg BID for 9 days
40 mg OD for 8 days
↑110%
↑128%
Boceprevir 800 mg TID for 6 days
40 mg single dose
↑63%
↑49%
Darunavir 600 mg BID/Ritonavir 100 mg BID for 7 days
40 mg single dose
↑81%
↑63%
Colestipol 10 g single dose
20 mg single dose
↓47%
↓53%
Cholestyramine 4 g single dose
Administered simultaneously
Administered 1 hour prior to cholestyramine
Administered 4 hours after cholestyramine20 mg single dose
↓40%
↑12%
↓12%
↓39%
↑30%
↓6.8%Cholestyramine 24 g OD for 4 weeks
20 mg BID for 8 weeks
5 mg BID for 8 weeks
10 mg BID for 8 weeks↓51%
↓38%
↓18%↑4.9%
↑23%
↓33%Fluconazole
200 mg IV for 6 days
200 mg PO for 6 days
20 mg PO+10 mg IV
20 mg PO+10 mg IV
↓34%
↓16%
↓33%
↓16%Kaletra 400 mg/100 mg BID for 14 days
20 mg OD for 4 days
↑33%
↑26%
Verapamil IR 120 mg for 1 day and
Verapamil ER 480 mg for 3 days40 mg single dose
↑31%
↑42%
Cimetidine 300 mg QID for 3 days
20 mg single dose
↑30%
↑9.8%
Antacids 15 mL QID for 3 days
20 mg single dose
↓28%
↓24%
Digoxin 0.2 mg OD for 9 days
20 mg OD for 9 days
↑23%
↑26%
Probucol 500 mg single dose
20 mg single dose
↑14%
↑24%
Warfarin 5 mg OD for 6 days
20 mg BID for 6 days
↓13%
↑6.7%
Itraconazole 200 mg OD for 30 days
40 mg OD for 30 days
↑11% (compared to Day 1)
↑17% (compared to Day 1)
Gemfibrozil 600 mg single dose
20 mg single dose
↓7%
↓20%
Aspirin 324 mg single dose
20 mg single dose
↑4.7%
↑8.9%
Niacin 1 g single dose
20 mg single dose
↓3.6%
↓8.2%
Diltiazem
20 mg single dose
↑2.7%
↑30%
Grapefruit juice
40 mg single dose
↓1.8%
↑3.7%
Table 6: Effect of Pravastatin on the Pharmacokinetics of Coadministered Drugs Pravastatin Dosing Regimen Name and Dose Change in AUC Change in Cmax BID = twice daily; OD = once daily 20 mg BID for 6 days
Warfarin 5 mg OD for 6 days
Change in mean prothrombin time↑17%
↑0.4 sec↑15%
20 mg OD for 9 days
Digoxin 0.2 mg OD for 9 days
↑4.6%
↑5.3%
20 mg BID for 4 weeks
10 mg BID for 4 weeks
5 mg BID for 4 weeksAntipyrine 1.2 g single dose
↑3%
↑1.6%
↑ Less than 1%Not Reported
20 mg OD for 4 days
Kaletra 400 mg/100 mg BID for 14 days
No change
No change
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year study in rats fed pravastatin at doses of 10, 30, or 100 mg/kg body weight, there was an increased incidence of hepatocellular carcinomas in males at the highest dose (p<0.01). These effects in rats were observed at approximately 12 times the human dose (HD) of 80 mg based on body surface area (mg/m2) and at approximately 4 times the HD, based on AUC.
In a 2-year study in mice fed pravastatin at doses of 250 and 500 mg/kg/day, there was an increased incidence of hepatocellular carcinomas in males and females at both 250 and 500 mg/kg/day (p<0.0001). At these doses, lung adenomas in females were increased (p=0.013). These effects in mice were observed at approximately 15 times (250 mg/kg/day) and 23 times (500 mg/kg/day) the HD of 80 mg, based on AUC. In another 2-year study in mice with doses up to 100 mg/kg/day (producing drug exposures approximately 2 times the HD of 80 mg, based on AUC), there were no drug-induced tumors.
No evidence of mutagenicity was observed in vitro, with or without rat-liver metabolic activation, in the following studies: microbial mutagen tests, using mutant strains of Salmonella typhimurium or Escherichia coli; a forward mutation assay in L5178Y TK +/− mouse lymphoma cells; a chromosomal aberration test in hamster cells; and a gene conversion assay using Saccharomyces cerevisiae. In addition, there was no evidence of mutagenicity in either a dominant lethal test in mice or a micronucleus test in mice.
In a fertility study in adult rats with daily doses up to 500 mg/kg, pravastatin did not produce any adverse effects on fertility or general reproductive performance. No adverse effects were seen in juvenile rats dosed with 5 mg/kg/day pravastatin (5 times plasma exposure at the maximum recommended human dose [MRHD] of 80 mg based on AUC) in a study of pravastatin administered from postnatal days (PND) 4 through 80 at 5, 15 and 45 mg/kg/day. A PND 4 rat is generally comparable to a 3rd trimester human fetus with regards to neurologic development/myelination. At ≥ 15 mg/kg/day (≥ 20 times the MRHD), decreased body-weight gain was observed during the pre-weaning period and slight thinning of the corpus callosum was observed at the end of the drug-free recovery period (PND 132). Thinning of the corpus callosum was not associated with any inflammatory or degenerative changes in the brain. Impacts on neurobehavioral and learning endpoints were detected only at very high exposures (43 times the MRHD). No thinning of the corpus callosum was observed in rats dosed with pravastatin for 3 months beginning on PND 35 at ≥ 250 mg/kg/day. PND 35 in a rat is approximately equivalent to an 8 to 12-year-old human child.
-
14 CLINICAL STUDIES
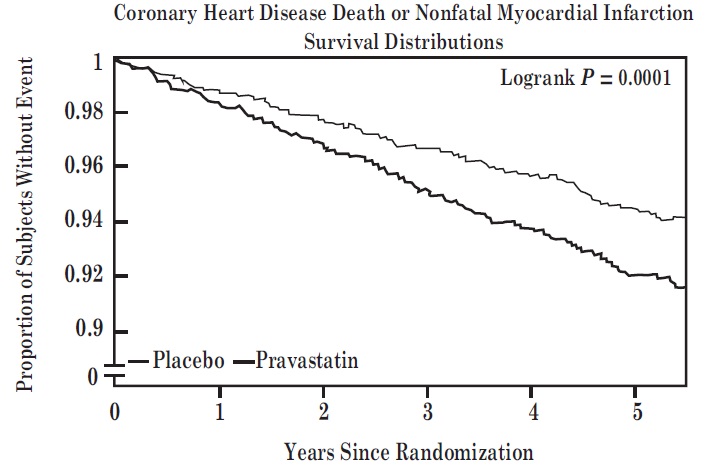
Prevention of Coronary Heart Disease
In the Pravastatin Primary Prevention Study (WOS), the effect of pravastatin sodium on fatal and nonfatal CHD was assessed in 6595 male patients 45 to 64 years of age, without a previous MI, and with LDL-C levels between 156 to 254 mg/dL. In this randomized, double-blind, placebo-controlled study, patients were treated with standard care, including dietary advice, and either pravastatin sodium 40 mg daily (N=3302) or placebo (N=3293) and followed for a median duration of 4.8 years. Median (25th, 75th percentile) percent changes from baseline after 6 months of pravastatin treatment in Total-C, LDL-C, TG, and HDL-C were −20.3 (−26.9, −11.7), −27.7 (−36, −16.9), −9.1 (−27.6, 12.5), and 6.7 (−2.1, 15.6), respectively.
Pravastatin sodium significantly reduced the rate of first coronary events (either CHD death or nonfatal MI) by 31% (248 events in the placebo group [CHD death=44, nonfatal MI=204] versus 174 events in the pravastatin sodium group [CHD death=31, nonfatal MI=143], p=0.0001 [see figure below]). The risk reduction with pravastatin sodium was similar across the age range studied and throughout the range of baseline LDL cholesterol levels.
Pravastatin sodium also decreased the risk for undergoing myocardial revascularization procedures (coronary artery bypass graft [CABG] surgery or percutaneous transluminal coronary angioplasty [PTCA]) by 37% (80 vs 51 patients). Cardiovascular deaths were decreased by 32% (73 vs 50) and there was no increase in death from non-cardiovascular causes.
Secondary Prevention of Cardiovascular Events
In the LIPID study, the effect of pravastatin sodium, 40 mg daily, was assessed in 9014 patients (7498 men; 1516 women; 3514 patients ≥65 years; 782 patients with diabetes) who had experienced either an MI (5754 patients) or had been hospitalized for unstable angina pectoris (3260 patients) in the preceding 3 to 36 months. Patients in this multicenter, double-blind, placebo-controlled study participated for an average of 5.6 years (median of 5.9 years) and at randomization had Total-C between 114 and 563 mg/dL (mean 219 mg/dL), LDL-C between 46 and 274 mg/dL (mean 150 mg/dL), TG between 35 and 2710 mg/dL (mean 160 mg/dL), and HDL-C between 1 and 103 mg/dL (mean 37 mg/dL). At baseline, 82% of patients were receiving aspirin and 76% were receiving antihypertensive medication. Treatment with pravastatin sodium significantly reduced the risk for total mortality by reducing coronary death (see Table 7). The risk reduction due to treatment with pravastatin sodium on CHD mortality was consistent regardless of age. Pravastatin sodium significantly reduced the risk for total mortality (by reducing CHD death) and CHD events (CHD mortality or nonfatal MI) in patients who qualified with a history of either MI or hospitalization for unstable angina pectoris.
Table 7: LIPID - Primary and Secondary Endpoints Number (%) of Subjects Event Pravastatin Sodium
40 mg
(N=4512)Placebo
(N=4502)Risk
Reductionp-value Primary Endpoint
CHD mortality
287 (6.4)
373 (8.3)
24%
0.0004
Secondary Endpoints
Total mortality
498 (11)
633 (14.1)
23%
<0.0001
CHD mortality or nonfatal MI
557 (12.3)
715 (15.9)
24%
<0.0001
Myocardial revascularization
procedures (CABG or PTCA)584 (12.9)
706 (15.7)
20%
<0.0001
Stroke
All-cause
169 (3.7)
204 (4.5)
19%
0.0477
Non-hemorrhagic
154 (3.4)
196 (4.4)
23%
0.0154
Cardiovascular mortality
331 (7.3)
433 (9.6)
25%
<0.0001
In the CARE study, the effect of pravastatin sodium, 40 mg daily, on CHD death and nonfatal MI was assessed in 4159 patients (3583 men and 576 women) who had experienced a MI in the preceding 3 to 20 months and who had normal (below the 75th percentile of the general population) plasma total cholesterol levels. Patients in this double-blind, placebo-controlled study participated for an average of 4.9 years and had a mean baseline Total-C of 209 mg/dL. LDL-C levels in this patient population ranged from 101 to 180 mg/dL (mean 139 mg/dL). At baseline, 84% of patients were receiving aspirin and 82% were taking antihypertensive medications. Median (25th, 75th percentile) percent changes from baseline after 6 months of pravastatin treatment in Total-C, LDL-C, TG, and HDL-C were −22 (−28.4, −14.9), −32.4 (−39.9, −23.7), −11 (−26.5, 8.6), and 5.1 (−2.9, 12.7), respectively. Treatment with pravastatin sodium significantly reduced the rate of first recurrent coronary events (either CHD death or nonfatal MI), the risk of undergoing revascularization procedures (PTCA, CABG), and the risk for stroke or TIA (see Table 8).
Table 8: CARE - Primary and Secondary Endpoints Number (%) of Subjects Event Pravastatin Sodium
40 mg
(N=2081)Placebo
(N=2078)Risk
Reductionp-value a The risk reduction due to treatment with pravastatin sodium was consistent in both sexes. Primary Endpoint
CHD mortality or nonfatal MIa
212 (10.2)
274 (13.2)
24%
0.003
Secondary Endpoints
Myocardial revascularization procedures (CABG or PTCA)
294 (14.1)
391 (18.8)
27%
<0.001
Stroke or TIA
93 (4.5)
124 (6)
26%
0.029
Primary Hyperlipidemia
In multicenter, double-blind, placebo-controlled studies of patients with primary hyperlipidemia, treatment with pravastatin sodium in daily doses ranging from 10 to 40 mg consistently and significantly decreased Total-C, LDL-C, and TG (see Table 9).
In a pooled analysis of 2 multicenter, double-blind, placebo-controlled studies of patients with primary hyperlipidemia, treatment with pravastatin sodium at a daily dose of 80 mg (N=277) significantly decreased Total-C, LDL-C, and TG. The 25th and 75th percentile changes from baseline in LDL-C for pravastatin sodium 80 mg were −43% and −30%. The efficacy results of the individual studies were consistent with the pooled data (see Table 9).
Table 9: Primary Hyperlipidemia Trials: Dose Response of Pravastatin Sodium Once Daily Administration a A multicenter, double-blind, placebo-controlled study.
b Pooled analysis of 2 multicenter, double-blind, placebo-controlled studies.Dose
Total-C
LDL-C
HDL-C
TG
Mean Percent Changes From Baseline After 8 Weeksa
Placebo (N=36)
−3%
−4%
+1%
−4%
10 mg (N=18)
−16%
−22%
+7%
−15%
20 mg (N=19)
−24%
−32%
+2%
−11%
40 mg (N=18)
−25%
−34%
+12%
−24%
Mean Percent Changes From Baseline After 6 Weeksb
Placebo (N=162)
0%
−1%
−1%
+1%
80 mg (N=277)
−27%
−37%
+3%
−19%
Hypertriglyceridemia
The response to pravastatin sodium in patients with hypertriglyceridemia (baseline TG >200 mg/dL and LDL-C <160 mg/dL) was evaluated in a subset of 429 patients from the CARE study. For pravastatin-treated subjects, the median (min, max) baseline TG level was 246 (200.5, 349.5) mg/dL (see Table 10).
Table 10: Patients with Hypertriglyceridemia Median (25th, 75th percentile) % Change from Baseline Pravastatin 40 mg (N=429)
Placebo (N=430)
TG
−21.1 (−34.8, 1.3)
−6.3 (−23.1, 18.3)
Total-C
−22.1 (−27.1, −14.8)
0.2 (−6.9, 6.8)
LDL-C
−31.7 (−39.6, −21.5)
0.7 (−9, 10)
HDL-C
7.4 (−1.2, 17.7)
2.8 (−5.7, 11.7)
Non-HDL-C
−27.2 (−34, −18.5)
−0.8 (−8.2, 7)
Dysbetalipoproteinemia
The response to pravastatin sodium in two double-blind crossover studies of 46 patients with genotype E2/E2 and dysbetalipoproteinemia is shown in Table 11.
Table 11: Patients with Dysbetalipoproteinemia Median (min, max) % Change from Baseline Median (min, max)
at Baseline (mg/dL)Median % Change (min, max)
Pravastatin 40 mg (N=20)Study 1
Total-C
386.5 (245, 672)
−32.7 (−58.5, 4.6)
TG
443 (275, 1299)
−23.7 (−68.5, 44.7)
VLDL-Ca
206.5 (110, 379)
−43.8 (−73.1, −14.3)
LDL-Ca
117.5 (80, 170)
−40.8 (−63.7, 4.6)
HDL-C
30 (18, 88)
6.4 (−45, 105.6)
Non-HDL-C
344.5 (215, 646)
−36.7 (−66.3, 5.8)
a N=14
Median (min, max)
at Baseline (mg/dL)Median % Change (min, max)
Pravastatin 40 mg (N=26)Study 2
Total-C
340.3 (230.1, 448.6)
−31.4 (−54.5, −13)
TG
343.2 (212.6, 845.9)
−11.9 (−56.5, 44.8)
VLDL-C
145 (71.5, 309.4)
−35.7 (−74.7, 19.1)
LDL-C
128.6 (63.8, 177.9)
−30.3 (−52.2, 13.5)
HDL-C
38.7 (27.1, 58)
5 (−17.7, 66.7)
Non-HDL-C
295.8 (195.3, 421.5)
−35.5 (−81, −13.5)
HeFH in Pediatric Patients Aged 8 Years and Above
A double-blind, placebo-controlled study in 214 pediatric patients (100 males and 114 females) with heterozygous familial hypercholesterolemia (HeFH), aged 8 to 18 years was conducted for 2 years. The pediatric patients aged 8 to 13 years were randomized to placebo (N=63) or 20 mg of pravastatin sodium daily (N=65) and the pediatric patients aged 14 to 18 years were randomized to placebo (N=45) or 40 mg of pravastatin daily (N=41). Inclusion in the study required an LDL-C level >95th percentile for age and sex and one parent with either a clinical or molecular diagnosis of familial hypercholesterolemia. The mean baseline LDL-C value was 239 mg/dL and 237 mg/dL in the pravastatin (range: 151 to 405 mg/dL) and placebo (range: 154 to 375 mg/dL) groups, respectively.
Pravastatin sodium significantly decreased plasma levels of LDL-C, Total-C, and ApoB in both pediatric age groups (see Table 12). The effect of pravastatin sodium treatment in the 2 age groups was similar.
Table 12: Lipid-Lowering Effects of Pravastatin Sodium in Pediatric Patients with Heterozygous Familial Hypercholesterolemia: Least-Squares Mean % Change from Baseline at Month 24 (Last Observation Carried Forward: Intent-to-Treat)a Pravastatin
20 mg
(Aged 8 to 13
years)
N=65Pravastatin
40 mg
(Aged 14 to 18
years)
N=41Combined
Pravastatin
(Aged 8 to 18
years)
N=106Combined
Placebo
(Aged 8 to 18
years)
N=10895% CI of the Difference Between Combined Pravastatin and Placebo a The above least-squares mean values were calculated based on log-transformed lipid values.
b Significant at p≤0.0001 when compared with placebo.LDL-C
−26.04b
−21.07b
−24.07b
−1.52
(−26.74, −18.86)
TC
−20.75b
−13.08b
−17.72b
−0.65
(−20.40, −13.83)
HDL-C
1.04
13.71
5.97
3.13
(−1.71, 7.43)
TG
−9.58
−0.30
−5.88
−3.27
(−13.95, 10.01)
ApoB
(N)−23.16b
(61)−18.08b
(39)−21.11b
(100)−0.97
(106)(−24.29, −16.18)
The mean achieved LDL-C was 186 mg/dL (range: 67 to 363 mg/dL) in the pravastatin sodium group k to 236 mg/dL (range: 105 to 438 mg/dL) in the placebo group.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Pravastatin Sodium Tablets, USP are supplied as:
Strength
How Supplied
NDC
Tablet Description
10 mg
of pravastatin sodium, USP
bottles of 90
68462-195-90
yellow colored, circular shaped, flat faced tablets with “G5” debossed on one side and “10” debossed on the other side. Bottles contain a desiccant canister.
bottles of 500
68462-195-05
20 mg
of pravastatin sodium, USP
bottles of 90
68462-196-90
yellow, rounded-rectangular, biconvex tablets with “G5” debossed on one side and “20” debossed on the other side. Bottles contain a desiccant canister.
bottles of 500
68462-196-05
40 mg
of pravastatin sodium, USP
bottles of 90
68462-197-90
green, rounded-rectangular, biconvex tablets with “G5” debossed on one side and “40” debossed on the other side. Bottles contain a desiccant canister.
bottles of 500
68462-197-05
80 mg
of pravastatin sodium, USP
bottles of 90
68462-198-90
yellow, oval, biconvex tablets with “G5” debossed on one side and “80” debossed on the other side. Bottles contain a desiccant canister.
bottles of 500
68462-198-05
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [See USP Controlled Room Temperature]. Keep tightly closed (protect from moisture). Protect from light.
-
17 PATIENT COUNSELING INFORMATION
Myopathy and Rhabdomyolysis
Advise patients that pravastatin sodium tablets may cause myopathy and rhabdomyolysis. Inform patients that the risk is increased when taking certain types of medication and they should discuss all medication, both prescription and over the counter, with their healthcare provider. Instruct patients to promptly report any unexplained muscle pain, tenderness or weakness particularly if accompanied by malaise or fever [see Warnings and Precautions (5.1), Drug Interactions (7.1)].
Hepatic Dysfunction
Inform patients that pravastatin sodium tablets may cause liver enzyme elevations and possibly liver failure. Advise patients to promptly report fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice [see Warnings and Precautions (5.3)].
Increases in HbA1c and Fasting Serum Glucose Levels Inform patients that increases in HbA1c and fasting serum glucose levels may occur with pravastatin sodium tablets. Encourage patients to optimize lifestyle measures, including regular exercise, maintaining a healthy body weight, and making healthy food choices [see Warnings and Precautions (5.4)].
Pregnancy
Advise pregnant patients and patients who may become pregnant of the potential risk to a fetus. Advise patients to inform their healthcare provider of a known or suspected pregnancy to discuss if pravastatin sodium tablets should be discontinued [see Use in Specific Populations (8.1)].
Lactation
Advise patients that breastfeeding is not recommended during treatment with pravastatin sodium tablets [see Use in Specific Populations (8.2)].
Manufactured for:
Glenmark Pharmaceuticals Inc., USA
Mahwah, NJ 07430Questions? 1 (888) 721-7115
www.glenmarkpharma-us.com
July 2022
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
PRAVASTATIN SODIUM
pravastatin sodium tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68462-195 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAVASTATIN SODIUM (UNII: 3M8608UQ61) (PRAVASTATIN - UNII:KXO2KT9N0G) PRAVASTATIN SODIUM 10 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MEGLUMINE (UNII: 6HG8UB2MUY) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) Product Characteristics Color YELLOW Score no score Shape ROUND Size 6mm Flavor Imprint Code G5;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68462-195-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 05/11/2007 2 NDC:68462-195-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 05/11/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077987 05/11/2007 PRAVASTATIN SODIUM
pravastatin sodium tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68462-196 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAVASTATIN SODIUM (UNII: 3M8608UQ61) (PRAVASTATIN - UNII:KXO2KT9N0G) PRAVASTATIN SODIUM 20 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MEGLUMINE (UNII: 6HG8UB2MUY) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) Product Characteristics Color YELLOW Score no score Shape RECTANGLE (Rounded rectangular) Size 8mm Flavor Imprint Code G5;20 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68462-196-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 05/11/2007 2 NDC:68462-196-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 05/11/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077987 05/11/2007 PRAVASTATIN SODIUM
pravastatin sodium tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68462-197 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAVASTATIN SODIUM (UNII: 3M8608UQ61) (PRAVASTATIN - UNII:KXO2KT9N0G) PRAVASTATIN SODIUM 40 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MEGLUMINE (UNII: 6HG8UB2MUY) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) Product Characteristics Color GREEN Score no score Shape RECTANGLE (Rounded rectangular) Size 10mm Flavor Imprint Code G5;40 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68462-197-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 05/11/2007 2 NDC:68462-197-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 05/11/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077987 05/11/2007 PRAVASTATIN SODIUM
pravastatin sodium tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68462-198 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAVASTATIN SODIUM (UNII: 3M8608UQ61) (PRAVASTATIN - UNII:KXO2KT9N0G) PRAVASTATIN SODIUM 80 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MEGLUMINE (UNII: 6HG8UB2MUY) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) Product Characteristics Color YELLOW Score no score Shape OVAL Size 18mm Flavor Imprint Code G5;80 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68462-198-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 12/28/2007 2 NDC:68462-198-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 12/28/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077987 12/28/2007 Labeler - Glenmark Pharmaceuticals Inc., USA (130597813) Establishment Name Address ID/FEI Business Operations Glenmark Pharmaceuticals Limited 677318665 ANALYSIS(68462-195, 68462-196, 68462-197, 68462-198) , MANUFACTURE(68462-195, 68462-196, 68462-197, 68462-198) Establishment Name Address ID/FEI Business Operations Glenmark Pharmaceuticals Limited 862603186 ANALYSIS(68462-195, 68462-196, 68462-197, 68462-198) , MANUFACTURE(68462-195, 68462-196, 68462-197, 68462-198) Establishment Name Address ID/FEI Business Operations Glenmark Pharmaceuticals Inc., USA 079922908 MANUFACTURE(68462-195, 68462-196, 68462-197, 68462-198) , ANALYSIS(68462-195, 68462-198, 68462-198, 68462-198)