Label: LETROZOLE tablet, film coated


-
NDC Code(s):
16729-034-01,
16729-034-10,
16729-034-15,
16729-034-16, view more16729-034-17, 16729-034-43
- Packager: Accord Healthcare, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated January 16, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to useletrozole tabletssafely and effectively. See full prescribing information forletrozole tablets.
LETROZOLE tablets, for oral use
Initial U.S. Approval:1997INDICATIONS AND USAGE
Letrozole tablet is an aromatase inhibitor indicated for:
- Adjuvant treatment of postmenopausal women with hormone receptor positive early breast cancer ( 1.1)
- Extended adjuvant treatment of postmenopausal women with early breast cancer who have received prior standard adjuvant tamoxifen therapy ( 1.2)
- First and second-line treatment of postmenopausal women with hormone receptor positive or unknown advanced breast cancer ( 1.3)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
2.5 mg tablets ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Decreases in bone mineral density may occur. Consider bone mineral density monitoring ( 5.1)
- Increases in total cholesterol may occur. Consider cholesterol monitoring. ( 5.2)
- Fatigue, dizziness and somnolence may occur. Exercise caution when operating machinery ( 5.4)
- Embryo-Fetal toxicity: Can cause fetal harm when administered to pregnant women. Obtain a pregnancy test in females of reproductive potential. Advise females of reproductive potential to use effective contraception ( 5.6, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (greater than 20%) were hot flashes, arthralgia; flushing, asthenia, edema, arthralgia, headache, dizziness, hypercholesterolemia, sweating increased, bone pain and musculoskeletal ( 6).
To report SUSPECTED ADVERSE REACTIONS, contact Accord Healthcare Inc. at 1-866-941-7875 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. ( 8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 10/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Adjuvant Treatment of Early Breast Cancer
1.2 Extended Adjuvant Treatment of Early Breast Cancer
1.3 First and Second-Line Treatment of Advanced Breast Cancer
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Use in Adjuvant Treatment of Early Breast Cancer
2.3 Use in Extended Adjuvant Treatment of Early Breast Cancer
2.4 Use in First and Second-Line Treatment of Advanced Breast Cancer
2.5 Use in Hepatic Impairment
2.6 Use in Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Bone Effects
5.2 Cholesterol
5.3 Hepatic Impairment
5.4 Fatigue and Dizziness
5.5 Laboratory Test Abnormalities
5.6 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 >Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Updated Adjuvant Treatment of Early Breast Cancer
14.2 Extended Adjuvant Treatment of Early Breast Cancer, Median Treatment Duration of 24 Months
14.3 Updated Analyses of Extended Adjuvant Treatment of Early Breast Cancer, Median Treatment Duration of 60 Months
14.4 First-Line Treatment of Advanced Breast Cancer
14.5 Second-Line Treatment of Advanced Breast Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Adjuvant Treatment of Early Breast Cancer
Letrozole tablets are indicated for the adjuvant treatment of postmenopausal women with hormone receptor positive early breast cancer.
1.2 Extended Adjuvant Treatment of Early Breast Cancer
Letrozole tablets are indicated for the extended adjuvant treatment of early breast cancer in postmenopausal women, who have received 5 years of adjuvant tamoxifen therapy. The effectiveness of letrozole tablets in extended adjuvant treatment of early breast cancer is based on an analysis of disease-free survival in patients treated with letrozole tablets for a median of 60 months [see Clinical Studies ( 14.2, 14.3)] .
1.3 First and Second-Line Treatment of Advanced Breast Cancer
Letrozole tablets are indicated for first-line treatment of postmenopausal women with hormone receptor positive or unknown, locally advanced or metastatic breast cancer. Letrozole tablets are also indicated for the treatment of advanced breast cancer in postmenopausal women with disease progression following antiestrogen therapy [see Clinical Studies ( 14.4, 14.5)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
The recommended dose of letrozole tablet is one 2.5 mg tablet administered once a day, without regard to meals.
2.2 Use in Adjuvant Treatment of Early Breast Cancer
In the adjuvant setting, the optimal duration of treatment with letrozole is unknown. In both the adjuvant study and the post approval adjuvant study, median treatment duration was 5 years. Treatment should be discontinued at relapse [see Clinical Studies ( 14.1)].
2.3 Use in Extended Adjuvant Treatment of Early Breast Cancer
In the extended adjuvant setting, the optimal treatment duration with letrozole tablet is not known. The planned duration of treatment in the study was 5 years. In the final updated analysis, conducted at a median follow-up of 62 months, the median treatment duration for letrozole tablets was 60 months. Seventy-one (71%) percent of patients were treated for at least 3 years and 58% of patients completed at least 4.5 years of extended adjuvant treatment. The treatment should be discontinued at tumor relapse [see Clinical Studies ( 14.2)].
2.4 Use in First and Second-Line Treatment of Advanced Breast Cancer
In patients with advanced disease, treatment with letrozole tablets should continue until tumor progression is evident [see Clinical Studies ( 14.4, 14.5)] .
2.5 Use in Hepatic Impairment
No dosage adjustment is recommended for patients with mild to moderate hepatic impairment, although letrozole tablets blood concentrations were modestly increased in subjects with moderate hepatic impairment due to cirrhosis. The dose of letrozole tablets in patients with cirrhosis and severe hepatic dysfunction should be reduced by 50% [see Warnings and Precautions ( 5.3)]. The recommended dose of letrozole tablets for such patients is 2.5 mg administered every other day. The effect of hepatic impairment on letrozole tablets exposure in noncirrhotic cancer patients with elevated bilirubin levels has not been determined.
2.6 Use in Renal Impairment
No dosage adjustment is required for patients with renal impairment if creatinine clearance is greater than or equal to 10 mL/min [see Clinical Pharmacology ( 12.3)] .
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
- Pregnancy: Letrozole can cause fetal harm [see Use in Specific Populations ( 8.1)] . Pregnancy: Letrozole can cause fetal harm [see Use in Specific Populations ( 8.1)] .
- Known hypersensitivity to the active substance, or to any of the excipients [see Adverse Reactions ( 6)] . Known hypersensitivity to the active substance, or to any of the excipients [see Adverse Reactions ( 6)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Bone Effects
Use of letrozole may cause decreases in bone mineral density (BMD). Consideration should be given to monitoring BMD. Results of a safety study to evaluate safety in the adjuvant setting comparing the effect on lumbar spine (L2-L4) BMD of adjuvant treatment with letrozole to that with tamoxifen showed at 24 months a median decrease in lumbar spine BMD of 4.1% in the letrozole arm compared to a median increase of 0.3% in the tamoxifen arm (difference = 4.4%) ( P<0.0001) [see Adverse Reactions ( 6)] . Updated results from the BMD substudy (MA-17B) in the extended adjuvant setting demonstrated that at 2 years patients receiving letrozole had a median decrease from baseline of 3.8% in hip BMD compared to a median decrease of 2.0% in the placebo group. The changes from baseline in lumbar spine BMD in letrozole and placebo treated groups were not significantly different [see Adverse Reactions ( 6)].
In the adjuvant trial (BIG 1-98) the incidence of bone fractures at any time after randomization was 14.7% for letrozole and 11.4% for tamoxifen at a median follow-up of 96 months. The incidence of osteoporosis was 5.1% for letrozole and 2.7% for tamoxifen [see Adverse Reactions ( 6)] . In the extended adjuvant trial (MA-17), the incidence of bone fractures at any time after randomization was 13.3% for letrozole and 7.8% for placebo at a median follow-up of 62 months. The incidence of new osteoporosis was 14.5% for letrozole and 7.8% for placebo [see Adverse Reactions ( 6)].
5.2 Cholesterol
Consideration should be given to monitoring serum cholesterol. In the adjuvant trial (BIG 1-98), hypercholesterolemia was reported in 52.3% of letrozole patients and 28.6% of tamoxifen patients. Grade 3 to 4 hypercholesterolemia was reported in 0.4% of letrozole patients and 0.1% of tamoxifen patients. Also in the adjuvant setting, an increase of greater than or equal to 1.5 x upper limit of normal (ULN) in total cholesterol (generally nonfasting) was observed in patients on monotherapy who had baseline total serum cholesterol within the normal range (i.e., less than=1.5 x ULN) in 155/1843 (8.4%) patients on letrozole vs 71/1840 (3.9%) patients on tamoxifen. Lipid lowering medications were required for 29% of patients on letrozole and 20% on tamoxifen [see Adverse Reactions ( 6)].
5.3 Hepatic Impairment
Subjects with cirrhosis and severe hepatic impairment who were dosed with 2.5 mg of letrozole experienced approximately twice the exposure to letrozole as healthy volunteers with normal liver function [see Clinical Pharmacology ( 12.3)] . Therefore, a dose reduction is recommended for this patient population. The effect of hepatic impairment on letrozole exposure in cancer patients with elevated bilirubin levels has not been determined [see Dosage and Administration ( 2.5)].
5.4 Fatigue and Dizziness
Because fatigue, dizziness, and somnolence have been reported with the use of letrozole, caution is advised when driving or using machinery until it is known how the patient reacts to letrozole use.
5.5 Laboratory Test Abnormalities
No dose-related effect of letrozole on any hematologic or clinical chemistry parameter was evident. Moderate decreases in lymphocyte counts, of uncertain clinical significance, were observed in some patients receiving letrozole 2.5 mg. This depression was transient in about half of those affected. Two patients on letrozole developed thrombocytopenia; relationship to the study drug was unclear. Patient withdrawal due to laboratory abnormalities, whether related to study treatment or not was infrequent.
5.6 Embryo-Fetal Toxicity
Based on post-marketing reports, findings from animal studies and the mechanism of action, letrozole can cause fetal harm and is contraindicated for use in pregnant women. In post-marketing reports, use of letrozole during pregnancy resulted in cases of spontaneous abortions and congenital birth defects. Letrozole caused embryo-fetal toxicities in rats and rabbits at maternal exposures that were below the maximum recommended human dose (MHRD) on a mg/m 2basis. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during therapy with letrozole and for at least 3 weeks after the last dose [see Adverse Reactions ( 6.2), Use in Specific Populations ( 8.1, 8.3) and Clinical Pharmacology ( 12.1)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling.
- Bone effects [see Warnings and Precautions ( 5.1)]
- Increases in cholesterol [see Warnings and Precautions ( 5.2)]
- Fatigue and Dizziness [see Warnings and Precautions ( 5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adjuvant Treatment of Early Breast Cancer
In study, BIG 1-98, the median treatment duration of adjuvant treatment was 60 months and the median duration of follow-up for safety was 96 months for patients receiving letrozole and tamoxifen.
Certain adverse reactions were prospectively specified for analysis (see Table 1), based on the known pharmacologic properties and side effect profiles of the two drugs.
Adverse reactions were analyzed irrespective of whether a symptom was present or absent at baseline. Most adverse reactions reported (approximately 75% of patients who reported AEs) were Grade 1 or Grade 2 applying the Common Toxicity Criteria (CTC) Version 2.0/ Common Terminology Criteria for Adverse Events (CTCAE), Version 3.0. Table 1 describes adverse reactions (Grades 1-4 and Grades 3-4) irrespective of relationship to study treatment in the adjuvant trial for the monotherapy arms analysis (safety population).
Table 1: Patients with Adverse Reactions (CTC Grades 1-4) in the Adjuvant Study – Monotherapy Arms Analysis (Median Follow-up 96 Months; Median Treatment 60 Months) Grades 1-4 Grades 3-4 Adverse Reactions Letrozole Tablets
N=2448
n (%)Tamoxifen
N=2447
n (%)Letrozole Tablets
N=2448
n (%)Tamoxifen
N=2447
n (%)Patients with any adverse reaction 2309 (93.4) 2212 (90.4) 636 (26.0) 606 (24.8) Hypercholesterolemia * 1280 (52.3) 700 (28.6) 11 ( 0.4) 6 ( 0.2) Hot flashes * 819 (33.5) 929 (38.0) - - - - Arthralgia/Arthritis * 621 (25.4) 504 (20.6) 84 ( 3.4) 50 ( 2.0) Bone fractures 1 361 (14.7) 280 (11.4) - - - - Night sweats * 356 (14.6) 426 (17.4) - - - - Weight Increase * 317 (12.9) 378 (15.4) 27 ( 1.1) 39 ( 1.6) Nausea * 284 (11.6) 277 (11.3) 6 ( 0.2) 9 ( 0.4) Bone Fractures **2 249 (10.2) 175 ( 7.2) - - - - Fatigue (lethargy, malaise, asthenia) **2 235 ( 9.6) 250 (10.2) 6 ( 0.2) 7 ( 0.3) Myalgia * 221 ( 9.0) 212 ( 8.7) 18 ( 0.7) 14 ( 0.6) Vaginal Bleeding * 129 ( 5.3) 320 (13.1) 1 (<0.1) 8 ( 0.3) Edema * 164 ( 6.7) 160 ( 6.5) 3 ( 0.1) 1 (<0.1) Weight decrease 140 ( 5.7) 129 ( 5.3) 8 ( 0.3) 5 ( 0.2) Osteoporosis ** 126 ( 5.1) 66 ( 2.7) 10 ( 0.4) 5 ( 0.2) Back Pain 125 ( 5.1) 136 ( 5.6) 7 ( 0.3) 11 ( 0.4) Bone pain 123 ( 5.0) 109 ( 4.5) 6 ( 0.2) 4 ( 0.2) Depression 119 ( 4.9) 114 ( 4.7) 16 ( 0.7) 14 ( 0.6) Vaginal irritation * 112 ( 4.6) 77 ( 3.1) 2 (<0.1) 2 (<0.1) Headache * 105 ( 4.3) 94 ( 3.8) 8 ( 0.3) 4 ( 0.2) Pain in extremity 103 ( 4.2) 79 ( 3.2) 6 ( 0.2) 4 ( 0.2) Osteopenia * 87 ( 3.6) 76 ( 3.1) 0 - 3 (0.1) Dizziness/Light-Headedness * 84 ( 3.4) 80 ( 3.3) 1 (<0.1) 6 (0.2) Alopecia 83 ( 3.4) 84 ( 3.4) - - - - Vomiting * 80 ( 3.3) 80 ( 3.3) 3 ( 0.1) 5 (0.2) Cataract * 49 ( 2.0) 54 ( 2.2) 16 ( 0.7) 17 ( 0.7) Constipation * 49 ( 2.0) 71 ( 2.9) 3 ( 0.1) 1 (<0.1) Myocardial infarction 1 42 (1.7) 28 (1.1) - - - - Breast pain * 37 ( 1.5) 43 ( 1.8) 1 (<0.1) - - Anorexia * 20 ( 0.8) 20 ( 0.8) 1 (<0.1) 1 (<0.1) Endometrial proliferation disorders * 14 (0.6) 86 (3.5) 0 - 14 (0.6) Ovarian cyst * 11 (0.4) 18 (0.7) 4 (0.2) 4 (0.2) Endometrial hyperplasia/ Cancer **1 11 (0.4) 72 (2.9) - - - - Endometrial hyperplasia/ Cancer **3 6/1909 ( 0.3) 57/1943 (2.9) - - - - Other endometrial disorders ** 2 (<0.1) 3 ( 0.1) 0 - 0 - Myocardial infarction **2 24 ( 1.0) 12 ( 0.5) - - - - Myocardial ischemia 6 ( 0.2) 9 ( 0.4) - - - - Cerebrovascular accident/TIA **1 74 (3.0) 68 (2.8) - - - - Cerebrovascular accident/TIA **2 51 ( 2.1) 47 ( 1.9) - - - - Angina requiring surgery **1 35 ( 1.4) 33 ( 1.3) - - - - Angina requiring surgery **2 25 ( 1.0) 25 ( 1.0) - - - - Thromboembolic event **1 79 ( 3.2) 113 ( 4.6) - - - - Thromboembolic event **2 51 ( 2.1) 89 ( 3.6) - - - - Cardiac failure 1 39 (1.6) 34 (1.4) - - - - Cardiac failure 2 27 (1.1) 15 (0.6) - - - - Hypertension 1 160 (6.5) 175 (7.2) - - - - Hypertension 2 138 (5.6) 139 (5.7) - - - - Other cardiovascular **1 172 (7.0) 174 (7.1) - - - - Other cardiovascular 2 120 (4.9) 119 (4.9) - - - - Second primary malignancy 1 129 (5.3) 150 (6.1) - - - - Second primary malignancy 2 54 ( 2.2) 79 ( 3.2) - - - - *Target events pre-specified for analysis
**Events pre-printed on CRF
1At median follow-up of 96 months (i.e. any time after randomization) for letrozole tablets (range up to 144 months) and 95 months for tamoxifen (range up to 143 months )
2At median treatment duration of 60 months (i.e. during treatment + 30 days after discontinuation of treatment) for letrozole tablets and tamoxifen (range up to 68 months)
3Excluding women who had undergone hysterectomy before study entry
TIA = Transient ischemic attackNote: Cardiovascular events (including cerebrovascular and thromboembolic events), skeletal and urogenital/endometrial events and second primary malignancies were collected life -long. All of these events were assumed to be of CTC Grade 3 to 5 and were not individually graded
When considering all grades during study treatment, a higher incidence of events was seen for letrozole regarding fractures (10.1% vs. 7.1%), myocardial infarctions (1.0% vs. 0.5%), and arthralgia (25.2% vs. 20.4%) (letrozole vs. tamoxifen respectively). A higher incidence was seen for tamoxifen regarding thromboembolic events (2.1% vs. 3.6%), endometrial hyperplasia/cancer (0.3% vs. 2.9%), and endometrial proliferation disorders (0.3% vs. 1.8%) (letrozole vs. tamoxifen respectively).
At a median follow up of 96 months, a higher incidence of events was seen for letrozole (14.7%) than for tamoxifen (11.4%) regarding fractures. A higher incidence was seen for tamoxifen compared to letrozole regarding thromboembolic events (4.6% vs. 3.2%), and endometrial hyperplasia or cancer (2.9% vs. 0.4%) (tamoxifen vs. letrozole, respectively).
Bone Study: Results of a safety trial in 263 postmenopausal women with resected receptor positive early breast cancer in the adjuvant setting comparing the effect on lumbar spine (L2-L4) BMD of adjuvant treatment with letrozole to that with tamoxifen showed at 24 months a median decrease in lumbar spine BMD of 4.1% in the letrozole arm compared to a median increase of 0.3% in the tamoxifen arm (difference = 4.4%) ( P<0.0001). No patients with a normal BMD at baseline became osteoporotic over the 2 years and only 1 patient with osteopenia at baseline (T score of -1.9) developed osteoporosis during the treatment period (assessment by central review). The results for total hip BMD were similar, although the differences between the two treatments were less pronounced. During the 2 year period, fractures were reported by 4 of 103 patients (4%) in the letrozole arm, and 6 of 97 patients (6%) in the tamoxifen arm.
Lipid Study: In a safety trial in 263 postmenopausal women with resected receptor positive early breast cancer at 24 months comparing the effects on lipid profiles of adjuvant letrozole to tamoxifen, 12% of patients on letrozole had at least one total cholesterol value of a higher CTCAE grade than at baseline compared with 4% of patients on tamoxifen. In another postapproval randomized, multicenter, open label, study of letrozole vs anastrozole in the adjuvant treatment of postmenopausal women with hormone receptor and node positive breast cancer (FACE, NCT00248170), the median duration of treatment was 60 months for both treatment arms. Table 2 describes adverse reactions (Grades 1-4 and Grades 3-4) irrespective of relationship to study treatment in the adjuvant study (safety population).
Table 2: Adverse Reactions (CTC Grades 1-4), Occurring in at least 5% of Patients in Either Treatment Arm, by Preferred Term (Safety set) Adverse Reactions Letrozole
N = 2049
n (%)Anastrozole
N = 2062
n (%)Grade 3/4
n (%)All grades
n (%)Grade 3/4
n (%)All grades
n (%)Patients with at least one AR 628 (30.6) 2049 (100.0) 591 (28.7) 2062 (100.0) Arthralgia 80 (3.9) 987 (48.2) 69 (3.3) 987 (47.9) Hot flush 17 (0.8) 666 (32.5) 9 (0.4) 666 (32.3) Fatigue 8 (0.4) 345 (16.8) 10 (0.5) 343 (16.6) Osteoporosis 5 (0.2) 223 (10.9) 11 (0.5) 225 (10.9) Myalgia 16 (0.8) 233 (11.4) 15 (0.7) 212 (10.3) Back pain 11 (0.5) 212 (10.3) 17 (0.8) 193 (9.4) Osteopenia 4 (0.2) 203 (9.9) 1 (0.0) 173 (8.4) Pain in extremity 9 (0.4) 168 (8.2) 3 (0.1) 174 (8.4) Lymphoedema 5 (0.2) 159 (7.8) 2 (0.1) 179 (8.7) Insomnia 7 (0.3) 160 (7.8) 3 (0.1) 149 (7.2) Hypercholesterolaemia 2 (0.1) 155 (7.6) 1 (0.0) 151 (7.3) 25 (1.2) 156 (7.6) 20 (1.0) 149 (7.2) Depression 16 (0.8) 147 (7.2) 13 (0.6) 137 (6.6) Bone pain 10 (0.5) 138 (6.7) 9 (0.4) 122 (5.9) Nausea 6 (0.3) 137 (6.7) 5 (0.2) 152 (7.4) Headache 3 (0.1) 130 (6.3) 5 (0.2) 168 (8.1) Alopecia 2 (0.1) 127 (6.2) 0 (0.0) 134 (6.5) Musculoskeletal pain 6 (0.3) 123 (6.0) 9 (0.4) 147 (7.1) Radiation skin injury 11 (0.5) 120 (5.9) 6 (0.3) 88 (4.3) Dyspnoea 16 (0.8) 118 (5.8) 10 (0.5) 96 (4.7) Cough 1 (0.0) 106 (5.2) 1 (0.0) 120 (5.8) Musculoskeletal stiffness 2 (0.1) 102 (5.0) 2 (0.1) 84 (4.1) Dizziness 2 (0.2) 94 (4.6) 7 (0.3) 109 (5.3) The following adverse reactions were also identified in less than 5% of the 2049 patients treated with letrozole and not included in the table: fall, vertigo, hyperbilirubinemia, jaundice, and chest pain.
Extended Adjuvant Treatment of Early Breast Cancer, Median Treatment Duration of 24 Months
In study MA-17, the median duration of extended adjuvant treatment was 24 months and the median duration of follow-up for safety was 28 months for patients receiving letrozole tablets and placebo.
Table 3 describes the adverse reactions occurring at a frequency of at least 5% in any treatment group during treatment. Most adverse reactions reported were Grade 1 and Grade 2 based on the CTC Version 2.0. In the extended adjuvant setting, the reported drug-related adverse reactions that were significantly different from placebo were hot flashes, arthralgia/arthritis, and myalgia.
Table 3: Adverse Reactions Occurring in at least 5% of Patients in either Treatment Arm Number (%) of Patients with Grade 1-4 Adverse Reactions Number (%) of Patients with Grade 3-4 Adverse Reactions Letrozole Tablets Placebo Letrozole Tablets Placebo N=2563 N=2573 N=2563 N=2573 Any Adverse Reaction 2232 (87.1) 2174 (84.5) 419 (16.3) 389 (15.1) Vascular Disorders 1375 (53.6) 1230 (47.8) 59 (2.3) 74 (2.9) Flushing 1273 (49.7) 1114 (43.3) 3 (0.1) 0 - General Disorders 1154 (45) 1090 (42.4) 30 (1.2) 28 (1.1) Asthenia 862 (33.6) 826 (32.1) 16 (0.6) 7 (0.3) Edema NOS 471 (18.4) 416 (16.2) 4 (0.2) 3 (0.1) Musculoskeletal Disorders 978 (38.2) 836 (32.5) 71 (2.8) 50 (1.9) Arthralgia 565 (22) 465 (18.1) 25 (1) 20 (0.8) Arthritis NOS 173 (6.7) 124 (4.8) 10 (0.4) 5 (0.2) Myalgia 171 (6.7) 122 (4.7) 8 (0.3) 6 (0.2) Back Pain 129 (5) 112 (4.4) 8 (0.3) 7 (0.3) Nervous System Disorders 863 (33.7) 819 (31.8) 65 (2.5) 58 (2.3) Headache 516 (20.1) 508 (19.7) 18 (0.7) 17 (0.7) Dizziness 363 (14.2) 342 (13.3) 9 (0.4) 6 (0.2) Skin Disorders 830 (32.4) 787 (30.6) 17 (0.7) 16 (0.6) Sweating Increased 619 (24.2) 577 (22.4) 1 (<0.1) 0 - Gastrointestinal Disorders 725 (28.3) 731 (28.4) 43 (1.7) 42 (1.6) Constipation 290 (11.3) 304 (11.8) 6 (0.2) 2 (<0.1) Nausea 221 (8.6) 212 (8.2) 3 (0.1) 10 (0.4) Diarrhea NOS 128 (5) 143 (5.6) 12 (0.5) 8 (0.3) Metabolic Disorders 551 (21.5) 537 (20.9) 24 (0.9) 32 (1.2) Hypercholesterolemia 401 (15.6) 398 (15.5) 2 (<0.1) 5 (0.2) Reproductive Disorders 303 (11.8) 357 (13.9) 9 (0.4) 8 (0.3) Vaginal Hemorrhage 123 (4.8) 171 (6.6) 2 (<0.1) 5 (0.2) Vulvovaginal Dryness 137 (5.3) 127 (4.9) 0 - 0 - Psychiatric Disorders 320 (12.5) 276 (10.7) 21 (0.8) 16 (0.6) Insomnia 149 (5.8) 120 (4.7) 2 (<0.1) 2 (<0.1) Respiratory Disorders 279 (10.9) 260 (10.1) 30 (1.2) 28 (1.1) Dyspnea 140 (5.5) 137 (5.3) 21 (0.8) 18 (0.7) Investigations 184 (7.2) 147 (5.7) 13 (0.5) 13 (0.5) Infections and Infestations 166 (6.5) 163 (6.3) 40 (1.6) 33 (1.3) Renal Disorders 130 (5.1) 100 (3.9) 12 (0.5) 6 (0.2) Based on a median follow-up of patients for 28 months,the incidence of clinical fractures from the core randomized study in patients who received letrozole was 5.9% (152) and placebo was 5.5% (142). The incidence of self-reported osteoporosis was higher in patients who received letrozole 6.9% (176) than in patients who received placebo 5.5% (141). Bisphosphonates were administered to 21.1% of the patients who received letrozole and 18.7% of the patients who received placebo.
The incidence of cardiovascular ischemic events from the core randomized study was comparable between patients who received letrozole 6.8% (175) and placebo 6.5% (167).
A patient-reported measure that captures treatment impact on important symptoms associated with estrogen deficiency demonstrated a difference in favor of placebo for vasomotor and sexual symptom domains.
Bone Substudy:[see Warnings and Precautions ( 5.1)].
Lipid Substudy :In the extended adjuvant setting, based on a median duration of follow-up of 62 months, there was no significant difference between letrozole and placebo in total cholesterol or in any lipid fraction at any time over 5 years. Use of lipid lowering drugs or dietary management of elevated lipids was allowed. [see Warnings and Precautions ( 5.2)].
Updated Analysis, Extended Adjuvant Treatment of Early Breast Cancer, Median Treatment Duration of 60 Months
The extended adjuvant treatment trial (MA-17) was unblinded early [see Adverse Reactions ( 6)] . At the updated (final analysis), overall the side effects seen were consistent to those seen at a median treatment duration of 24 months.
During treatment or within 30 days of stopping treatment (median duration of treatment 60 months) a higher rate of fractures was observed for letrozole (10.4%) compared to placebo (5.8%), as also a higher rate of osteoporosis (letrozole 12.2% vs placebo 6.4%).
Based on 62 months median duration of follow-up in the randomized letrozole arm in the safety population the incidence of new fractures at any time after randomization was 13.3% for letrozole and 7.8% for placebo. The incidence of new osteoporosis was 14.5% for letrozole and 7.8% for placebo.
During treatment or within 30 days of stopping treatment (median duration of treatment 60 months) the incidence of cardiovascular events was 9.8% for letrozole and 7.0% for placebo.
Based on 62 months median duration of follow-up in the randomized letrozole arm in the safety population the incidence of cardiovascular disease at any time after randomization was 14.4% for letrozole and 9.8% for placebo.
Lipid Substudy: In the extended adjuvant setting (MA-17), based on a median duration of follow-up of 62 months, there was no significant difference between letrozole and placebo in total cholesterol or in any lipid fraction over 5 years. Use of lipid lowering drugs or dietary management of elevated lipids was allowed. [see Warnings and Precautions( 5.2)]
First-Line Treatment of Advanced Breast Cancer
In study P025 a total of 455 patients were treated for a median time of exposure of 11 months in the letrozole arm (median 6 months in the tamoxifen arm). The incidence of adverse reactions was similar for letrozole and tamoxifen. The most frequently reported adverse reactions were bone pain, hot flushes, back pain, nausea, arthralgia and dyspnea. Discontinuations for adverse reactions other than progression of tumor occurred in 10/455 (2%) of patients on letrozole and in 15/455 (3%) of patients on tamoxifen.
Adverse reactions that were reported in at least 5% of the patients treated with letrozole 2.5 mg or tamoxifen 20 mg in the first-line treatment study are shown in Table 4.
Table 4: Adverse Reactions Occurring in at least 5% of Patients in either Treatment Arm Adverse Letrozole Tamoxifen Reactions 2.5 mg 20 mg (N=455) (N=455) % % General Disorders Fatigue 13 13 Chest Pain 8 9 Edema Peripheral 5 6 Pain NOS 5 7 Weakness 6 4 Investigations Weight Decreased 7 5 Vascular Disorders Hot Flushes 19 16 Hypertension 8 4 Gastrointestinal Disorders Nausea 17 17 Constipation 10 11 Diarrhea 8 4 Vomiting 7 8 Infections/Infestations Influenza 6 4 Urinary Tract Infection NOS 6 3 Injury, Poisoning and Procedural Complications Post-Mastectomy Lymphedema 7 7 Metabolism and Nutrition Disorders Anorexia 4 6 Musculoskeletal and Connective Tissue Disorders Bone Pain 22 21 Back Pain 18 19 Arthralgia 16 15 Pain in Limb 10 8 Nervous System Disorders Headache NOS 8 7 Psychiatric Disorders Insomnia 7 4 Reproductive System and Breast Disorders Breast Pain 7 7 Respiratory, Thoracic and Mediastinal Disorders Dyspnea 18 17 Cough 13 13 Chest Wall Pain 6 6 Other less frequent (less than or equal to 2%) adverse reactions considered consequential for both treatment groups, included peripheral thromboembolic events, cardiovascular events, and cerebrovascular events. Peripheral thromboembolic events included venous thrombosis, thrombophlebitis, portal vein thrombosis and pulmonary embolism. Cardiovascular events included angina, myocardial infarction, myocardial ischemia, and coronary heart disease. Cerebrovascular events included transient ischemic attacks, thrombotic or hemorrhagic strokes and development of hemiparesis.
Second- Line Treatment of Advanced Breast Cancer
Study discontinuations in the megestrol acetate comparison study (AR/BC2) for adverse reactions other than progression of tumor were 5/188 (2.7%) on letrozole 0.5 mg, in 4/174 (2.3%) on letrozole 2.5 mg, and in 15/190 (7.9%) on megestrol acetate. There were fewer thromboembolic events at both letrozole doses than on the megestrol acetate arm (0.6% vs 4.7%). There was also less vaginal bleeding (0.3% vs 3.2%) on letrozole than on megestrol acetate. In the aminoglutethimide comparison study (AR/BC3), discontinuations for reasons other than progression occurred in 6/193 (3.1%) on 0.5 mg letrozole, 7/185 (3.8%) on 2.5 mg letrozole, and 7/178 (3.9%) of patients on aminoglutethimide.
Comparisons of the incidence of adverse reactions revealed no significant differences between the high and low dose letrozole groups in either study. Most of the adverse reactions observed in all treatment groups were mild to moderate in severity and it was generally not possible to distinguish adverse reactions due to treatment from the consequences of the patient’s metastatic breast cancer, the effects of estrogen deprivation, or intercurrent illness.
Adverse reactions that were reported in at least 5% of the patients treated with letrozole 0.5 mg, letrozole 2.5 mg, megestrol acetate, or aminoglutethimide in the two controlled trials AR/BC2 and AR/BC3 are shown in Table 5.
Table 5: Adverse Reactions Occurring at a Frequency of at Least 5% of Patients in Either Treatment Arm Adverse Pooled Pooled megestrol Reactions Letrozole Letrozole acetate aminoglutethimide 2.5 mg 0.5 mg 160 mg 500 mg (N=359) (N=380) (N=189) (N=178) % % % % Body as a Whole Chest Pain 6 3 7 3 Peripheral Edema 1 5 5 8 3 Asthenia 4 5 4 5 Weight Increase 2 2 9 3 Cardiovascular Hypertension 5 7 5 6 Digestive System Nausea 13 15 9 14 Vomiting 7 7 5 9 Constipation 6 7 9 7 Diarrhea 6 5 3 4 Pain-Abdominal 6 5 9 8 Anorexia 5 3 5 5 Dyspepsia 3 4 6 5 Infections/Infestations Viral Infection 6 5 6 3 Lab Abnormality Hypercholesterolemia 3 3 0 6 Musculoskeletal System Musculoskeletal 2 21 22 30 14 Arthralgia 8 8 8 3 Nervous System Headache 9 12 9 7 Somnolence 3 2 2 9 Dizziness 3 5 7 3 Respiratory System Dyspnea 7 9 16 5 Coughing 6 5 7 5 Skin and Appendages Hot Flushes 6 5 4 3 Rash 3 5 4 3 12 Pruritus 1 2 5 3 1Includes peripheral edema, leg edema, dependent edema, edema
2Includes musculoskeletal pain, skeletal pain, back pain, arm pain, leg pain
3Includes rash, erythematous rash, maculopapular rash, psoriasiform rash, vesicular rash
Other less frequent (less than 5%) adverse reactions considered consequential and reported in at least 3 patients treated with letrozole, included hypercalcemia, fracture, depression, anxiety, pleural effusion, alopecia, increased sweating and vertigo.
First and Second-Line Treatment of Advanced Breast Cancer
In the combined analysis of the first- and second-line metastatic trials and postmarketing experiences other adverse reactions that were reported were cataract, eye irritation, palpitations, cardiac failure, tachycardia, dysesthesia (including hypesthesia/paresthesia), arterial thrombosis, memory impairment, irritability, nervousness, urticaria, increased urinary frequency, leukopenia, stomatitis cancer pain, pyrexia, vaginal discharge, appetite increase, dryness of skin and mucosa (including dry mouth), and disturbances of taste and thirst.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of letrozole. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Eye Disorders: blurred vision
Hepatobiliary Disorders: increased hepatic enzymes, hepatitis
Immune System Disorders: anaphylactic reactions, hypersensitivity reactions
Nervous System Disorders: carpal tunnel syndrome, trigger finger
Pregnancy: spontaneous abortions, congenital birth defects
Skin and subcutaneous disorders: angioedema, toxic epidermal necrolysis, erythema multiforme
-
7 DRUG INTERACTIONS
Tamoxifen
Coadministration of letrozole and tamoxifen 20 mg daily resulted in a reduction of letrozole plasma levels of 38% on average (study P015). Clinical experience in the second-line breast cancer trials (AR/BC2 and AR/BC3) indicates that the therapeutic effect of letrozole therapy is not impaired if letrozole is administered immediately after tamoxifen.
Cimetidine
A pharmacokinetic interaction study with cimetidine (study P004) showed no clinically significant effect on letrozole pharmacokinetics.
Warfarin
An interaction study (P017) with warfarin showed no clinically significant effect of letrozole on warfarin pharmacokinetics.
Other anticancer agents
There is no clinical experience to date on the use of letrozole in combination with other anticancer agents.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on postmarketing reports, findings from animal studies and the mechanism of action, letrozole can cause fetal harm and is contraindicated for use in pregnant women. In post-marketing reports, use of letrozole during pregnancy resulted in cases of spontaneous abortions and congenital birth defects; however, the data are insufficient to inform a drug-associated risk [see Contraindications ( 4), Warnings and Precautions ( 5.6), Adverse Reactions ( 6.2) and Clinical Pharmacology ( 12.1)].
In animal reproduction studies, administration of letrozole to pregnant animals during organogenesis resulted in increased post-implantation pregnancy loss and resorption, fewer live fetuses, and fetal malformation affecting the renal and skeletal systems in rats and rabbits at doses approximately 0.1 times the daily maximum recommended human dose (MRHD) on a mg/m 2basis (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. However, the background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
In a fertility and early embryonic development toxicity study in female rats, oral administration of letrozole starting 2 weeks before mating until pregnancy day 6 resulted in an increase in pre-implantation loss at doses ≥ 0.003 mg/kg/day (approximately 0.01 times the maximum recommended human dose on a mg/m 2basis).
In an embryo-fetal developmental toxicity study in rats, daily administration of oral letrozole during the period of organogenesis at doses ≥ 0.003 mg/kg (approximately 0.01 time the maximum recommended human dose on a mg/m 2basis) resulted in embryo-fetal toxicity including intrauterine mortality, increased resorptions and postimplantation loss, decreased numbers of live fetuses and fetal anomalies including absence and shortening of renal papilla, dilation of ureter, edema and incomplete ossification of frontal skull and metatarsals. Letrozole was teratogenic to rats at a dose of 0.03 mg/kg (approximately 0.01 times the maximum recommended human dose on a mg/m 2basis) and caused fetal domed head and cervical/centrum vertebral fusion.
In the embryo-fetal development toxicity study in rabbits, daily administration of oral letrozole during the period of organogenesis at doses ≥ 0.002 mg/kg (approximately 0.01 times the maximum recommended human dose on a mg/m 2basis) resulted in embryo-fetal toxicity including intrauterine mortality, increased resorption, increased postimplantation loss and decreased numbers of live fetuses. Fetal anomalies included incomplete ossification of the skull, sternebrae, and fore- and hind legs.
8.2 Lactation
Risk Summary
It is not known if letrozole is present in human milk. There are no data on the effects of letrozole on the breastfed infant or milk production. Exposure of lactating rats to letrozole was associated with impaired reproductive performance of the male offspring (see Data). Because of the potential for serious adverse reactions in breastfed infants from letrozole tablets, advise lactating women not to breastfeed while taking letrozole tablets and for at least 3 weeks after the last dose.
Data
Animal Data
In a postnatal developmental toxicity study in lactating rats, letrozole was administered orally at doses of 1, 0.003, 0.03 or 0.3 mg/kg/day on day 0 through day 20 of lactation. The reproductive performance of the male offspring was impaired at letrozole dose as low as 0.003 mg/kg/day (approximately 0.01 times the maximum recommended human dose on a mg/m 2basis), as reflected by decreased mating and pregnancy ratios. There were no effects on the reproductive performance of female offspring.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Based on animal studies, letrozole can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations ( 8.1)]. Females of reproductive potential should have a pregnancy test prior to starting treatment with letrozole.
Contraception
Females
Based on animal studies, letrozole tablets can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations ( 8.1)]. . Advise females of reproductive potential to use effective contraception during treatment with letrozole tablets and for at least 3 weeks after the last dose.
Infertility
Females
Based on studies in female animals, letrozole tablets may impair fertility in females of reproductive potential [see Nonclinical Toxicology ( 13.1)].
Males
Based on studies in male animals, letrozole tablets may impair fertility in males of reproductive potential [see Nonclinical Toxicology ( 13.1)].
8.4 Pediatric Use
The safety and effectiveness in pediatric patients have not been established.
Letrozole administration to young (postnatal day 7) rats for 12 weeks duration at 0.003, 0.03, 0.3 mg/kg/day by oral gavage resulted in adverse skeletal/growth effects (bone maturation, bone mineral density) and neuroendocrine and reproductive developmental perturbations of the hypothalamic-pituitary axis. Administration of 0.3 mg/kg/day resulted in AUC values that were similar to the AUC in adult patients receiving the recommended dose of 2.5 mg/day. Decreased fertility was accompanied by hypertrophy of the hypophysis and testicular changes that included degeneration of the seminiferous tubular epithelium and atrophy of the female reproductive tract. Young rats in this study were allowed to recover following discontinuation of letrozole treatment for 42 days. Histopathological changes were not reversible at clinically relevant exposures.
8.5 Geriatric Use
The median age of patients in all studies of first-line and second-line treatment of metastatic breast cancer was 64 to 65 years. About 1/3 of the patients were greater than or equal to 70 years old. In the first-line study, patients greater than or equal to 70 years of age experienced longer time to tumor progression and higher response rates than patients less than 70.
For the extended adjuvant setting (MA-17), more than 5,100 postmenopausal women were enrolled in the clinical study. In total, 41% of patients were aged 65 years or older at enrollment, while 12% were 75 or older. In the extended adjuvant setting, no overall differences in safety or efficacy were observed between these older patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
In the adjuvant setting (BIG 1-98), more than 8,000 postmenopausal women were enrolled in the clinical study. In total, 36 % of patients were aged 65 years or older at enrollment, while 12% were 75 or older. More adverse reactions were generally reported in elderly patients irrespective of study treatment allocation. However, in comparison to tamoxifen, no overall differences with regards to the safety and efficacy profiles were observed between elderly patients and younger patients.
-
10 OVERDOSAGE
Isolated cases of letrozole overdose have been reported. In these instances, the highest single dose ingested was 62.5 mg or 25 tablets. While no serious adverse reactions were reported in these cases, because of the limited data available, no firm recommendations for treatment can be made. However, emesis could be induced if the patient is alert. In general, supportive care and frequent monitoring of vital signs are also appropriate. In single-dose studies, the highest dose used was 30 mg, which was well tolerated; in multiple-dose trials, the largest dose of 10 mg was well tolerated.
Lethality was observed in mice and rats following single oral doses that were equal to or greater than 2,000 mg/kg (about 4,000 to 8,000 times the daily maximum recommended human dose on a mg/m 2basis); death was associated with reduced motor activity, ataxia and dyspnea. Lethality was observed in cats following single IV doses that were equal to or greater than 10 mg/kg (about 50 times the daily maximum recommended human dose on a mg/m 2basis); death was preceded by depressed blood pressure and arrhythmias.
-
11 DESCRIPTION
Letrozole tablet USP for oral administration contains 2.5 mg of letrozole, a nonsteroidal aromatase inhibitor (inhibitor of estrogen synthesis). It is chemically described as 4,4'-(1H-1,2,4-Triazol-1-ylmethylene) dibenzonitrile, and its structural formula is
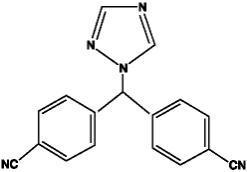
Letrozole is a white to yellowish crystalline powder, practically odorless, freely soluble in dichloromethane, slightly soluble in ethanol, and practically insoluble in water. It has a molecular weight of 285.31, empirical formula C 17H 11N 5, and a melting range of 184°C to 185°C.
Letrozole is available as 2.5 mg tablets for oral administration.
InactiveIngredients: Colloidal anhydrous silica, hypromellose, iron oxide yellow , lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, sodium starch glycolate, starch (corn), talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The growth of some cancers of the breast is stimulated or maintained by estrogens. Treatment of breast cancer thought to be hormonally responsive (i.e., estrogen and/or progesterone receptor positive or receptor unknown) has included a variety of efforts to decrease estrogen levels (ovariectomy, adrenalectomy, hypophysectomy) or inhibit estrogen effects (antiestrogens and progestational agents). These interventions lead to decreased tumor mass or delayed progression of tumor growth in some women.
In postmenopausal women, estrogens are mainly derived from the action of the aromatase enzyme, which converts adrenal androgens (primarily androstenedione and testosterone) to estrone and estradiol. The suppression of estrogen biosynthesis in peripheral tissues and in the cancer tissue itself can therefore be achieved by specifically inhibiting the aromatase enzyme.
Letrozole is a nonsteroidal competitive inhibitor of the aromatase enzyme system; it inhibits the conversion of androgens to estrogens. In adult nontumor- and tumor-bearing female animals, letrozole is as effective as ovariectomy in reducing uterine weight, elevating serum LH, and causing the regression of estrogen-dependent tumors. In contrast to ovariectomy, treatment with letrozole does not lead to an increase in serum FSH. Letrozole selectively inhibits gonadal steroidogenesis but has no significant effect on adrenal mineralocorticoid or glucocorticoid synthesis.
Letrozole inhibits the aromatase enzyme by competitively binding to the heme of the cytochrome P450 subunit of the enzyme, resulting in a reduction of estrogen biosynthesis in all tissues. Treatment of women with letrozole significantly lowers serum estrone, estradiol and estrone sulfate and has not been shown to significantly affect adrenal corticosteroid synthesis, aldosterone synthesis, or synthesis of thyroid hormones.
12.2 >Pharmacodynamics
In postmenopausal patients with advanced breast cancer, daily doses of 0.1 mg to 5 mg letrozole suppress plasma concentrations of estradiol, estrone, and estrone sulfate by 75% to 95% from baseline with maximal suppression achieved within two-three days. Suppression is dose-related, with doses of 0.5 mg and higher giving many values of estrone and estrone sulfate that were below the limit of detection in the assays. Estrogen suppression was maintained throughout treatment in all patients treated at 0.5 mg or higher.
Letrozole is highly specific in inhibiting aromatase activity. There is no impairment of adrenal steroidogenesis. No clinically-relevant changes were found in the plasma concentrations of cortisol, aldosterone, 11-deoxycortisol, 17-hydroxy-progesterone, ACTH or in plasma renin activity among postmenopausal patients treated with a daily dose of letrozole 0.1 mg to 5 mg. The ACTH stimulation test performed after 6 and 12 weeks of treatment with daily doses of 0.1, 0.25, 0.5, 1, 2.5, and 5 mg did not indicate any attenuation of aldosterone or cortisol production. Glucocorticoid or mineralocorticoid supplementation is, therefore, not necessary.
No changes were noted in plasma concentrations of androgens (androstenedione and testosterone) among healthy postmenopausal women after 0.1, 0.5, and 2.5 mg single doses of letrozole or in plasma concentrations of androstenedione among postmenopausal patients treated with daily doses of 0.1 mg to 5 mg. This indicates that the blockade of estrogen biosynthesis does not lead to accumulation of androgenic precursors. Plasma levels of LH and FSH were not affected by letrozole in patients, nor was thyroid function as evaluated by TSH levels, T3 uptake, and T4 levels.
12.3 Pharmacokinetics
Absorption and Distribution: Letrozole is rapidly and completely absorbed from the gastrointestinal tract and absorption is not affected by food. It is metabolized slowly to an inactive metabolite whose glucuronide conjugate is excreted renally, representing the major clearance pathway. About 90% of radiolabeled letrozole is recovered in urine. Letrozole’s terminal elimination half-life is about 2 days and steady-state plasma concentration after daily 2.5 mg dosing is reached in 2 to 6 weeks. Plasma concentrations at steady state are 1.5 to 2 times higher than predicted from the concentrations measured after a single dose, indicating a slight non-linearity in the pharmacokinetics of letrozole upon daily administration of 2.5 mg. These steady-state levels are maintained over extended periods, however, and continuous accumulation of letrozole does not occur. Letrozole is weakly protein bound and has a large volume of distribution (approximately 1.9 L/kg).
Elimination
Metabolism and Excretion: Metabolism to a pharmacologically-inactive carbinol metabolite (4,4'-methanol-bisbenzonitrile) and renal excretion of the glucuronide conjugate of this metabolite is the major pathway of letrozole clearance. Of the radiolabel recovered in urine, at least 75% was the glucuronide of the carbinol metabolite, about 9% was two unidentified metabolites, and 6% was unchanged letrozole.
In human microsomes with specific CYP isozyme activity, CYP3A4 metabolized letrozole to the carbinol metabolite while CYP2A6 formed both this metabolite and its ketone analog. In human liver microsomes, letrozole inhibited CYP2A6 and CYP2C19, however, the clinical significance of these findings is unknown.
Specific Populations
Pediatric, Geriatric and Race: In the study populations (adults ranging in age from 35 to greater than 80 years), no change in pharmacokinetic parameters was observed with increasing age. Differences in letrozole pharmacokinetics between adult and pediatric populations have not been studied. Differences in letrozole pharmacokinetics due to race have not been studied.
Renal Impairment: In a study of volunteers with varying renal function (24-hour creatinine clearance: 9 to 116 mL/min), no effect of renal function on the pharmacokinetics of single doses of 2.5 mg of letrozole was found. In addition, in a study (AR/BC2) of 347 patients with advanced breast cancer, about half of whom received 2.5 mg letrozole and half 0.5 mg letrozole, renal impairment (calculated creatinine clearance: 20 to 50 mL/min) did not affect steady-state plasma letrozole concentrations.
HepaticImpairment: In a study of subjects with mild to moderate non-metastatic hepatic dysfunction (e.g., cirrhosis, Child-Pugh classification A and B), the mean area under curve (AUC) values of the volunteers with moderate hepatic impairment were 37% higher than in normal subjects, but still within the range seen in subjects without impaired function.
In a pharmacokinetic study, subjects with liver cirrhosis and severe hepatic impairment (Child-Pugh classification C, which included bilirubins about 2 to 11 times ULN with minimal to severe ascites) had two-fold increase in exposure (AUC) and 47% reduction in systemic clearance. Breast cancer patients with severe hepatic impairment are thus expected to be exposed to higher levels of letrozole than patients with normal liver function receiving similar doses of this drug. [see Dosage and Administration ( 2.5)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A conventional carcinogenesis study in mice at doses of 0.6 to 60 mg/kg/day (about 1 to 100 times the daily maximum recommended human dose on a mg/m 2basis) administered by oral gavage for up to 2 years revealed a dose-related increase in the incidence of benign ovarian stromal tumors. The incidence of combined hepatocellular adenoma and carcinoma showed a significant trend in females when the high dose group was excluded due to low survival. In a separate study, plasma AUC 0-12hrlevels in mice at 60 mg/kg/day were 55 times higher than the AUC 0-24hrlevel in breast cancer patients at the recommended dose. The carcinogenicity study in rats at oral doses of 0.1 to 10 mg/kg/day (about 0.4 to 40 times the daily maximum recommended human dose on a mg/m 2basis) for up to 2 years also produced an increase in the incidence of benign ovarian stromal tumors at 10 mg/kg/day. Ovarian hyperplasia was observed in females at doses equal to or greater than 0.1 mg/kg/day. At 10 mg/kg/day, plasma AUC 0-24hrlevels in rats were 80 times higher than the level in breast cancer patients at the recommended dose.The benign ovarian stromal tumors observed in mice and rats were considered to be related to the pharmacological inhibition of estrogen synthesis and may be due to increased luteinizing hormone resulting from the decrease in circulating estrogen.
Letrozole was not mutagenic in in vitrotests (Ames and E.coli bacterial tests) but was observed to be a potential clastogen in in vitroassays (CHO K1 and CCL 61 Chinese hamster ovary cells). Letrozole was not clastogenic in vivo(micronucleus test in rats).
In a fertility and early embryonic development toxicity study in female rats, oral administration of letrozole starting 2 weeks before mating until pregnancy day 6 resulted in an increase in pre-implantation loss at doses ≥ 0.03 mg/kg/day (approximately 0.1 times the maximum recommended human dose on a mg/m 2basis). In repeat-dose toxicity studies, administration of letrozole caused sexual inactivity in females and atrophy of the reproductive tract in males and females at doses of 0.6, 0.1 and 0.03 mg/kg in mice, rats and dogs, respectively (approximately 1, 0.4 and 0.4 times the daily maximum recommended human dose on a mg/m 2basis, respectively).
-
14 CLINICAL STUDIES
14.1 Updated Adjuvant Treatment of Early Breast Cancer
In a multicenter study (BIG 1-98, NCT00004205) enrolling over 8,000 postmenopausal women with resected, receptor-positive early breast cancer, one of the following treatments was randomized in a double-blind manner:
Option 1:
- tamoxifen for 5 years
- Letrozole for 5 years
- tamoxifen for 2 years followed by letrozole for 3 years
- Letrozole for 2 years followed by tamoxifen for 3 years
Option 2:
- tamoxifen for 5 years
- Letrozole for 5 years
The study in the adjuvant setting, BIG 1-98 was designed to answer two primary questions: whether letrozole for 5 years was superior to Tamoxifen for 5 years (Primary Core Analysis) and whether switching endocrine treatments at 2 years was superior to continuing the same agent for a total of 5 years (Sequential Treatments Analysis). Selected baseline characteristics for the study population are shown in Table 6.
The primary endpoint of this trial was disease-free survival (DFS) (i.e., interval between randomization and earliest occurrence of a local, regional, or distant recurrence, or invasive contralateral breast cancer, or death from any cause). The secondary endpoints were overall survival (OS), systemic disease-free survival (SDFS), invasive contralateral breast cancer, time to breast cancer recurrence (TBR) and time to distant metastasis (TDM).
The Primary Core Analysis (PCA) included all patients and all follow-up in the monotherapy arms in both randomization options, but follow-up in the two sequential treatments arms was truncated 30 days after switching treatments. The PCA was conducted at a median treatment duration of 24 months and a median follow-up of 26 months. Letrozole was superior to tamoxifen in all endpoints except overall survival and contralateral breast cancer [e.g., DFS: hazard ratio, HR 0.79; 95% CI (0.68, 0.92); P=0.002; SDFS: HR 0.83; 95% CI (0.70, 0.97); TDM: HR 0.73; 95% CI (0.60, 0.88); OS: HR 0.86; 95% CI (0.70, 1.06).
In 2005, based on recommendations by the independent Data Monitoring Committee, the tamoxifen arms were unblinded and patients were allowed to complete initial adjuvant therapy with letrozole (if they had received tamoxifen for at least 2 years) or to start extended adjuvant treatment with letrozole (if they had received tamoxifen for at least 4.5 years) if they remained alive and disease-free. In total, 632 patients crossed to letrozole or another aromatase inhibitor. Approximately 70% (448) of these 632 patients crossed to letrozole to complete initial adjuvant therapy and most of these crossed in years 3 to 4. All of these patients were in Option 1. A total of 184 patients started extended adjuvant therapy with letrozole (172 patients) or with another aromatase inhibitor (12 patients). To explore the impact of this selective crossover, results from analyses censoring follow-up at the date of the selective crossover (in the tamoxifen arm) are presented for the MAA.
The PCA allowed the results of letrozole for 5 years compared with tamoxifen for 5 years to be reported in 2005 after a median follow-up of only 26 months. The design of the PCA is not optimal to evaluate the effect of letrozole after a longer time (because follow-up was truncated in two arms at around 25 months). The MAA (ignoring the two sequential treatment arms) provided follow-up equally as long in each treatment and did not over-emphasize early recurrences as the PCA did. The MAA thus provides the clinically appropriate updated efficacy results in answer to the first primary question, despite the confounding of the tamoxifen reference arm by the selective crossover to letrozole. The updated results for the MAA are summarized in Table 7. Median follow-up for this analysis is 73 months.
The Sequential Treatments Analysis (STA) addresses the second primary question of the study. The primary analysis for the STA was from switch (or equivalent time-point in monotherapy arms) + 30 days (STA-S) with a two-sided test applied to each pair-wise comparison at the 2.5% level. Additional analyses were conducted from randomization (STA-R) but these comparisons (added in light of changing medical practice) were under-powered for efficacy.
Table 6: Adjuvant Study - Patient and Disease Characteristics (ITT Population) Primary Core Analysis (PCA) Monotherapy Arms Analysis (MAA) Letrozole Tamoxifen Letrozole Tamoxifen N=4003 N=4007 N=2463 N=2459 Characteristic n (%) n (%) n (%) n (%) Age (median, years) 61 61 61 61 Age range (years) 38 to 89 39 to 90 38 to 88 39 to 90 Hormone receptor status (%) ER+ and/or PgR+ 99.7 99.7 99.7 99.7 Both unknown 0.3 0.3 0.3 0.3 Nodal status (%) Node negative 52 52 50 52 Node positive 41 41 43 41 Nodal status unknown 7 7 7 7 Prior adjuvant chemotherapy (%) 24 24 24 24 Table 7: Updated Adjuvant Study Results - Monotherapy Arms Analysis (Median Follow-up 73 Months) Letrozole
N=2463Tamoxifen
N=2459Hazard ratio Events
(%)5-year rate Events
(%)5-year rate (95% CI) P Disease-free survival 1 ITT 445 (18.1) 87.4 500 (20.3) 84.7 0.87 (0.76, 0.99) 0.03 Censor 445 87.4 483 84.2 0.84 (0.73, 0.95) 0 positive nodes ITT 165 92.2 189 90.3 0.88 (0.72, 1.09) 1 to 3 positive nodes ITT 151 85.6 163 83.0 0.85 (0.68, 1.06) >=4 positive nodes ITT 123 71.2 142 62.6 0.81 (0.64, 1.03) Adjuvant chemotherapy ITT 119 86.4 150 80.6 0.77 (0.60, 0.98) No chemotherapy ITT 326 87.8 350 86.1 0.91 (0.78, 1.06) Systemic DFS 2 ITT 401 88.5 446 86.6 0.88 (0.77,1.01) Time to distant metastasis 3 ITT 257 92.4 298 90.1 0.85 (0.72, 1.00) Adjuvant chemotherapy ITT 84 - 109 - 0.75(0.56 to 1.00) No chemotherapy ITT 173 - 189 - 0.90 (0.73,1.11) Distant DFS 4 ITT 385 89.0 432 87.1 0.87 (0.76,1.00) Contralateral breast cancer ITT 34 99.2 44 98.6 0.76 (0.49, 1.19) Overall survival ITT 303 91.8 343 90.9 0.87 (0.75, 1.02) Censor 303 91.8 338 90.1 0.82 (0.70, 0.96) 0 positive nodes ITT 107 95.2 121 94.8 0.90 (0.69,1.16) 1 to 3 positive nodes ITT 99 90.8 114 90.6 0.81 (0.62,1.06) >=4 positive nodes ITT 92 80.2 104 73.6 0.86 (0.65, 1.14) Adjuvant chemotherapy ITT 76 91.5 96 88.4 0.79 (0.58, 1.06) No chemotherapy ITT 227 91.9 247 91.8 0.91 (0.76, 1.08) Definition of:
1 Disease-free survival: Interval from randomization to earliest event of invasive loco-regional recurrence, distant metastasis, invasive contralateral breast cancer, or death without a prior event
2 Systemic disease-free survival: Interval from randomization to invasive regional recurrence, distant metastasis, or death without a prior cancer event
3 Time to distant metastasis: Interval from randomization to distant metastasis
4 Distant disease-free survival: Interval from randomization to earlier event of relapse in a distant site or death from any causeITT analysis ignores selective crossover in tamoxifen arms Censored analysis censors follow-up at the date of selective crossover in 632 patients who crossed to letrozole or another aromatase inhibitor after the tamoxifen arms were unblinded in 2005
Figure 1 shows the Kaplan-Meier curves for Disease-Free Survival Monotherapy Analysis
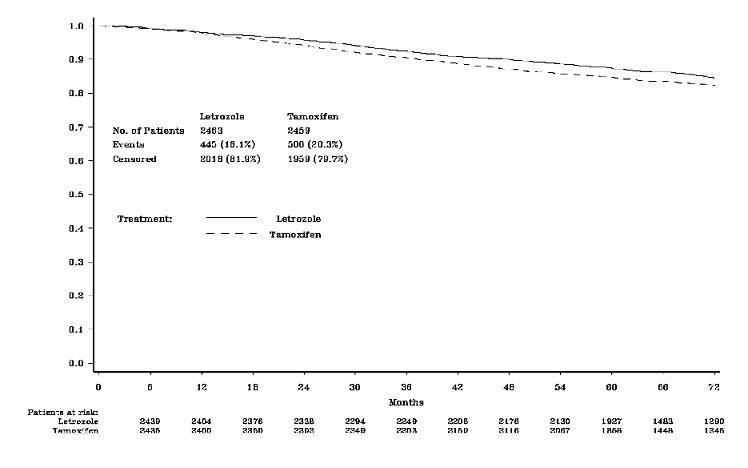
DFS events defined as loco-regional recurrence, distant metastasis, invasive contralateral breast cancer, or death from any cause (i.e., definition excludes second non-breast primary cancers).
The medians of overall survival for both arms were not reached for the MAA. There was no statistically significant difference in overall survival. The hazard ratio for survival in the letrozole arm compared to the tamoxifen arm was 0.87, with 95% CI (0.75, 1.02) (see Table 7). There were no significant differences in DFS, OS, SDFS, and Distant DFS from switch in the Sequential Treatments Analysis with respect to either monotherapy (e.g., [tamoxifen 2 years followed by] letrozole 3 years versus tamoxifen beyond 2 years, DFS HR 0.89; 97.5% CI 0.68, 1.15 and [letrozole 2 years followed by] tamoxifen 3 years versus letrozole beyond 2 years, DFS HR 0.93; 97.5% CI 0.71, 1.22). There were no significant differences in DFS, OS, SDFS, and Distant DFS from randomization in the Sequential Treatments Analyses.
14.2 Extended Adjuvant Treatment of Early Breast Cancer, Median Treatment Duration of 24 Months
A double-blind, randomized, placebo-controlled trial (MA-17, NCT00003140) of letrozole was performed in over 5,100 postmenopausal women with receptor-positive or unknown primary breast cancer who were disease free after 5 years of adjuvant treatment with tamoxifen.
The planned duration of treatment for patients in the study was 5 years, but the trial was terminated early because of an interim analysis showing a favorable letrozole effect on time without recurrence or contralateral breast cancer. At the time of unblinding, women had been followed for a median of 28 months, 30% of patients had completed 3 or more years of follow-up and less than 1% of patients had completed 5 years of follow-up.
Selected baseline characteristics for the study population are shown in Table 8.
Table 8: Selected Study Population Demographics (Modified ITT Population) Baseline Status Letrozole Placebo N=2582 N=2586 Hormone Receptor Status (%) ER+ and/or PgR+ 98 98 Both Unknown 2 2 Nodal Status (%) Node Negative 50 50 Node Positive 46 46 Nodal Status Unknown 4 4 Chemotherapy 46 46 Table 9 shows the study results. Disease-free survival was measured as the time from randomization to the earliest event of loco-regional or distant recurrence of the primary disease or development of contralateral breast cancer or death. DFS by hormone receptor status, nodal status and adjuvant chemotherapy were similar to the overall results. Data were premature for an analysis of survival.
Table 9: Extended Adjuvant Study Results Letrozole
N = 2582Placebo
N = 2586Hazard Ratio
(95% CI)P-Value Disease Free Survival (DFS)1Events 122 (4.7%) 193 (7.5%) 0.62 (0.49, 0.78) 2 0.00003 Local Breast Recurrence 9 22 Local Chest Wall Recurrence 2 8 Regional Recurrence 7 4 Distant Recurrence 55 92 0.61 (0.44 to 0.84) 0.003 Contralateral Breast Cancer 19 29 Deaths Without Recurrence or Contralateral Breast Cancer 30 38 CI = confidence interval for hazard ratio. Hazard ratio of less than 1.0 indicates difference in favor of letrozole (lesser risk of recurrence); hazard ratio greater than 1.0 indicates difference in favor of placebo (higher risk of recurrence with letrozole).
1 First event of loco-regional recurrence, distant relapse, contralateral breast cancer or death from any cause
2 Analysis stratified by receptor status, nodal status and prior adjuvant chemotherapy (stratification factors as at randomization). P-value based on stratified log-rank test.14.3 Updated Analyses of Extended Adjuvant Treatment of Early Breast Cancer, Median Treatment Duration of 60 Months
Table 10: Update of Extended Adjuvant Study Results Letrozole
N = 2582
(%)Placebo
N = 2586
(%)Hazard Ratio1
(95% CI)P-Value2 Disease Free Survival (DFS) events3 344 (13.3) 402 (15.5) 0.89 (0.77, 1.03) 0.12 Breast cancer recurrence
(Protocol definition of DFS events 4)209 286 0.75 (0.63, 0.89) 0.001 Local Breast Recurrence 15 44 Local Chest Wall Recurrence 6 14 Regional Recurrence 10 8 Distant Recurrence 140 167 Distant Recurrence (first or subsequent events)
Contralateral Breast Cancer142
37169
530.88 (0.70,1.10) 0.246 Deaths Without Recurrence or Contralateral Breast Cancer 135 116 1 Adjusted by receptor status, nodal status and prior chemotherapy
2Stratified log-rank test, stratified by receptor status, nodal status and prior chemotherapy
3DFS events defined as earliest of loco-regional recurrence, distant metastasis, contralateral breast cancer or death from any cause, and ignoring switches to letrozole in 60% of the placebo arm.
4Protocol definition does not include deaths from any causeUpdated analyses were conducted at a median follow-up of 62 months. In the letrozole arm, 71% of the patients were treated for a least 3 years and 58% of patients completed at least 4.5 years of extended adjuvant treatment. After the unblinding of the study at a median follow-up of 28 months, approximately 60% of the selected patients in the placebo arm opted to switch to letrozole.
In this updated analysis shown in Table 10 letrozole significantly reduced the risk of breast cancer recurrence or contralateral breast cancer compared with placebo (HR 0.75; 95% CI 0.63, 0.89; P=0.001). However, in the updated DFS analysis (interval between randomization and earliest event of loco-regional recurrence, distant metastasis, contralateral breast cancer, or death from any cause) the treatment difference was heavily diluted by 60% of the patients in the placebo arm switching to letrozole and accounting for 64% of the total placebo patient- years of follow-up. Ignoring these switches, the risk of DFS event was reduced by a non-significant 11% (HR 0.89; 95% CI 0.77, 1.03). There was no significant difference in distant disease-free survival or overall survival.
14.4 First-Line Treatment of Advanced Breast Cancer
A randomized, double-blind, multinational trial (P025) compared letrozole 2.5 mg with tamoxifen 20 mg in 916 postmenopausal patients with locally advanced (Stage IIIB or loco-regional recurrence not amenable to treatment with surgery or radiation) or metastatic breast cancer. Time to progression (TTP) was the primary endpoint of the trial. Selected baseline characteristics for this study are shown in Table 11.
Table 11: Selected Study Population Demographics Baseline Status Letrozole Tamoxifen N=458 N=458 Stage of Disease IIIB 6% 7% IV 93% 92% Receptor Status ER and PgR Positive 38% 41% ER or PgR Positive 26% 26% Both Unknown 34% 33% ER -or PgR -/Other Unknown <1% 0 Previous Antiestrogen Therapy Adjuvant 19% 18% None 81% 82% Dominant Site of Disease Soft Tissue 25% 25% Bone 32% 29% Viscera 43% 46% Letrozole was superior to tamoxifen in TTP and rate of objective tumor response (see Table 12).
Table 12 summarizes the results of the trial, with a total median follow-up of approximately 32 months. (All analyses are unadjusted and use 2-sided P-values.)
Table 12: Results of First-Line Treatment of Advanced Breast Cancer Letrozole Tamoxifen Hazard or Odds 2.5 mg 20 mg Ratio (95% CI) N=453 N=454 P-Value (2-Sided) Median Time to Progression 9.4 months 6.0 months 0.72 (0.62, 0.83) 1 P<0.0001 Objective Response Rate (CR + PR) 145 (32%) 95 (21%) 1.77 (1.31, 2.39) 2 P=0.0002 (CR) 42 (9%) 15 (3%) 2.99 (1.63, 5.47) 2 P=0.0004 Duration of Objective Response Median 18 months 16 months (N=145) (N=95) Overall Survival 35 months 32 months (N=458) (N=458) P=0.5136 3 1Hazard ratio
2Odds ratio
3Overall log-rank test
Figure 2 shows the Kaplan-Meier curves for TTP.
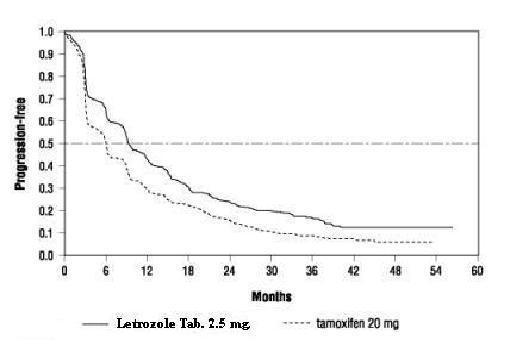
Table 13 shows results in the subgroup of women who had received prior antiestrogen adjuvant therapy, Table 14 , results by disease site and Table 15, the results by receptor status.
Table 13: Efficacy in Patients Who Received Prior Antiestrogen Therapy Variable Letrozole Tamoxifen 2.5 mg 20 mg N=84 N=83 Median Time to Progression(95% CI) 8.9 months (6.2, 12.5) 5.9 months (3.2, 6.2) Hazard Ratio for TTP (95% CI) 0.60 (0.43, 0.84) Objective Response Rate (CR + PR) 22 (26%) 7 (8%) Odds Ratio for Response (95% CI) 3.85 (1.50, 9.60) Hazard ratio less than 1 or odds ratio greater than 1 favors letrozole; hazard ratio greater than 1 or odds ratio less than 1 favors tamoxifen.
Table 14: Efficacy by Disease Site Letrozole Tamoxifen 2.5 mg 20 mg Dominant Disease Site Soft Tissue: N=113 N=115 Median TTP 12.1 months 6.4 months Objective Response Rate 50% 34% Bone: N=145 N=131 Median TTP 9.5 months 6.3 months Objective Response Rate 23% 15% Viscera: N=195 N=208 Median TTP 8.3 months 4.6 months Objective Response Rate 28% 17% Table 15: Efficacy by Receptor Status Variable Letrozole Tamoxifen 2.5 mg 20 mg Receptor Positive N=294 N=305 Median Time to Progression (95% CI) 9.4 months (8.9, 11.8) 6.0 months (5.1, 8.5) Hazard Ratio for TTP (95% CI) 0.69 (0.58, 0.83) Objective Response Rate (CR+PR) 97 (33%) 66 (22%) Odds Ratio for Response 95% CI) 1.78 (1.20, 2.60) Receptor Unknown N=159 N=149 Median Time to Progression (95% CI) 9.2 months (6.1, 12.3) 6.0 months (4.1, 6.4) Hazard Ratio for TTP (95% CI) 0.77 (0.60, 0.99) Objective Response Rate (CR+PR) 48 (30%) 29 (20%) Odds Ratio for Response (95% CI) 1.79 (1.10, 3.00) Hazard ratio less than 1 or odds ratio greater than 1 favors letrozole; hazard ratio greater than 1 or odds ratio less than 1 favors tamoxifen.
Figure 3: shows the Kaplan-Meier curves for survival.
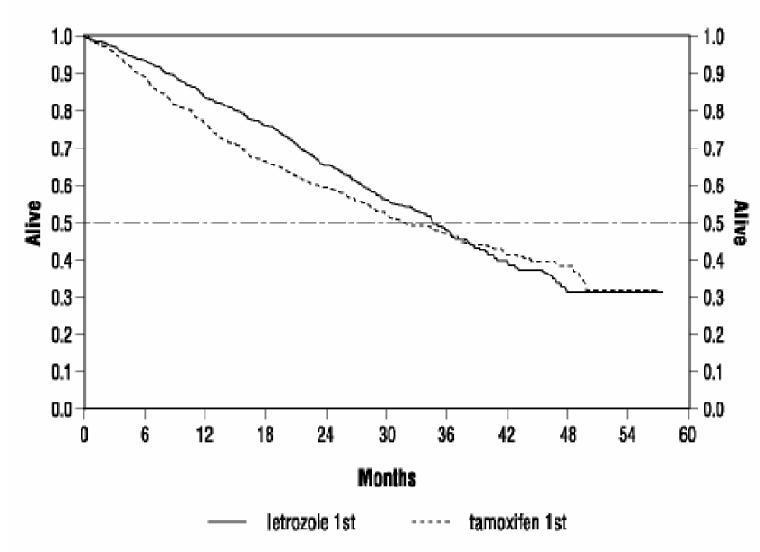
Legend: Randomized letrozole: n=458, events 57%, median overall survival 35 months (95% CI 32 to 38 months)
Randomized tamoxifen: n=458, events 57%, median overall survival 32 months (95% CI 28 to 37 months)
Overall log-rank P=0.5136 (i.e., there was no significant difference between treatment arms in overall survival).
The median overall survival was 35 months for the letrozole group and 32 months for the tamoxifen group, with a P-value 0.5136. Study design allowed patients to cross over upon progression to the other therapy. Approximately 50% of patients crossed over to the opposite treatment arm and almost all patients who crossed over had done so by 36 months. The median time to crossover was 17 months (letrozole to tamoxifen) and 13 months (tamoxifen to letrozole). In patients who did not cross over to the opposite treatment arm, median survival was 35 months with letrozole (n=219, 95% Cl 29 to 43 months) vs. 20 months with tamoxifen (n=229, 95% Cl 16 to 26 months).
14.5 Second-Line Treatment of Advanced Breast Cancer
Letrozole was initially studied at doses of 0.1 mg to 5.0 mg daily in six noncomparative trials (AR/BC1, P01, AR/ST1, AR/PS1, AR/ES1 and NJO-03) in 181 postmenopausal estrogen/progesterone receptor positive or unknown advanced breast cancer patients previously treated with at least antiestrogen therapy. Patients had received other hormonal therapies and also may have received cytotoxic therapy. Eight (20%) of forty patients treated with letrozole 2.5 mg daily in trials achieved an objective tumor response (complete or partial response).
Two large randomized, controlled, multinational (predominantly European) trials (AR/BC2, AR/BC3) were conducted in patients with advanced breast cancer who had progressed despite antiestrogen therapy. Patients were randomized to letrozole 0.5 mg daily, letrozole 2.5 mg daily, or a comparator [megestrol acetate 160 mg daily in one study (AR/BC2); and aminoglutethimide 250 mg twice a day with corticosteroid supplementation in the other study (AR/BC3)]. In each study over 60% of the patients had received therapeutic antiestrogens, and about one-fifth of these patients had an objective response. The megestrol acetate controlled study was double-blind; the other study was open label. Selected baseline characteristics for each study are shown in Table 16.
Table 16:Selected Study Population Demographics Parameter megestrol acetate aminoglutethimide study study No. of Participants 552 557 Receptor Status ER/PR Positive 57% 56% ER/PR Unknown 43% 44% Previous Therapy Adjuvant Only 33% 38% Therapeutic +/- Adj. 66% 62% Sites of Disease Soft Tissue 56% 50% Bone 50% 55% Viscera 40% 44% Confirmed objective tumor response (complete response plus partial response) was the primary endpoint of the trials. Responses were measured according to the Union Internationale Contre le Cancer (UICC) criteria and verified by independent, blinded review. All responses were confirmed by a second evaluation 4 to 12 weeks after the documentation of the initial response.
Table 17 shows the results for the first trial (AR/BC2), with a minimum follow-up of 15 months, that compared letrozole 0.5 mg, letrozole 2.5 mg, and megestrol acetate 160 mg daily. (All analyses are unadjusted.)
Table 17: Megestrol Acetate Study Results Letrozole Letrozole megestrol 0.5 mg 2.5 mg acetate N=188 N=174 N=190 Objective Response (CR + PR) 22 (11.7%) 41 (23.6%) 31 (16.3%) Median Duration of Response 552 days (Not reached) 561 days Median Time to Progression 154 days 170 days 168 days Median Survival 633 days 730 days 659 days Odds Ratio for Response letrozole 2.5 mg: letrozole 0.5 mg=2.33 letrozole 2.5 mg: megestrol=1.58 (95% CI: 1.32, 4.17); P=0.004* (95% CI: 0.94, 2.66); P=0.08* Relative Risk of Progression letrozole 2.5 mg: letrozole 0.5 mg=0.81 letrozole 2.5 mg : megestrol=0.77 (95% CI: 0.63, 1.03); P=0.09* (95% CI: 0.60, 0.98); P=0.03* * Two-sided P-value
The Kaplan-Meier curves for progression for the megestrol acetate study are shown in Figure 4.
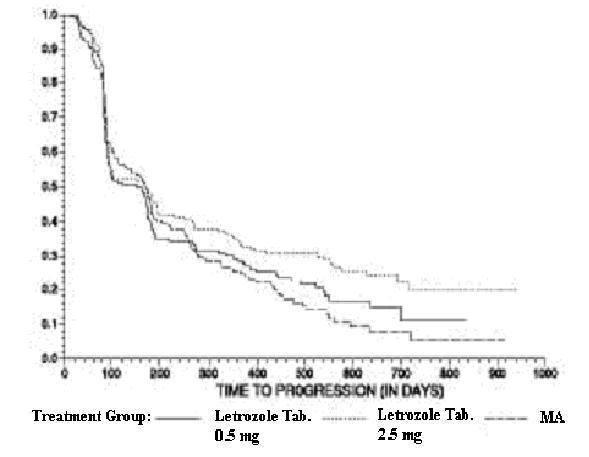
The results for the study comparing letrozole to aminoglutethimide (AR/BC3), with a minimum follow-up of 9 months, are shown in Table 18. (Unadjusted analyses are used).
Table 18: Aminoglutethimide Study Results Letrozole Letrozole 0.5 mg 2.5 mg aminoglutethimide N=193 N=185 N=179 Objective Response (CR+PR) 34 (17.6%) 34 (18.4%) 22 (12.3%) Median Duration of Response 619 days 706 days 450 days Median Time to Progression 103 days 123 days 112 days Median Survival 636 days 792 days 592 days Odds Ratio for Response letrozole 2.5 mg: letrozole 2.5 mg: letrozole 0.5 mg=1.05 aminoglutethimide=1.61 (95% CI: 0.62, 1.79); P=0.85* (95% CI: 0.90, 2.87); P=0.11* Relative Risk of Progression letrozole 2.5 mg: letrozole 2.5 mg: letrozole 0.5 mg=0.86 aminoglutethimide=0.74 (95% CI: 0.68, 1.11); P=0.25* (95% CI: 0.57, 0.94); P=0.02* *Two-sided P-value
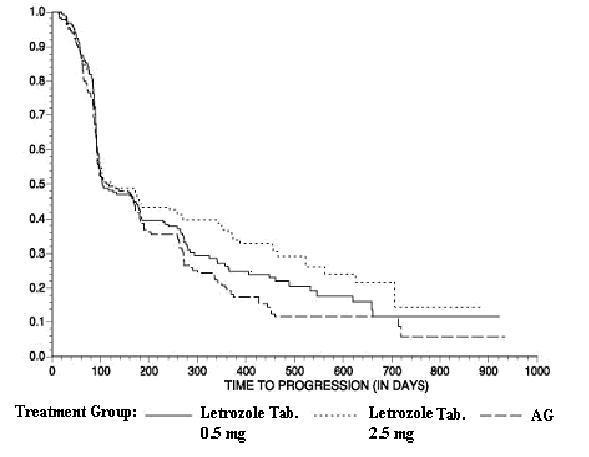
The Kaplan-Meier curves for progression for the aminoglutethimide study is shown in Figure 5.
- tamoxifen for 5 years
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Packaged in HDPE bottles and unit dose blister package.
2.5 mg tablets – yellow round, biconvex, film coated tablets imprinted with 'LT' on one side and plain on other side.
Bottles of 30 tablets with a child-resistant closure ...................................................................................NDC 16729-034-10
Bottles of 90 tablets with a child-resistant closure ...................................................................................NDC 16729-034-15
Bottles of 100 tablets with a child-resistant closure..................................................................................NDC 16729-034-01
Bottles of 500 tablets.................................................................................................................NDC 16729-034-16
Bottles of 1000 tablets................................................................................................................NDC 16729-034-17
Unit dose blister package 100 (10x10)...................................................................................................NDC 16729-034-43
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during letrozole therapy and for at least 3 weeks after the last dose. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with letrozole tablets [see Warnings and Precautions ( 5.6), and Use in Specific Populations ( 8.1), ( 8.3)]
Lactation
Advise women not to breastfeed during letrozole tablets treatment and for at least 3 weeks after the last dose [see Use in Specific Populations ( 8.2) .
Infertility
Advise females and males of reproductive potential of the potential for reduced fertility from letrozole tablets [see Use in Specific Populations ( 8.3) .
Fatigue and Dizziness
Since fatigue and dizziness have been observed with the use of letrozole tablets and somnolence was uncommonly reported, caution is advised when driving or using machinery.
Bone EffectsConsideration should be given to monitoring bone mineral density.
Manufactured For:
Accord Healthcare, Inc.,
8041 Arco Corporate Drive,
Suite 200,
Raleigh, NC 27617,
USA.Manufactured By:
Intas Pharmaceuticals Limited,
Plot No. : 457, 458,
Village – Matoda,
Bavla Road, Ta.- Sanand,
Dist.- Ahmedabad – 382 210.
India.10 2293 2 6025926
Issued October 2023
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LETROZOLE
letrozole tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:16729-034 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LETROZOLE (UNII: 7LKK855W8I) (LETROZOLE - UNII:7LKK855W8I) LETROZOLE 2.5 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, CORN (UNII: O8232NY3SJ) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color yellow Score no score Shape ROUND (biconvex) Size 6mm Flavor Imprint Code LT Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:16729-034-10 30 in 1 BOTTLE; Type 0: Not a Combination Product 06/02/2011 2 NDC:16729-034-15 90 in 1 BOTTLE; Type 0: Not a Combination Product 12/31/2014 3 NDC:16729-034-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2026 4 NDC:16729-034-16 500 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2026 5 NDC:16729-034-17 1000 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2026 6 NDC:16729-034-43 100 in 1 CARTON 01/01/2026 6 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090934 06/02/2011 Labeler - Accord Healthcare, Inc. (604222237) Establishment Name Address ID/FEI Business Operations Intas Pharmaceuticals Ltd. 725927649 manufacture(16729-034) , analysis(16729-034)

