Label: OJEMDA- tovorafenib kit
OJEMDA- tovorafenib tablet, film coated


-
NDC Code(s):
82950-001-04,
82950-001-06,
82950-001-16,
82950-001-20, view more82950-001-24, 82950-012-01, 82950-112-10
- Packager: Day One Biopharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated May 23, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OJEMDA safely and effectively. See full prescribing information for OJEMDA.
OJEMDA (tovorafenib) tablets, for oral use
OJEMDA (tovorafenib) for oral suspension
Initial U.S. Approval: 2024INDICATIONS AND USAGE
OJEMDA is a kinase inhibitor indicated for the treatment of patients 6 months of age and older with relapsed or refractory pediatric low-grade glioma (LGG) harboring a BRAF fusion or rearrangement, or BRAF V600 mutation. (1)
This indication is approved under accelerated approval based on response rate and duration of response [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
DOSAGE AND ADMINISTRATION
- Confirm the presence of BRAF fusion or rearrangement, or BRAF V600 mutation prior to initiation of treatment with OJEMDA. (2.1)
- Recommended dosage of OJEMDA is based on body surface area (see Tables 1 and 2). (2.3)
- Administer OJEMDA orally, once weekly, with or without food. (2.3, 2.4).
- Tablets: Swallow tablets whole with water. Do not chew, cut, or crush. (2.4)
- For Oral Suspension: See full prescribing information for preparation and administration instructions. (2.4)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Hemorrhage: Major hemorrhagic events can occur during treatment with OJEMDA. Withhold, resume at reduced dose, or permanently discontinue based on severity. (5.1)
- Skin Toxicity Including Photosensitivity: Advise patients to monitor for new or worsening skin reactions. Advise patients to limit direct ultraviolet exposure and use precautionary measures such as sunscreen, sunglasses and/or protective clothing during treatment with OJEMDA. Withhold, reduce the dose or permanently discontinue based on severity. (5.2)
- Hepatotoxicity: OJEMDA can cause hepatotoxicity. Monitor liver function tests prior to administration and during treatment. Withhold, reduce the dose or permanently discontinue based on severity. (5.3)
- Effect on Growth: Reductions in growth velocity have been reported. Routinely monitor growth in pediatric patients. (5.4)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise of the potential risk to a fetus and to use effective nonhormonal contraception. (5.5, 8.1, 8.3)
- NF1 Associated Tumors: Increased tumor growth may occur with OJEMDA. (5.6, 13.2)
ADVERSE REACTIONS
The most common adverse reactions (≥30%) were rash, hair color changes, fatigue, viral infection, vomiting, headache, hemorrhage, pyrexia, dry skin, constipation, nausea, dermatitis acneiform, and upper respiratory tract infection. (6)
The most common Grade 3 or 4 laboratory abnormalities (≥2%) were decreased phosphate, decreased hemoglobin, increased creatine phosphokinase, increased alanine aminotransferase, decreased albumin, decreased lymphocytes, decreased leukocytes, increased aspartate aminotransferase, decreased potassium, and decreased sodium.
To report SUSPECTED ADVERSE REACTIONS, contact Day One Biopharmaceuticals at toll-free phone # 1-877-204-2820 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Moderate and Strong CYP2C8 Inhibitors: Avoid coadministration with OJEMDA. (7.1).
- Moderate and Strong CYP2C8 Inducers: Avoid coadministration with OJEMDA. (7.1).
- Certain CYP3A Substrates: Avoid coadministration of OJEMDA with CYP3A substrates where minimal concentration changes can cause reduced efficacy. (7.2).
- Hormonal contraceptives: Avoid coadministration with OJEMDA. (7.2).
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Testing Before Initiating OJEMDA
2.3 Recommended Dosage
2.4 Administration
2.5 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
5.2 Skin Toxicity Including Photosensitivity
5.3 Hepatotoxicity
5.4 Effect on Growth
5.5 Embryo-Fetal Toxicity
5.6 NF1 Associated Tumors
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on OJEMDA
7.2 Effects of OJEMDA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
OJEMDA is indicated for the treatment of patients 6 months of age and older with relapsed or refractory pediatric low-grade glioma (LGG) harboring a BRAF fusion or rearrangement, or BRAF V600 mutation.
This indication is approved under accelerated approval based on response rate and duration of response [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Confirm the presence of BRAF fusion or rearrangement, or BRAF V600 mutation prior to initiation of treatment with OJEMDA [see Warnings and Precautions (5.6), Clinical Studies (14)].
An FDA approved test for the detection of BRAF fusion or rearrangement, or BRAF V600 mutation in relapsed or refractory pediatric LGG is not currently available.
2.2 Recommended Testing Before Initiating OJEMDA
Before initiating OJEMDA, evaluate liver function tests, including ALT, AST and bilirubin [see Warnings and Precautions (5.3)].
2.3 Recommended Dosage
The recommended dosage of OJEMDA based on body surface area (BSA) is 380 mg/m2 orally once weekly (the maximum recommended dosage is 600 mg orally once weekly) with or without food [see Administration (2.4) and Clinical Pharmacology (12.3)] until disease progression or intolerable toxicity. OJEMDA may be administered as an immediate release tablet (see Table 1) or as an oral suspension (see Table 2). A recommended dosage for patients with BSA less than 0.3 m2 has not been established.
Table 1 Recommended OJEMDA Tablets Dosage Based on Body Surface Area Body Surface Area (m2) Recommended Dosage 0.30-0.89 Administer OJEMDA oral suspension once weekly
(see Table 2)0.90-1.12 400 mg once weekly 1.13-1.39 500 mg once weekly ≥ 1.40 600 mg once weekly Table 2 Recommended Dosage for OJEMDA for Oral Suspension Based on Body Surface Area Body Surface Area (m2) Dose Volume (mL)* Dosage - *
- OJEMDA for oral suspension has a concentration of 25 mg/mL. Each bottle of OJEMDA for oral suspension delivers 300 mg/12 mL.
0.30-0.35 5 125 mg once weekly 0.36-0.42 6 150 mg once weekly 0.43-0.48 7 175 mg once weekly 0.49-0.54 8 200 mg once weekly 0.55-0.63 9 225 mg once weekly 0.64-0.77 11 275 mg once weekly 0.78-0.83 12 300 mg once weekly 0.84-0.89 14 350 mg once weekly 0.90-1.05 15 375 mg once weekly 1.06-1.25 18 450 mg once weekly 1.26-1.39 21 525 mg once weekly ≥1.40 24 600 mg once weekly Continue once weekly dosing until disease progression or intolerable toxicity.
2.4 Administration
- Take OJEMDA at a regularly scheduled time once weekly.
- OJEMDA may be taken with or without food [see Clinical Pharmacology (12.3)].
If a dose is missed by:
- 3 days or less, take the missed dose as soon as possible, and take the next dose on its regularly scheduled day.
- more than 3 days, skip the missed dose and take the next dose on its regularly scheduled day.
If vomiting occurs immediately after taking a dose, repeat that dose.
OJEMDA for oral suspension
- Prior to first time use of OJEMDA for oral suspension, ensure that caregivers (and if appropriate, patients) read and understand the "Instructions for Use" before preparing, measuring, and administering OJEMDA.
Preparation and Administration
- Reconstitute the powder in each supplied bottle with exactly 14 mL of room temperature water to form the OJEMDA for oral suspension. After reconstitution each mL contains 25 mg of tovorafenib. Product foaming after reconstitution reduces the deliverable volume.
- Each bottle delivers 300 mg of tovorafenib in 12 mL. For doses greater than 300 mg, reconstitute two bottles to achieve the dose. Split the dose as equally as possible between the two bottles (e.g., 6 mL and 7 mL for a 325 mg dose).
- Administer OJEMDA for oral suspension using the supplied oral dosing syringe or feeding tube (minimum 12 French) immediately after preparation.
- If the OJEMDA for oral suspension is not administered within 15 minutes after preparation, instruct the patient to discard it.
2.5 Dosage Modifications for Adverse Reactions
The recommended dosage reductions for adverse reactions for OJEMDA tablets are provided in Table 3 and OJEMDA for oral suspension in Table 4.
Table 3 OJEMDA Tablets: Recommended Dosage Reductions for Adverse Reactions BSA (m2) First Dosage Reduction Second Dosage Reduction 0.30-1.12 Administer the oral suspension once weekly (see Table 4) 1.13-1.39 400 mg once weekly Administer OJEMDA oral suspension once weekly
(see Table 4)≥1.40 500 mg once weekly 400 mg once weekly Table 4 OJEMDA for Oral Suspension: Recommended Dosage Reductions for Adverse Reactions BSA (m2) First Dosage Reduction Second Dosage Reduction Volume (mL) Dose (mg) Volume (mL) Dose (mg) 0.30-0.35 4 100 mg once weekly 3 75 mg once weekly 0.36-0.42 5 125 mg once weekly 4 100 mg once weekly 0.43-0.48 6 150 mg once weekly 5 125 mg once weekly 0.49-0.54 7 175 mg once weekly 6 150 mg once weekly 0.55-0.63 8 200 mg once weekly 6 150 mg once weekly 0.64-0.77 9 225 mg once weekly 8 200 mg once weekly 0.78-0.83 10 250 mg once weekly 8 200 mg once weekly 0.84-0.89 12 300 mg once weekly 10 250 mg once weekly 0.90-1.05 13 325 mg once weekly 11 275 mg once weekly 1.06-1.25 15 375 mg once weekly 13 325 mg once weekly 1.26-1.39 18 450 mg once weekly 15 375 mg once weekly ≥1.40 20 500 mg once weekly 16 400 mg once weekly The recommended dosage modifications of OJEMDA for adverse reactions are in Table 5.
Table 5 Recommended Dosage Modifications for Adverse Reactions Severity of ADR* Dosage Modification† Hemorrhage [see Warnings and Precautions (5.1)] - Intolerable Grade 2
- Any Grade 3
Withhold OJEMDA. - If improved to Grade 0-1, resume at lower dosage.
- If not improved, consider permanent discontinuation of OJEMDA.
- First occurrence of any Grade 4
Withhold OJEMDA. - If improved to Grade 0-1, resume at lower dosage.
- Permanently discontinue OJEMDA.
- Recurrent Grade 4
Permanently discontinue OJEMDA. Skin Toxicity including Photosensitivity [see Warnings and Precautions (5.2)] - Intolerable Grade 2
- Grade 3 or 4
Withhold OJEMDA. - If improved to Grade 0-1, resume at lower dosage.
- If not improved, consider permanent discontinuation of OJEMDA.
Hepatotoxicity [see Warnings and Precautions (5.3)] - Grade 3 AST or ALT
- Grade 3 bilirubin
Withhold OJEMDA.
If improved to Grade ≤ 2 or baseline, resume as follows:- If laboratory abnormality resolves within 8 days, resume OJEMDA at the same dose.
- If laboratory abnormality does not resolve within 8 days, resume OJEMDA at lower dosage.
- First occurrence of any Grade 4
Withhold OJEMDA. - If improved to Grade 0-1, resume at lower dosage.
- Permanently discontinue OJEMDA
- Recurrent Grade 4
Permanently discontinue OJEMDA. Other Adverse Reactions [see Adverse Reactions (6.1)] - Intolerable Grade 2
- Any Grade 3
Withhold OJEMDA. - If improved to Grade 0-1, resume at lower dosage.
- If not improved, consider permanent discontinuation of OJEMDA.
- First occurrence of any Grade 4
Withhold OJEMDA. - If improved to Grade 0-1, resume at lower dosage.
- Permanently discontinue OJEMDA.
- Recurrent Grade 4
Permanently discontinue OJEMDA. -
3 DOSAGE FORMS AND STRENGTHS
Tablets:
- 100 mg: orange, film-coated, oval tablets debossed with "100" on one side and "D101" on the opposite side. Each tablet contains 100 mg of tovorafenib.
For Oral Suspension:
- 25 mg/mL: white to off white powder. After reconstitution, each mL of strawberry flavored tovorafenib suspension contains 25 mg of tovorafenib. Each bottle delivers 300 mg of tovorafenib in 12 mL.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
Hemorrhage, including major hemorrhage defined as symptomatic bleeding in a critical area or organ, can occur with OJEMDA. In the pooled safety population [see Adverse Reactions (6.1)], hemorrhagic events occurred in 37% of patients, including epistaxis in 26% and intratumoral hemorrhage in 9%. Serious events of bleeding occurred in 5% of patients including Grade 5 tumor hemorrhage in 1 patient (0.6%). OJEMDA was permanently discontinued for hemorrhage in 2% of patients. Advise patients and caregivers of the risk of hemorrhage during treatment with OJEMDA. Monitor for signs and symptoms of hemorrhage and evaluate as clinically indicated. Withhold and resume at reduced dose upon improvement, or permanently discontinue based on severity [see Dosage and Administration (2.5)].
5.2 Skin Toxicity Including Photosensitivity
OJEMDA can cause rash, including maculopapular rash and photosensitivity. In the pooled safety population [see Adverse Reactions (6.1)], rash occurred in 67% of patients treated with OJEMDA, including Grade 3 rash in 12%. Rash resulted in dose interruption in 15% of patients and dose reduction in 7% of patients. OJEMDA was permanently discontinued due to rash in 1% of patients (n=2). In the pooled safety population, dermatitis acneiform occurred in 26% of patients treated with OJEMDA, including Grade 3 dermatitis acneiform in 0.6% of patients (n=1). Dose reduction was required in 2% of patients (n=3) due to dermatitis acneiform. Monitor for new or worsening skin reactions. Consider dermatologic consultation and initiate supportive care as clinically indicated. Withhold, reduce the dose, or permanently discontinue OJEMDA based on severity of adverse reaction [see Dosage and Administration (2.5)].
Photosensitivity
In the pooled safety population [see Adverse Reactions (6.1)], photosensitivity occurred in 12% of patients treated with OJEMDA, including Grade 3 events in 0.6% of patients (n=1). Advise patients to use precautionary measures against ultraviolet exposure such as use of sunscreen, sunglasses, and/or protective clothing during treatment with OJEMDA. Withhold, reduce the dose, or permanently discontinue OJEMDA based on severity of adverse reaction [see Dosage and Administration (2.5)].
5.3 Hepatotoxicity
OJEMDA can cause hepatotoxicity. In the pooled safety population [see Adverse Reactions (6.1)], increased alanine aminotransferase (ALT) occurred in 42% and increased aspartate aminotransferase (AST) occurred in 74%, including Grade 3 ALT in 4% and increased AST in 2% of patients treated with OJEMDA. The median time to onset of increased ALT or AST was 14 days (range: 3 to 280 days). Increased ALT or AST leading to dose interruption occurred in 5% of patients and dose reductions were required in 1.2% of patients. Increased bilirubin occurred in 23% of patients, including Grade 3 increased bilirubin in 0.6% of patients (n=1) treated with OJEMDA. Hyperbilirubinemia leading to dose discontinuation occurred in a single adult patient with an advanced non-CNS solid tumor.
Monitor liver function tests, including ALT, AST and bilirubin, before initiation of OJEMDA, one month after initiation and then every three months thereafter and as clinically indicated. Withhold and resume at the same or reduced dose upon improvement, or permanently discontinue OJEMDA based on the severity [see Dosage and Administration (2.5)].
5.4 Effect on Growth
OJEMDA can cause reductions in growth velocity. In FIREFLY-1 [see Adverse Reactions (6.1)], treatment-emergent adverse effects on growth occurred in 15% of patients 18 years of age or younger, including Grade 3 events in 5% of patients. OJEMDA was permanently discontinued for reduction in growth velocity in 2% of patients (n=2). Growth velocity recovered after interruption of treatment with OJEMDA. Routinely monitor patient growth during treatment with OJEMDA [see Adverse Reactions (6), Use in Specific Populations (8.4)].
5.5 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, OJEMDA may cause fetal harm when administered to a pregnant woman. Tovorafenib was embryo lethal in rats at doses approximately 0.8-fold the human exposure at the recommended dose based on area under the curve (AUC). Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective nonhormonal contraception during treatment with OJEMDA and for 28 days after the last dose, since OJEMDA can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)]. Advise male patients with female partners of reproductive potential to use effective nonhormonal contraception during treatment with OJEMDA and for 2 weeks after the last dose [see Use in Specific Populations (8.1, 8.3)].
5.6 NF1 Associated Tumors
Based on nonclinical data in NF1 models without BRAF alterations, tovorafenib may promote tumor growth in patients with NF1 tumors [see Nonclinical Toxicology (13.2)]. Confirm evidence of a BRAF alteration prior to initiation of treatment with OJEMDA.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hemorrhage [see Warnings and Precautions (5.1)]
- Skin Toxicity Including Photosensitivity [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Effect on Growth [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety population described in WARNINGS AND PRECAUTIONS reflects exposure to OJEMDA taken orally once weekly at a dose based on body surface area [see Clinical Studies (14)] in 140 patients with relapsed or refractory pediatric LGG or advanced solid tumors harboring a RAF alteration and a flat dose of 600 mg in 32 adult patients with advanced solid tumors until disease progression or intolerable toxicity. Among 172 patients treated with OJEMDA, 86% were exposed for 6 months or longer and 49% were exposed for 1 year or longer.
Pediatric Low-grade Glioma
The safety of OJEMDA was evaluated in 137 patients with relapsed or refractory pediatric LGG harboring a BRAF alteration in FIREFLY-1 (Arms 1 and 2) [see Clinical Studies (14)]. Patients received OJEMDA at a dose based on body surface area [see Dosage and Administration (2.3] orally once weekly until disease progression or intolerable toxicity.
The median age of patients was 9 years (range 1 to 24 years); 53% male; 58% White, 7% Asian, 2% Black or African American, 6% other races, 25% race was not reported; 2.9% were Hispanic or Latino; and 90% Karnofsky/Lansky performance status of 80 to 100.
Serious adverse reactions occurred in 45% of patients who received OJEMDA. Serious adverse reactions in >2% of patients included viral infection (9%), pneumonia (4%), and sepsis (4%). A fatal adverse reaction of tumor hemorrhage occurred in 1 patient (1%).
Permanent discontinuation of OJEMDA due to an adverse reaction occurred in 7% of patients. Adverse reactions which resulted in permanent discontinuation of OJEMDA in more than one patient were tumor hemorrhage and reduction in growth velocity.
Dosage interruptions of OJEMDA due to an adverse reaction occurred in 57% of patients. Adverse reactions which required dose interruption in ≥5% of patients included rash, pyrexia, vomiting, and hemorrhage.
Dosage reductions of OJEMDA due to an adverse reaction occurred in 24% of patients. Adverse reactions which required dose reduction in ≥2% of patients included rash, and fatigue.
The most common adverse reactions (≥30%) were rash, hair color changes, fatigue, viral infection, vomiting, headache, hemorrhage, pyrexia, dry skin, constipation, nausea, dermatitis acneiform, and upper respiratory tract infection.
The most common Grade 3 or 4 laboratory abnormalities (≥2%) were decreased phosphate, decreased hemoglobin, increased creatine phosphokinase, increased alanine aminotransferase, decreased albumin, decreased lymphocytes, decreased leukocytes, increased aspartate transferase, decreased potassium, and decreased sodium.
Table 6 and Table 7 present adverse reactions and laboratory abnormalities, respectively, identified in FIREFLY-1 (Arms 1 and 2).
Table 6 Adverse Reactions (≥20%) in Patients with Pediatric LGG Who Received OJEMDA in FIREFLY-1 (Arms 1 and 2) Adverse Reaction OJEMDA
(N=137)All Grades (%) Grade 3 or 4 (%) - *
- Includes terms erythema multiforme, eczema, rash erythematous, rash macular, rash follicular, rash pruritic, rash maculopapular, rash, rash popular, rash pustular, skin exfoliation, drug eruption, dermatitis, dermatitis bullous.
- †
- Includes terms lip edema, periorbital edema, edema peripheral, localized edema, face edema, vulval edema.
- ‡
- Includes terms viral infection, rhinovirus infection, enterovirus infection, viral upper respiratory tract infection, enterocolitis viral, oral herpes, gastroenteritis viral, influenza, influenza like illness, respiratory syncytial virus infection, enterovirus infection, coronavirus infection, COVID-19, SARS-COV-2 test positive, herpes simplex, parainfluenza virus infection, adenoviral upper respiratory infection, viraemia, adenovirus infection, conjunctivitis viral, eye infection viral, metapneumovirus infection, parvovirus infection, respiratory syncytial virus bronchiolitis, respiratory tract infection viral, viral pharyngitis, viral rhinitis, viral tonsillitis.
- §
- Includes terms retching, hematemesis.
- ¶
- Includes terms colitis, enterocolitis.
- #
- Includes terms mouth ulceration, mucosal inflammation, aphthous ulcer, cheilitis.
- Þ
- Includes terms tumor hemorrhage, gastrointestinal hemorrhage, subdural hemorrhage, epistaxis, intracranial tumor hemorrhage, upper gastrointestinal hemorrhage, lower gastrointestinal hemorrhage, vaginal hemorrhage, gingival bleeding, post procedural hemorrhage, hemoptysis, anal hemorrhage.
- ß
- Includes one Grade 5 event.
Skin and Subcutaneous Tissue Disorders Rash* 77 12 Hair color changes 76 0 Dry skin 36 0 Dermatitis acneiform 31 1 Pruritus 26 1 General Disorders Fatigue 55 4 Pyrexia 39 4 Edema† 26 0 Infections and Infestations Viral infection‡ 55 7 Upper respiratory tract infection 31 1.5 Paronychia 26 1.5 Gastrointestinal Disorders Vomiting§ 50 4 Constipation 33 0 Nausea 33 0 Abdominal pain 28 0 Diarrhea¶ 22 1.5 Stomatitis# 20 0 Nervous system disorders Headache 45 1 Vascular Disorders HemorrhageÞ 42 5ß Other clinically important adverse reactions observed in less than 20% of patients treated with OJEMDA were reductions in growth velocity [see Warnings and Precautions (5.4)] and photosensitivity [see Warnings and Precautions (5.2)].
Table 7 Select Laboratory Abnormalities (≥20%) that Worsened from Baseline in Patients with Pediatric LGG Who Received OJEMDA in FIREFLY-1 (Arms 1 and 2) Laboratory Abnormality* OJEMDA† All Grades (%) Grade 3 or 4 (%) Hematology Decreased hemoglobin 90 15 Decreased lymphocytes 50 2 Decreased leukocytes 31 2 Increased lymphocytes 23 0 Chemistry Decreased phosphate 87 25 Increased AST 83 2 Increased creatine phosphokinase 83 11 Increased LDH 73 0 Decreased potassium 51 2 Increased ALT 50 5 Increased bilirubin 22 1 Decreased albumin 24 5 Decreased sodium 20 2 Increased creatine phosphokinase was a clinically important laboratory abnormality that worsened from baseline in patients treated with OJEMDA.
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on OJEMDA
Table 8 describes drug interactions where coadministration with another drug affects OJEMDA.
Table 8 Coadministration with Other Drugs that Affect the Use of OJEMDA Strong or Moderate CYP2C8 Inhibitors Prevention or Management - Avoid coadministration of OJEMDA with a strong or moderate CYP2C8 inhibitor.
Mechanism and Clinical Effect(s) - Tovorafenib is a CYP2C8 substrate. Strong or moderate CYP2C8 inhibitors are predicted to increase tovorafenib exposure based on a mechanistic understanding of its elimination [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions with OJEMDA.
Strong or Moderate CYP2C8 Inducers Prevention or Management - Avoid coadministration of OJEMDA with a strong or moderate CYP2C8 inducer.
Mechanism and Clinical Effect(s) - Tovorafenib is a CYP2C8 substrate. Strong or moderate CYP2C8 inducers are predicted to decrease tovorafenib exposure based on a mechanistic understanding of its elimination [see Clinical Pharmacology (12.3)], which may reduce the effectiveness of OJEMDA.
7.2 Effects of OJEMDA on Other Drugs
Table 9 describes drug interactions where coadministration with OJEMDA affects another drug.
Table 9 Coadministration with OJEMDA that Affects the Use of Other Drugs CYP3A Substrates Prevention or Management - Hormonal Contraceptives: Avoid coadministration of hormonal contraceptives with OJEMDA. If coadministration is unavoidable, use an additional effective nonhormonal contraceptive method during coadministration and for 28 days after discontinuation of OJEMDA.
- Other CYP3A Substrates: Avoid coadministration of OJEMDA with certain CYP3A substrates where minimal concentration changes may lead to serious therapeutic failures. If coadministration is unavoidable, monitor patients for loss of efficacy unless otherwise recommended in the Prescribing Information for CYP3A substrates.
Mechanism and Clinical Effect(s) - Tovorafenib is a CYP3A inducer.
- Tovorafenib is predicted to decrease exposure of certain CYP3A substrates where minimal concentration changes may lead to serious therapeutic failures [see Clinical Pharmacology (12.3)], which may reduce the effectiveness of these substrates.
- Coadministration with hormonal contraceptives (CYP3A substrate) may decrease progestin-x and ethinyl estradiol exposure, which may lead to contraceptive failure and/or an increase in breakthrough bleeding [see Warnings and Precautions (5.5), Use in Specific Populations (8.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], OJEMDA can cause fetal harm when administered to a pregnant woman. There are no available data on the use of OJEMDA in pregnant women. Oral administration of tovorafenib to pregnant rats during the period of organogenesis resulted in embryo lethality at exposures 0.8 times the human exposure at the recommended dose based on AUC (see Data). Advise pregnant women of the potential risk to a fetus. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study, once daily oral administration of tovorafenib to pregnant rats during the period of organogenesis from gestation days 7 through 17 at doses of 37.5, 75, and 150 mg/kg resulted in early resorptions and total litter loss at all doses. The dose of 37.5 mg/kg/day is approximately 0.8-fold the human exposure at the recommended dose based on AUC.
8.2 Lactation
Risk Summary
There are no data on the presence of tovorafenib or its metabolites in human milk, their effects on the breastfed child, or on milk production. Due to the potential for serious adverse reactions in breastfed children from OJEMDA, advise lactating women not to breastfeed during treatment with OJEMDA and for 2 weeks following the last dose.
8.3 Females and Males of Reproductive Potential
OJEMDA can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating OJEMDA [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective nonhormonal contraception during treatment with OJEMDA and for 28 days after the last dose. OJEMDA can render hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Infertility
Based on findings in animals, OJEMDA may impact fertility in males and females of reproductive potential. The effects on male fertility were reversible. The effects on female fertility were not reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of OJEMDA in pediatric patients 6 months of age and older with relapsed or refractory pediatric LGG harboring a BRAF fusion or rearrangement, or BRAF V600 mutation have been established based on data from a multicenter, open-label, single-arm clinical trial [see Clinical Studies (14)].
The efficacy of OJEMDA was evaluated in 76 patients with relapsed or refractory pediatric LGG. The safety of OJEMDA was evaluated in 137 patients with relapsed or refractory pediatric LGG in FIREFLY-1 (Arms 1 and 2). Of these 137 patients, 2% (n=3) were 6 month to < 2 years of age, 67% (n=92) were 2 years to < 12 years of age, and 31% (n=42) were >12 years of age [see Adverse Reactions (6.1)]. Cmax and AUC in pediatric patients aged 11 months to 17 years were within the range of values observed in adults given the same dose per body surface area.
The safety and effectiveness of OJEMDA in patients younger than 6 months of age have not been established.
Effect on Growth
Patients with pediatric LGG treated with OJEMDA for up to 24 months showed reductions from baseline in Z-scores for height compared to age and sex-matched normative data. Among 19 patients who experienced reductions in growth velocity who had hand radiographs taken to assess bone age, there was no evidence of premature closure of the epiphyseal growth plates or advancement of bone age. Patients followed after interruption of treatment with OJEMDA showed recovery of growth and increase in Z-scores. Monitor growth routinely during treatment [see Warnings and Precautions (5.4)].
8.6 Hepatic Impairment
No dose adjustment is recommended for patients with mild (bilirubin ≤ upper limit of normal (ULN) and aspartate aminotransferase (AST) > ULN or bilirubin > 1× to 1.5× ULN and any AST) hepatic impairment. OJEMDA has not been studied in patients with moderate (bilirubin > 1.5× to 3× ULN and any AST) to severe (bilirubin > 3× ULN and any AST) hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dose adjustment is recommended for patients with mild-to-moderate renal impairment (eGFR ≥ 30 mL/min/1.73 m2 calculated by Schwartz equation or MDRD equation). OJEMDA has not been studied in patients with severe renal impairment (eGFR <30 mL/min/1.73 m2) [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
OJEMDA contains tovorafenib, a kinase inhibitor. Tovorafenib has the molecular formula C17H12Cl2F3N7O2S and a molecular weight of 506.29. The chemical name for tovorafenib is 6-amino-5-chloro-N-[(1R)-1-[5-[[[5-chloro-4-(trifluoromethyl)-2-pyridinyl]amino]carbonyl]-2-thiazolyl]ethyl]-4-pyrimidinecarboxamide. Tovorafenib has the following chemical structure:
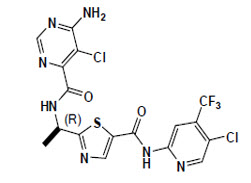
It is a white to off-white powder. The solubility of tovorafenib at 37ºC is ≤ 3 micrograms/mL from pH 1.2 to 8 in aqueous media.
OJEMDA (tovorafenib) tablets are supplied as 100 mg strength tablets for oral administration. Each tablet contains 100 mg tovorafenib and the following inactive ingredients: copovidone, colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and orange film coating (hypromellose, polyethylene glycol 8000, titanium dioxide, ferric oxide yellow, ferric oxide orange).
OJEMDA (tovorafenib) for oral suspension is a white to off white powder which produces a white suspension when reconstituted with water. Each mL of reconstituted tovorafenib suspension contains 25 mg of tovorafenib and the following inactive ingredients: artificial strawberry flavor, colloidal silicon dioxide, copovidone, maltodextrin, mannitol, microcrystalline cellulose, simethicone, sodium lauryl sulfate, and sucralose.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tovorafenib is a Type II RAF kinase inhibitor of mutant BRAF V600E, wild-type BRAF, and wild-type CRAF kinases.
Tovorafenib exhibited antitumor activity in cultured cells and xenograft tumor models harboring BRAF V600E and V600D mutations, and in a xenograft model harboring a BRAF fusion.
12.2 Pharmacodynamics
Exposure Response Relationships
Tovorafenib exposure is associated with reduction in height-for-age z-scores in pediatric patients. Reduced height-for-age risk persists during treatment with tovorafenib.
Higher tovorafenib exposure is associated with increased risk of skin rash, elevated liver enzymes (AST and ALT), and elevated creatine phosphokinase.
The exposure-response relationship for overall response rate based on RAPNO-LGG (Response Assessment in Pediatric Neuro-Oncology), and RANO-LGG (Response Assessment in Neuro-Oncology) were not clinically significant over the dosage range of 290 to 476 mg/m2 (0.76-1.25 times the approved recommended dosage) [see Dosage and Administration (2.3) and Clinical Studies (14)].
12.3 Pharmacokinetics
Tovorafenib pharmacokinetic parameters are presented as mean (CV%) unless otherwise indicated. Tovorafenib steady state maximum concentration (Cmax) is 6.9 µg/mL (23%) and the area under the concentration-time curve (AUC) is 508 µg*h/mL (31%). Time to reach steady state of tovorafenib is 12 days (33%). Tovorafenib exposure increases in a dose-proportional manner. No clinically significant tovorafenib accumulation occurs.
Absorption
Tovorafenib median (minimum, maximum) time to achieve peak plasma concentration (Tmax) is 3 hours (1.5, 4 hours), following a single dose with tablets or oral suspension.
Distribution
Tovorafenib apparent volume of distribution is 60 L/m2 (23%). Tovorafenib is 97.5% bound to human plasma proteins in vitro.
Elimination
Tovorafenib terminal half-life is approximately 56 hours (33%) and the apparent clearance is 0.7 L/h/m2 (31%).
Specific Populations
No clinically significant differences of tovorafenib were observed based on age (range: 1 to 94 years), sex, race (White, Black, Asian), mild hepatic impairment [bilirubin ≤ upper limit of normal (ULN) and AST > ULN or bilirubin > 1 to 1.5× ULN and any AST], and mild-to-moderate renal impairment (eGFR) ≥ 30 mL/min/1.73 m2 calculated by Schwartz equation or MDRD equation.
Drug Interaction Studies
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with tovorafenib have not been conducted.
Tovorafenib was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay. Tovorafenib was not genotoxic in cultured human lymphocytes without metabolic activation. Tovorafenib induced chromosomal aberrations in cultured human lymphocytes with metabolic activation at a single concentration in vitro. Tovorafenib was not genotoxic in an in vivo rat bone marrow micronucleus assay.
In a fertility and early embryonic development study in rats, animals were administered tovorafenib doses of 37.5, 75, or 150 mg/kg/day orally. Female animals, paired with untreated males, were dose for 14 days prior to pairing, during the mating period, and up to Gestation Day 6. Tovorafenib decreased the number of pregnancies, corpora lutea, and live embryos, as well as increased post-implantation losses at all doses. The dose of 37.5 mg/kg/day is approximately 0.8-fold the human exposure at the recommended dose based on AUC.
In repeat- dose toxicology studies in rats of up to 3 months duration, tovorafenib-related findings in female rats included reversible increased thickness of the vaginal mucosa, increased size and/or numbers of corpora hemorrhagicum and hemorrhage, and non-reversible cystic follicles, decreased corpora lutea, and interstitial cell hyperplasia were observed in ovaries at doses ≥ 50 mg/kg once every other day (approximately 0.4-fold the human exposure at the recommended dose based on AUC). In male rats, tovorafenib reduced weights of epididymis and testes, which correlated with reversible tubular degeneration/atrophy of the testes and reduced epididymal sperm at doses ≥ 50 mg/kg once every other day (approximately 0.3-fold the human exposure at the recommended dose based on AUC).
13.2 Animal Toxicology and/or Pharmacology
In vitro, tovorafenib increased phosphorylation of ERK at clinically relevant concentrations in cells with neurofibromatosis Type 1-loss of function (NF1-LOF) suggesting activation, rather than inhibition, of the MAP kinase pathway. In an NF1 genetically engineered mouse model of plexiform neurofibroma without BRAF alteration, tovorafenib did not have antitumor activity, and while not statistically significant, an increase in tumor volume was noted in 2/12 mice (approximately 17%).
-
14 CLINICAL STUDIES
The efficacy of OJEMDA was evaluated in a multicenter, open-label, single-arm clinical trial (FIREFLY-1; NCT04775485). Eligible patients (N=76) were required to have a relapsed or refractory pediatric low-grade glioma (LGG) harboring an activating BRAF alteration based on local laboratory testing. Patients were also required to have at least one measurable lesion as defined by RANO 2010 criteria. All patients had received at least one line of prior systemic therapy and had documented evidence of radiographic progression. Patients with tumors harboring additional activating molecular alteration(s) (e.g., IDH1/2 mutations, FGFR mutations, etc.) or patients with known or suspected diagnosis of neurofibromatosis type 1 (NF1) were excluded.
Patients received OJEMDA approximately 420 mg/m2 orally once weekly (range: 290 to 476 mg/m2, 0.76-1.25 times the approved recommended dosage) according to body surface area with a maximum dose of 600 mg until disease progression or unacceptable toxicity. Although the OJEMDA dosages administered in FIREFLY-1 were between 290 mg/m2 to 476 mg/m2, the recommended OJEMDA dosage is 380 mg/m2 orally once weekly because this dosage was determined to be safe and effective for the treatment of patients 6 months of age and older with relapsed or refractory pediatric LGG harboring a BRAF fusion or rearrangement, or BRAF V600 mutation [see Dosage and Administration (2.3)].
Tumor assessments were performed every 12 weeks.
The major efficacy outcome measure was overall response rate (ORR), defined as the proportion of patients with complete response (CR), partial response (PR), or minor response (MR) by independent review based on RAPNO-LGG (Response Assessment in Pediatric Neuro-Oncology) criteria. Additional efficacy outcome measures were duration of response, time to response, and ORR by independent review based on RANO-LGG (2011) criteria.
The efficacy population included 76 patients who had measurable disease at baseline and who received OJEMDA. The median age was 8.5 years (range 2 to 21 years); 53% were male; 53% White, 7% Asian, 2.6% Black or African American, 3.9% multiple races, 8% other race, 26% where race was not reported; 3.9% were Hispanic or Latino, and 93% had Karnofsky/Lansky performance status of 80 to 100. Patients received a median of 3 prior systemic regimens (range: 1 to 9). Forty-five patients (59%) received prior treatment with a MAP kinase pathway inhibitor. The most common tumor locations were the optic pathway (51%), deep midline structures (12%), brain stem (8%), cerebral hemisphere and cerebellum (7% each). Fifty-six patients (74%) had a KIAA1549:BRAF fusion, twelve patients (16%) had a V600E mutation, and eight patients (11%) had a BRAF alteration classified as "other" including BRAF duplication or BRAF rearrangement. Efficacy results are shown in Table 10.
Table 10 Efficacy Results Based on Independent Review in FIREFLY-1 (Arm-1) Efficacy Parameter RAPNO-LGG
N=76*Abbreviations: LGG = low-grade glioma; RAPNO = Response Assessment in Pediatric Neuro-Oncology; CI = confidence interval; NE = not estimable. Overall Response Rate ORR (95% CI)† 51% (40, 63) Complete Response (CR), n (%) 0 (0) Partial Response (PR), n (%) 28 (37) Minor Response (MR), n (%) 11 (14) Duration of Response (DoR) N=39 Median (95% CI)‡, Months 13.8 (11.3, NE) % with observed DoR ≥ 6 months 85% % with observed DoR ≥ 12 months 23% Among responders, the median time to response was 5.3 months (range 1.6, 11.2). In exploratory analyses of BRAF alteration status, the ORR was 52% among patients with BRAF fusion or rearrangement (n=64), and 50% among patients with BRAF V600E mutation (n=12), respectively. In exploratory analyses of prior therapies, the ORR was 49% among patients who had received prior MAPK-targeted therapy (n=45), and 55% among patients who had not received prior MAPK-targeted therapy (n=31).
Based on RANO-LGG (2011) criteria (n=76), the ORR was 53% [95% CI: (41, 64)], including 20 patients each with PR and MR, respectively.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
OJEMDA tablets:
100 mg: orange, film-coated, oval tablets debossed with "100" on one side and "D101" on the opposite side and supplied as follows:
4 blister cards (4 tablets each) per box, NDC 82950-001-16.
5 blister cards (4 tablets each) per box, NDC 82950-001-20.
4 blister cards (6 tablets each) per box, NDC 82950-001-24.
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Dispense product in the original package. Tablets should not be removed from blisters until immediately before use.
OJEMDA for oral suspension:
25 mg/mL: white to off white powder in a clear glass bottle, co-packaged with a press-in bottle adaptor and a 20 mL oral dosing syringe (NDC# 82950-012-01).
Each mL of reconstituted, strawberry flavored tovorafenib suspension contains 25 mg of tovorafenib. Each bottle delivers 300 mg of tovorafenib in 12 mL.
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Do not use if safety seal under cap is broken or missing.
Suspension must be used immediately after reconstitution.
Discard the bottle (including any unused portion) and syringe after dosing.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Hemorrhage
Advise patients that OJEMDA can cause bleeding and to contact their healthcare provider for signs or symptoms of bleeding [see Warnings and Precautions (5.1)].
Skin Toxicities
Advise patients that OJEMDA can cause skin toxicities and to contact their healthcare provider for worsening or intolerable rash [see Warnings and Precautions (5.2)].
Photosensitivity
Advise patients that OJEMDA can cause photosensitivity. Advise patients to limit direct ultraviolet exposure during treatment with OJEMDA. Recommend that patients use precautionary measures such as use of sunscreen, sunglasses, and/or protective clothing during treatment with OJEMDA [see Warnings and Precautions (5.2)].
Hepatotoxicity
Advise patients that OJEMDA can cause liver toxicity and to contact their healthcare provider for signs or symptoms of liver dysfunction. Advise patients that serial testing of serum liver tests (ALT, AST, bilirubin) is recommended during treatment with OJEMDA [see Warnings and Precautions (5.3)].
Effect on Growth
Advise patients and caregivers that treatment with OJEMDA may cause a reduction in growth velocity, and that growth will be monitored during treatment with OJEMDA [see Warnings and Precautions (5.4)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus [see Warnings and Precautions (5.5), Use in Specific Populations (8.1, 8.3)].
- Advise females to inform their healthcare provider of a known or suspected pregnancy during treatment with OJEMDA.
- Advise females of reproductive potential to use effective nonhormonal contraception during treatment and for 28 days after discontinuation of treatment with OJEMDA.
- Advise male patients with female partners of reproductive potential to use effective nonhormonal contraception during treatment with OJEMDA and for 2 weeks after the last dose.
Lactation
Advise women not to breastfeed during treatment with OJEMDA and for 2 weeks after the last dose of OJEMDA [see Use in Specific Populations (8.2)].
Infertility
Advise males and females of reproductive potential of the potential risk for impaired fertility with OJEMDA [see Nonclinical Toxicology (13.1)].
Dosing and Administration
Inform patients and caregivers on how to take OJEMDA and what to do for missed or vomited doses [see Dosage and Administration (2.4)].
Prior to use of the oral suspension, ensure patients or caregivers receive training on proper dosing, preparation, and administration [see Dosage and Administration (2.4) and Instructions for Use].
-
SPL UNCLASSIFIED SECTION
Manufactured for:
Day One Biopharmaceuticals, Inc.
Brisbane CA 94005Manufactured by (tablets):
Quotient Sciences – Philadelphia LLC
3 Chelsea Parkway, Suite 305
Boothwyn PA 19061Manufactured by (oral suspension):
Quotient Sciences – Philadelphia LLC
3080 McCann Farm Dr.
Garnet Valley, PA 19060DAY101-USPI-052024 v02
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration.
DAY101-PPI-052024 v02Issued: 05/2024 PATIENT INFORMATION OJEMDA (oh-JEM-dah)
(tovorafenib)
tablets, for oral useOJEMDA (oh-JEM-dah)
(tovorafenib)
for oral suspensionWhat is OJEMDA?
OJEMDA is a prescription medicine used to treat certain types of brain tumors (cancers) called gliomas in patients 6 months and older:- that is a pediatric low-grade glioma (LGG), and
- that has come back after previous treatment or has not responded to previous treatment and
- that has a certain type of abnormal "BRAF" gene.
It is not known if OJEMDA is safe and effective in children younger than 6 months of age.Before taking or giving OJEMDA, tell your healthcare provider about all of your or your child's medical conditions, including if you: - have bleeding problems
- have skin problems
- have liver problems
- are pregnant or plan to become pregnant. OJEMDA can harm your unborn baby.
Females who are able to become pregnant:- Your healthcare provider will do a test to see if you are pregnant before starting treatment with OJEMDA.
- You should use effective non-hormonal birth control (contraception) during treatment with OJEMDA and for 28 days after your last dose of OJEMDA.
- Birth control methods that contain hormones (such as birth control pills, injections, or transdermal systems) may not work as well during treatment with OJEMDA and you could become pregnant. You should use another non-hormonal, effective method of birth control during treatment with OJEMDA.
- Talk to your healthcare provider about birth control methods that may be right for you during this time.
- Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with OJEMDA.
- are breastfeeding or plan to breastfeed. It is not known if OJEMDA passes into your breast milk. Do not breastfeed during treatment and for 2 weeks after your last dose of OJEMDA. Talk to your healthcare provider about the best way to feed your baby during this time.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get any new medicine.How should I take or give OJEMDA? - Take or give OJEMDA exactly as your healthcare provider tells you to. Do not change your or your child's dose or stop taking OJEMDA unless your healthcare provider tells you.
- Your healthcare provider may change your or your child's dose of OJEMDA, temporarily stop, or completely stop your treatment with OJEMDA if you develop certain side effects.
- Your healthcare provider will either prescribe you or your child OJEMDA tablets or OJEMDA for oral suspension.
- Take or give OJEMDA 1 time each week.
- Take or give OJEMDA with or without food.
- If you miss a weekly dose of OJEMDA by:
- 3 days or less, take or give the missed dose as soon as you remember. Take or give the next dose of OJEMDA on the next regularly scheduled day.
- more than 3 days, skip the missed dose and take or give the next dose of OJEMDA on the next regularly scheduled day.
- If vomiting happens right after taking a dose of OJEMDA, take or give the dose again. If you are not sure if you should take or give another dose, contact your healthcare provider.
- Swallow tablets whole with water. Do not chew, cut, or crush the tablets.
- See the Instructions for Use for instructions on how to prepare, measure and take or give a dose of OJEMDA for oral suspension. If you have any questions, talk to your healthcare provider or pharmacist.
- The OJEMDA oral suspension can be taken or given by mouth, or through a feeding tube (minimum 12 French).
- Your or your child's dose may require preparing 2 bottles of OJEMDA for oral suspension.
Limit the amount of time you spend in sunlight. OJEMDA can make your skin sensitive to the sun (photosensitivity). Use sun protection measures, such as sunscreen, sunglasses and wear protective clothes that cover your skin during your treatment with OJEMDA. What are the possible side effects of OJEMDA?
OJEMDA may cause serious side effects, including:-
bleeding problems (hemorrhage) are common during treatment with OJEMDA and can also be serious. Tell your healthcare provider if you develop any signs or symptoms of bleeding, including:
- headache, dizziness or feeling weak
- coughing up blood or blood clots
- vomiting blood or your vomit looks like "coffee grounds"
- red or black stools that look like tar
- skin reactions, including sensitivity to sunlight (photosensitivity). OJEMDA can cause skin reactions that can become severe. Tell your healthcare provider if you get new or worsening skin reactions, including:
- rash
- bumps or tiny papules
- acne
- peeling, redness, or irritation
- blisters
See "What should I avoid while taking OJEMDA?" - liver problems. Your healthcare provider will do blood tests to check your liver function before and during treatment with OJEMDA. Tell your healthcare provider right away if you develop any of the following symptoms:
- yellowing of your skin or your eyes
- dark or brown (tea-colored) urine
- nausea or vomiting
- loss of appetite
- tiredness
- bruising
- bleeding
- pain in your upper right stomach area
- slowed growth in children. Growth will be checked routinely during treatment with OJEMDA.
- rash
- hair color changes
- tiredness
- viral infection
- vomiting
- headache
- fever
- dry skin
- constipation
- nausea
- acne
- upper respiratory tract infection
- decreased phosphate
- decreased red and white blood cells
- increased creatinine phosphokinase
- increase in liver function test
- decreased albumin
- decreased potassium
- decreased sodium
OJEMDA may cause fertility problems in males and females, which may affect your ability to have children. Talk to your healthcare provider if this is a concern for you.
These are not all the possible side effects of OJEMDA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store OJEMDA?
OJEMDA tablets:- Store OJEMDA tablets at room temperature between 68˚F to 77˚F (20˚C to 25˚C).
- Keep OJEMDA tablets in its original package. Tablets should not be removed from blisters until right before use.
- Store the glass bottle containing OJEMDA for oral suspension at room temperature between 68˚F to 77˚F (20˚C to 25˚C).
- Do not use OJEMDA for oral suspension if the safety seal under the cap is broken or missing.
- Throw away your bottle(s), including any unused or expired OJEMDA for oral suspension, and oral dosing syringe after taking or giving a dose.
General information about the safe and effective use of OJEMDA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use OJEMDA for a condition for which it was not prescribed. Do not give OJEMDA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for more information about OJEMDA that is written for health professionals.What are the ingredients in OJEMDA?
Active ingredient: tovorafenib
Inactive ingredients:
Tablet: copovidone, colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and orange film coating (hypromellose, polyethylene glycol 8000, titanium dioxide, ferric oxide yellow, ferric oxide orange).
For oral suspension: artificial strawberry flavor, colloidal silicon dioxide, copovidone, maltodextrin, mannitol, microcrystalline cellulose, simethicone, sodium lauryl sulfate, and sucralose.
Manufactured for:
Day One Biopharmaceuticals, Inc.,
Brisbane CA 94005
For more information, go to www.OJEMDA.com or call 1-855-DAY-1BIO (1-855-329-1246). -
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
OJEMDA (oh-JEM-dah)
(tovorafenib)
for oral suspensionThis Instructions for Use has been approved by the U.S. Food and Drug Administration. Issued: 05/2024 This Instructions for Use contains information on how to prepare, measure, and take or give a dose of OJEMDA for oral suspension. Important information you need to know before preparing, measuring, and taking or giving a dose of OJEMDA for oral suspension. - Read this Instructions for Use carefully before you prepare, measure, and take or give a dose of OJEMDA for oral suspension for the first time and each time you get a refill. There may be new information. This information does not take the place of talking with your or your child's healthcare provider about your or your child's medical treatment or condition.
- Your healthcare provider or pharmacist should show you how to prepare, measure, and take or give a dose of OJEMDA for oral suspension correctly. Talk to your healthcare provider or pharmacist if you have questions.
- Take or give OJEMDA for oral suspension exactly as your healthcare provider tells you to.
- You will receive the OJEMDA prescription in a box that contains a bottle with powder, a 20 mL oral dosing syringe, and a bottle adaptor. Contact your healthcare provider or pharmacist if you do not have one or more of these items.
- The bottle is made of glass. Do not use the bottle if it is broken or damaged. Contact your healthcare provider or pharmacist for a new bottle.
- Check the expiration date on the bottle and box. Contact your pharmacist if the expiration date has passed. Expired or unused product can be thrown away (disposed of) in your household trash.
- Only use room temperature water for preparing OJEMDA for oral suspension.
- Each dose of OJEMDA for oral suspension must be given within 15 minutes after the medicine has been prepared.
- Each bottle of OJEMDA for oral suspension is for single use only.
- See "How to throw away used bottles, expired or unused OJEMDA for oral suspension, and oral dosing syringes?" at the end of this Instructions for Use.
The OJEMDA box contains: Included in the box: 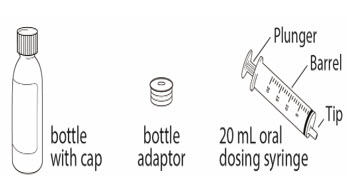
Not included in the box: - 1 empty clean cup
- room temperature water
- ENFIT syringe and ENFIT adaptor (if taking or giving OJEMDA oral suspension through a feeding tube)
- Always use the oral dosing syringe provided to make sure that you correctly measure your prescribed dose.
- The 20 mL oral dosing syringe is marked to help you correctly measure your prescribed dose of OJEMDA for oral suspension. The barrel of the oral dosing syringe has markings in milliliters (mL).
- Add exactly 14 mL of room temperature water to the bottle to prepare the OJEMDA for oral suspension. Only 12 mL of OJEMDA oral suspension will be taken or given from each prepared bottle.
- always add exactly 14 mL of room temperature water to each bottle, and
- prepare, take or give the dose of OJEMDA for oral suspension from the first bottle and then repeat the same steps to prepare, take or give the dose of OJEMDA for oral suspension from the second bottle.
- OJEMDA for oral suspension can be taken or given by mouth using the 20 mL oral dosing syringe, or through a feeding tube with a minimum size of 12 French using an ENFIT syringe.
- If you are taking or giving OJEMDA for oral suspension by mouth, follow Section A, Steps 1 to 19.
- If taking or giving OJEMDA for oral suspension by a feeding tube, follow Section B, Steps 20 to 25.
Section A: Preparing, measuring, and taking or giving a dose of OJEMDA for oral suspension Step 1. Wash and dry your hands before preparing, measuring, and taking or giving a dose of OJEMDA for oral solution. Step 2. Place your supplies on a clean, flat work surface. Step 3. Fill a cup half-way with room temperature water. Do not use cold water. 
Step 4. Remove air from the oral dosing syringe.
Pull the plunger up into the oral dosing syringe as far as it will go, and then push the plunger back down into the oral dosing syringe as far as it will go. This will help to remove all the air inside.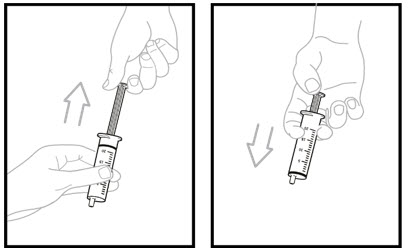
Step 5. Place the oral dosing syringe tip in the water. Pull up on the plunger to draw water into the oral dosing syringe to the 14 mL mark.
Note: Add exactly 14 ml of water to the bottle with powder.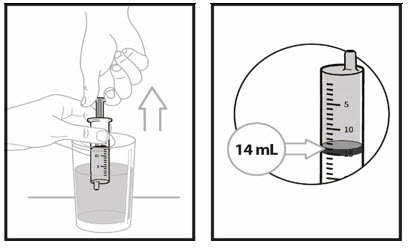
Step 6. Remove the oral dosing syringe from the cup. Turn the oral dosing syringe tip upward and check for air bubbles.
If large air bubbles appear in the oral dosing syringe, push the water back into the cup and then pull up on the plunger again to draw up the water to the 14 mL mark.
Repeat Step 6 until there are no large air bubbles present. Small air bubbles are ok.
Set the oral dosing syringe aside.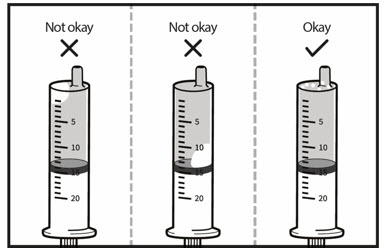
Step 7. Open the bottle with powder by pushing down firmly on the cap and turning it to the left (counter-clockwise). - Do not throw away the cap.
- Remove the Safety seal.
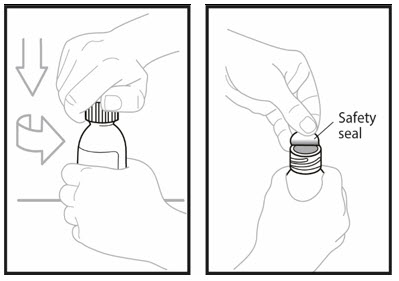
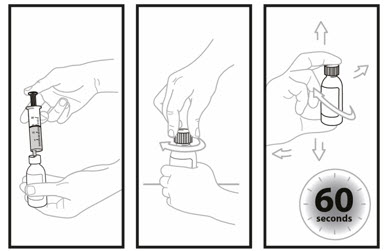
Step 8. Insert the tip of the oral dosing syringe into the opening of the bottle. Push down on the plunger and inject 14 mL of water into the bottle. - Remove the tip of the emptied oral dosing syringe from the bottle and set it aside.
- Right away, replace the cap back onto the bottle by pushing down while twisting the cap to the right (clockwise).
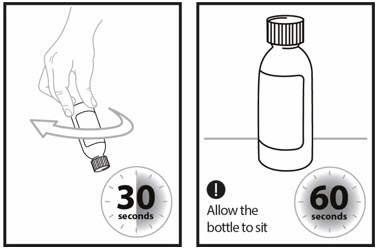
- Shake the bottle well for 60 seconds in all directions.
Step 9: Turn bottle upside down to check for any powder stuck to the inside of the bottle. - If you still see powder in the bottle, continue to shake the bottle for another 15 seconds until you no longer see the powder inside the bottle.
- Do not shake the bottle for more than 2 minutes total time.
- Check the bottle to make sure all of the powder is no longer visible.
- If you still see powder in the bottle, contact your healthcare provider or pharmacist and ask for a new bottle.
Step 10. Turn the bottle upside down again and swirl for 30 seconds. - Place the bottle on a flat, clean, work surface.
- Remove the cap and check that no solids are stuck in the bottle neck.
- If you see solids in the bottle neck, recap the bottle, turn the bottle upside down, and swirl for an additional 15 seconds.
- Allow the bottle to sit for 60 seconds to allow most of the foam to settle.
Step 11. Open the bottle by firmly pressing down on the cap and turning it to the left (counterclockwise). Do not throw away the cap.
Firmly insert the bottle adaptor into the bottle by pushing it tightly into the top of the bottle. The top edge of the bottle adaptor should be even with the bottle top.
Do not remove the bottle adaptor after it is inserted into the bottle.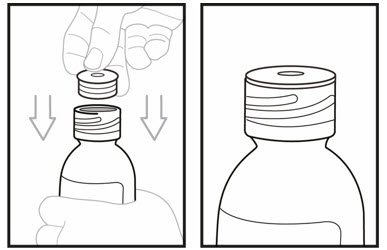
Step 12. Check your or your child's dose in milliliters (mL) as prescribed by your healthcare provider. Pick up the oral dosing syringe again. Each mark on the oral dosing syringe is equal to 1 mL. Draw air into the oral dosing syringe by pulling the plunger out to your prescribed dose. For example, if your prescribed dose is 12 mL, you would draw the oral dosing syringe by pulling the plunger out to the 12 mL mark. 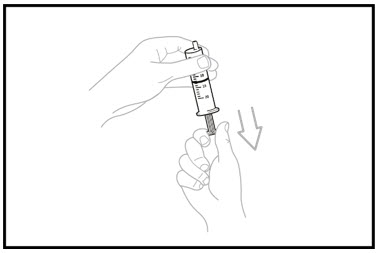
Step 13. Insert the tip of the oral dosing syringe into the bottle adaptor. - The tip of the oral dosing syringe should fit snugly into the hole of the bottle adaptor.
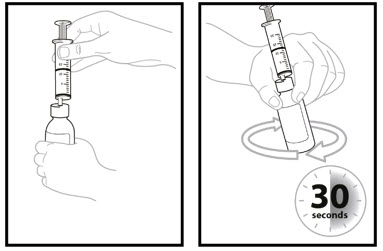
- Keep the oral dosing syringe attached to the bottle. With the oral dosing syringe in place and holding the bottle where the oral dosing syringe tip inserts into bottle adaptor, swirl the oral suspension for 30 seconds.
Step 14. Inject the air from the oral dosing syringe into the bottle. Hold the oral dosing syringe in place and turn the bottle upside down. To measure the prescribed dose, keep the tip of the oral dosing syringe facing up and pull down on the plunger until the top of the plunger lines up with the prescribed dose in mLs. 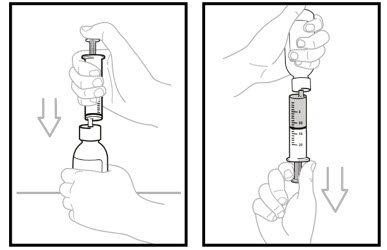
Step 15. While the syringe is still in the bottle, remove any air bubbles in the oral dosing syringe by gently pushing the OJEMDA oral suspension back into the bottle and then pulling down on the plunger again to draw up your prescribed dose.
Repeat Step 15 until you see that few or no air bubbles remain or if you draw up the wrong dose in the oral dosing syringe.
Note: Only use up to 12 mL of OJEMDA oral suspension from each prepared bottle.- If the prescribed dose is more than 12 mL (300 mg), split the dose as equally as possible between each prepared bottle.
- For example, if your dose is 13 mL, draw 6 mL from the first prepared bottle, and 7 mL from the second prepared bottle.
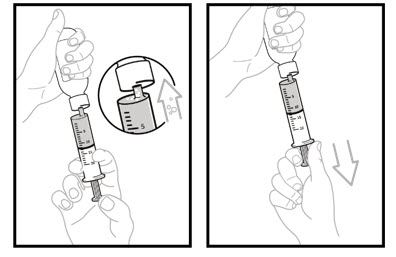
Step 16: Leave the tip of the oral dosing syringe in the bottle adaptor and carefully turn the bottle upright. Put the bottle onto your flat work surface again.
Slowly remove the oral dosing syringe tip from the bottle adaptor by gently pulling straight up. Do not hold the oral dosing syringe by the plunger because the plunger may come out.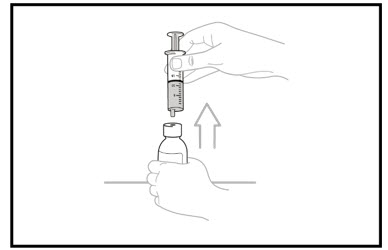
Step 17: Check again to be sure the top of the black stopper on the barrel of the oral dosing syringe is at your prescribed mL dose mark. If you do not have the correct prescribed mL dose, repeat Steps 15 to 17.
If you are taking or giving a dose of OJEMDA for oral suspension by mouth, continue to Step 18.
If you are taking or giving a dose of OJEMDA for oral suspension through a feeding tube, go to Section B.
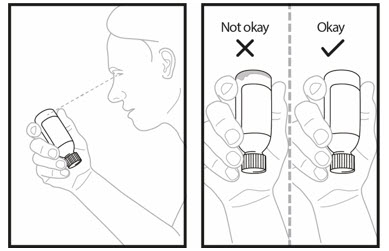
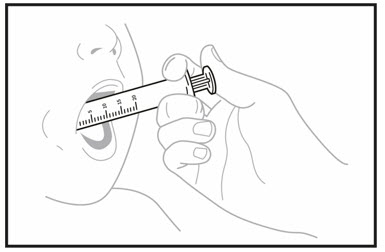
OJEMDA for oral suspension must be given within 15 minutes after prepared for use.Step 18. You or your child should sit upright to take or give a dose of OJEMDA oral suspension. Place the tip of the oral dosing syringe towards the inside of the cheek in your or your child's mouth. - Slowly push the medicine into the mouth by pressing down on the plunger.
- Do not forcefully push the plunger. This may cause choking.
- Allow the child to swallow while giving OJEMDA. You or your child may drink liquids right away after swallowing the OJEMDA for oral suspension.
- Be sure to take or give the entire dose of OJEMDA for oral suspension.
- If 2 bottles of OJEMDA for oral suspension are required to take or give your prescribed dose, repeat Section A, Steps 1 to 18 for the second bottle.
- Throw away the prepared OJEMDA for oral suspension if it is not taken or given within 15 minutes.
Step 19: See Section C for instructions on "How to throw away used bottles, expired or unused OJEMDA for oral suspension, and oral dosing syringes" Section B: Taking or giving a dose of OJEMDA or oral suspension through a feeding tube Before giving a dose of OJEMDA for oral suspension through a feeding tube, read the following information and talk to your or your child's healthcare provider before continuing to STEP 20: - OJEMDA for oral suspension may be given through a feeding tube, as directed by your healthcare provider.
- Only use a feeding tube with a minimum size of 12 French.
- Always use the 20 mL oral dosing syringe (included in the box) to prepare each dose of OJEMDA for oral suspension in the bottle.
- Always use a 20 mL ENFIT syringe and an ENFIT adaptor (neither included in the box) to measure and give each dose of OJEMDA for oral suspension through the feeding tube.
Step 20. Flush the feeding tube according to the manufacturer's instructions before giving a dose of OJEMDA for oral suspension. Step 21: Follow Steps 1 to 11 in Section A to prepare the OJEMDA oral suspension using the 20 mL oral dosing syringe.
Follow Steps 12 to 17 in Section A to draw up your or your child's dose of OJEMDA for oral suspension using the ENFIT syringe and ENFIT adaptor.Step 22: Connect the 20 mL ENFIT syringe containing OJEMDA for oral suspension to the feeding tube. Step 23: Apply steady pressure to the plunger to give the entire dose of OJEMDA for oral suspension through the feeding tube. Step 24: Flush the feeding tube after giving each dose of OJEMDA for oral suspension according to the manufacturer's instructions. If 2 bottles are required, repeat Step 21 and give the remainder of the dose right away. Step 25: Go to Section C for instructions on "How to throw away used bottles, expired or unused OJEMDA for oral suspension, and oral dosing syringes" Section C: How to throw away used bottles, expired or unused OJEMDA for oral suspension, and oral dosing syringes
- Throw away your used bottle(s), expired or unused OJEMDA for oral suspension, and oral dosing syringe(s) in your household trash.
- Do not re-use the oral dosing syringe(s).
How should I store OJEMDA? - Store the glass bottle containing OJEMDA for oral suspension at room temperature between 68˚F to 77˚F (20˚C to 25˚C).
Manufactured for:
Day One Biopharmaceuticals, Inc.,
Brisbane CA 94005
For more information, go to www.OJEMDA.com or call 1-855-DAY-1BIO (1-855-329-1246).DAY101-IFU-052024 v02
-
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC 82950-012-01
Rx onlyojemda™
(tovorafenib)
for oral suspension25 mg/mL
After reconstitution with 14 mL of
water, each bottle delivers a maximum
of 300 mg/12 mL (25mg/mL)
of tovorafenib suspension.Contents:
1 bottle of OJEMDA
1 oral dosing syringe
1 bottle adapter
1 Instructions for UseDay One
BIOPHARMACEUTICALS
- PRINCIPAL DISPLAY PANEL - 100 mg Tablet Blister Pack Carton
-
INGREDIENTS AND APPEARANCE
OJEMDA
tovorafenib kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:82950-012 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:82950-012-01 1 in 1 CARTON 04/30/2024 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BOTTLE, GLASS 12 mL Part 1 of 1 OJEMDA
tovorafenib powder, for suspensionProduct Information Item Code (Source) NDC:82950-112 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOVORAFENIB (UNII: ZN90E4027M) (TOVORAFENIB - UNII:ZN90E4027M) TOVORAFENIB 25 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) COPOVIDONE K25-31 (UNII: D9C330MD8B) SUCRALOSE (UNII: 96K6UQ3ZD4) MALTODEXTRIN (UNII: 7CVR7L4A2D) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM LAURYL SULFATE (UNII: 368GB5141J) DIMETHICONOL/TRIMETHYLSILOXYSILICATE CROSSPOLYMER (40/60 W/W; 1000000 PA.S) (UNII: 83D19O7250) Product Characteristics Color WHITE Score Shape Size Flavor STRAWBERRY Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:82950-112-10 12 mL in 1 BOTTLE, GLASS; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218033 04/30/2024 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218033 04/30/2024 OJEMDA
tovorafenib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:82950-001 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOVORAFENIB (UNII: ZN90E4027M) (TOVORAFENIB - UNII:ZN90E4027M) TOVORAFENIB 100 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) COPOVIDONE K25-31 (UNII: D9C330MD8B) MAGNESIUM STEARATE (UNII: 70097M6I30) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) Product Characteristics Color ORANGE Score no score Shape OVAL Size 16mm Flavor Imprint Code 100;D101 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:82950-001-24 4 in 1 CARTON 04/30/2024 1 NDC:82950-001-06 6 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC:82950-001-20 5 in 1 CARTON 04/30/2024 2 NDC:82950-001-04 4 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC:82950-001-16 4 in 1 CARTON 04/30/2024 3 NDC:82950-001-04 4 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA217700 04/30/2024 Labeler - Day One Biopharmaceuticals, Inc. (118074187) Establishment Name Address ID/FEI Business Operations Shangzhou SynTheAll Pharmaceuticals Co. Ltda (STA) 544385021 API MANUFACTURE(82950-012, 82950-001) Establishment Name Address ID/FEI Business Operations Quotient Sciences - Philadelphia LLC 126874135 MANUFACTURE(82950-012, 82950-001) Establishment Name Address ID/FEI Business Operations Sharp Packaging Services, LLC 002346625 PACK(82950-012, 82950-001) , LABEL(82950-012, 82950-001) Establishment Name Address ID/FEI Business Operations PPD, L.P. 838082055 ANALYSIS(82950-012, 82950-001)
