Label: LEQVIO- inclisiran injection, solution

- NDC Code(s): 0078-1000-60, 0078-1000-98
- Packager: Novartis Pharmaceuticals Corporation
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated June 21, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LEQVIO safely and effectively. See full prescribing information for LEQVIO.
LEQVIO® (inclisiran) injection, for subcutaneous use
Initial U.S. Approval: 2021INDICATIONS AND USAGE
LEQVIO is a small interfering RNA (siRNA) directed to proprotein convertase subtilisin kexin type 9 (PCSK9) mRNA indicated as an adjunct to diet and statin therapy for the treatment of adults with primary hyperlipidemia, including heterozygous familial hypercholesterolemia (HeFH), to reduce low-density lipoprotein cholesterol (LDL-C). (1)
DOSAGE AND ADMINISTRATION
- The recommended dosage of LEQVIO, in combination with statin therapy, is 284 mg administered as a single subcutaneous injection initially, again at 3 months, and then every 6 months. (2.1)
- LEQVIO should be administered by a healthcare professional. (2.2)
- Inject LEQVIO subcutaneously into the abdomen, upper arm, or thigh. (2.2)
DOSAGE FORMS AND STRENGTHS
Injection: 284 mg/1.5 mL (189 mg/mL) in a single-dose prefilled syringe. (3)
CONTRAINDICATIONS
Prior serious hypersensitivity to inclisiran or any of the excipients in LEQVIO. (4)
ADVERSE REACTIONS
Common adverse reactions in clinical trials (≥ 3%): injection site reaction, arthralgia, and bronchitis. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Important Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
- The recommended dosage of LEQVIO, in combination with statin therapy, is 284 mg administered as a single subcutaneous injection initially, again at 3 months, and then every 6 months.
- If a planned dose is missed by less than 3 months, administer LEQVIO and maintain dosing according to the patient’s original schedule.
- If a planned dose is missed by more than 3 months, restart with a new dosing schedule - administer LEQVIO initially, again at 3 months, and then every 6 months.
- Assess LDL-C when clinically indicated. The LDL-lowering effect of LEQVIO may be measured as early as 30 days after initiation and anytime thereafter without regard to timing of the dose.
2.2 Important Administration Instructions
- LEQVIO should be administered by a healthcare professional.
- Inject LEQVIO subcutaneously into the abdomen, upper arm, or thigh. Do not inject in areas of active skin disease or injury, such as sunburns, skin rashes, inflammation, or skin infections.
- Inspect LEQVIO visually before use. It should appear clear and colorless to pale yellow. Do not use if particulate matter or discoloration is seen.
For more detailed instruction on administration of the prefilled syringe, see Instructions for Use.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in Table 1 are derived from 3 placebo-controlled trials that included 1,833 patients treated with LEQVIO, including 1,682 exposed for 18 months (median treatment duration of 77 weeks) [see Clinical Studies (14)]. The mean age of the population was 64 years, 32% of the population were women, 92% were White, 6% were Black or African American, 1% were Asian, and < 1% were other races; 6% identified as Hispanic or Latino ethnicity. At baseline, 12% of patients had a diagnosis of HeFH and 85% had clinical atherosclerotic cardiovascular disease (ASCVD).
Adverse reactions reported in at least 3% of LEQVIO-treated patients, and more frequently than in placebo-treated patients, are shown in Table 1.
Table 1: Adverse Reactions Occurring in Greater Than or Equal to 3% of LEQVIO-treated Patients and More Frequently than with Placebo (Studies 1, 2, and 3) Adverse Reactions Placebo (N = 1,822)
%LEQVIO (N = 1,833)
%†includes related terms such as: injection site pain, erythema and rash Injection site reaction† 2 8 Arthralgia 4 5 Bronchitis 3 4 Adverse reactions led to discontinuation of treatment in 2.5% of patients treated with LEQVIO and 1.9% of patients treated with placebo. The most common adverse reactions leading to treatment discontinuation in patients treated with LEQVIO were injection site reactions (0.2% versus 0% for LEQVIO and placebo, respectively).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of LEQVIO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity: angioedema, rash, and urticaria.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Discontinue LEQVIO when pregnancy is recognized. Alternatively, consider the ongoing therapeutic needs of the individual patient. Inclisiran increases LDL-C uptake and lowers LDL-C levels in the circulation, thus decreasing cholesterol and possibly other biologically active substances derived from cholesterol; therefore, LEQVIO may cause fetal harm when administered to pregnant patients based on the mechanism of action [see Clinical Pharmacology (12.1)]. In addition, treatment of hyperlipidemia is not generally necessary during pregnancy. Atherosclerosis is a chronic process and the discontinuation of lipid-lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hyperlipidemia for most patients.
There are no available data on the use of LEQVIO in pregnant patients to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes.
In animal reproduction studies, no adverse developmental effects were observed in rats and rabbits with subcutaneous administration of inclisiran during organogenesis at doses up to 5 to 10 times the maximum recommended human dose (MRHD) based on body surface area (BSA) comparison (see Data). No adverse developmental outcomes were observed in offspring of rats administered inclisiran from organogenesis through lactation at 5 times the MRHD based on BSA comparison (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%–4% and 15%–20%, respectively.
Data
Animal Data
In embryo-fetal development studies conducted in Sprague-Dawley rats and New Zealand White rabbits, inclisiran was administered by subcutaneous injection at dose levels of 50, 100, and 150 mg/kg once daily during organogenesis (rats: Gestation Days 6 to 17; rabbits: Gestation Days 7 to 19). There was no evidence of embryo-fetal toxicity or teratogenicity at doses up to 5 and 10 times, respectively, the MRHD based on BSA comparison/dose. Inclisiran crosses the placenta and was detected in rat fetal plasma at concentrations that were 65 to 154 times lower than maternal levels.
In a pre- and postnatal development study conducted in Sprague-Dawley rats, inclisiran was administered once daily by subcutaneous injection at levels of 50, 100, and 150 mg/kg from Gestation Day 6 through Lactation Day 20. Inclisiran was well-tolerated in maternal rats, with no evidence of maternal toxicity and no effects on maternal performance. There were no effects on the development of the F1 generation, including survival, growth, physical and reflexological development, behavior, and reproductive performance at doses up to 5 times the MRHD, based on BSA comparison/dose.
8.2 Lactation
Risk Summary
There is no information on the presence of inclisiran in human milk, the effects on the breastfed infant, or the effects on milk production. Inclisiran was present in the milk of lactating rats in all dose groups. When a drug is present in animal milk, it is likely that the drug will be present in human milk (see Data). Oligonucleotide-based products typically have poor oral bioavailability; therefore, it is considered unlikely that low levels of inclisiran present in milk will adversely impact an infant’s development during lactation. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for LEQVIO and any potential adverse effects on the breastfed infant from LEQVIO or from the underlying maternal condition.
Data
In lactating rats, inclisiran was detected in milk at mean maternal plasma:milk ratios that ranged between 0.361 and 1.79. However, there is no evidence of systemic absorption in the suckling rat neonates.
8.4 Pediatric Use
The safety and effectiveness of LEQVIO have not been established in pediatric patients.
8.5 Geriatric Use
Of the 1,833 patients treated with LEQVIO in clinical studies, 981 (54%) patients were 65 years of age and older, while 239 (13%) patients were 75 years of age and older. No overall differences in safety or effectiveness were observed between patients 65 years of age and older and younger adult patients.
-
11 DESCRIPTION
LEQVIO contains inclisiran sodium, a small interfering RNA (siRNA) directed to proprotein convertase subtilisin kexin type 9 (PCSK9) mRNA. Inclisiran contains a covalently linked ligand containing three N-acetylgalactosamine (GalNAc) residues to facilitate delivery to hepatocytes. With one exception, the 2'ribose moieties of the inclisiran sodium are present as 2'-F or 2'-OMe ribonucleotide. In addition, six of the terminal phosphodiester backbones are present as phosphorothioate linkages as indicated below.
The molecular formula of inclisiran sodium is C529H664F12N176Na43O316P43S6 and its molecular weight is 17,284.72 g/mol. It has the following structural formula:
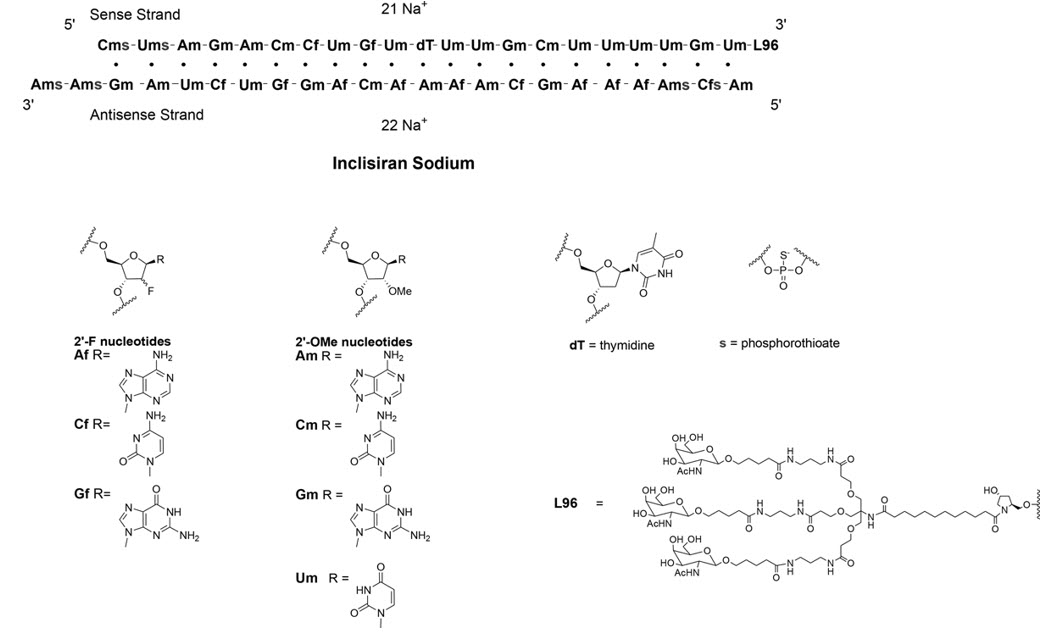
Abbreviations: Af = adenine 2'-F ribonucleotide; Cf = cytosine 2'-F ribonucleotide; Gf = guanine 2'-F ribonucleotide; Am = adenine 2'-OMe ribonucleotide; Cm = cytosine 2'-OMe ribonucleotide; Gm = guanine 2'-OMe ribonucleotide; Um = uracil 2'-OMe ribonucleotide; L96 = triantennary GalNAc (N-acetyl-galactosamine)
LEQVIO is a sterile, preservative-free, clear, and colorless to pale yellow solution for subcutaneous use in a prefilled syringe. Each syringe contains 1.5 mL of solution containing the equivalent of 284 mg inclisiran (present as 300 mg inclisiran sodium salt). LEQVIO is formulated in Water for Injection and may also contain sodium hydroxide and/or phosphoric acid for pH adjustment to a target pH of 7.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Inclisiran is a double-stranded small interfering ribonucleic acid (siRNA), conjugated on the sense strand with triantennary N-Acetylgalactosamine (GalNAc) to facilitate uptake by hepatocytes. In hepatocytes, inclisiran utilizes the RNA interference mechanism and directs catalytic breakdown of mRNA for PCSK9. This increases LDL-C receptor recycling and expression on the hepatocyte cell surface, which increases LDL-C uptake and lowers LDL-C levels in the circulation.
12.2 Pharmacodynamics
Following a single subcutaneous administration of 284 mg of inclisiran, LDL-C reduction was apparent within 14 days post dose. Mean reductions of 38% to 51% for LDL-C were observed 30 to 180 days post dose. At Day 180, LDL-C levels were still reduced by approximately 53%.
Following a dose at Day 1 and Day 90 of 284 mg of inclisiran, mean serum PCSK9 levels were reduced by approximately 75% and 69% at Day 120, and Day 180, respectively.
In the clinical studies, following four doses of LEQVIO at Day 1, Day 90 (3 months), Day 270 (~6 months) and Day 450 (~12 months), LDL-C, total cholesterol, ApoB, and non-HDL-C were reduced [see Clinical Studies (14)].
Cardiac Electrophysiology
At a dose 3 times the maximum recommended dose, inclisiran does not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
Absorption
Following a single subcutaneous administration, systemic exposure to inclisiran increased in a linear and dose proportional manner over a range from 25 mg to 800 mg of inclisiran sodium. At the recommended dosing regimen of 284 mg of LEQVIO, plasma concentrations reached peak in approximately 4 hours post dose with a mean Cmax of 509 ng/mL. Concentrations reached undetectable levels after 24 to 48 hours post dosing. The mean area under the plasma concentration-time curve from dosing extrapolated to infinity was 7,980 ng*h/mL. Pharmacokinetic findings following multiple subcutaneous administrations of LEQVIO were similar to single-dose administration.
Distribution
Inclisiran is 87% protein bound in vitro at the relevant clinical plasma concentrations. Following a single subcutaneous 284 mg dose of LEQVIO to healthy adults, the apparent volume of distribution is approximately 500 L. Inclisiran has been shown to have high uptake into, and selectively for the liver, the target organ for cholesterol lowering.
Elimination
The terminal elimination half-life of LEQVIO is approximately 9 hours, and no accumulation occurs with multiple dosing. Approximately 16% of LEQVIO is cleared through the kidney.
Metabolism
Inclisiran is primarily metabolized by nucleases to shorter nucleotides of varying length. Inclisiran is not a substrate for CYP450 or transporters.
Specific Populations
Male and Female Patients and Racial or Ethnic Groups
A population pharmacodynamic analysis was conducted on data from 4,328 patients. Age, body weight, gender, race, and creatinine clearance were found not to significantly influence inclisiran pharmacokinetics.
Patients with Renal Impairment
Pharmacokinetic analysis of data from a dedicated renal impairment study reported increases in inclisiran Cmax and AUC of approximately 2.3 to 3.3-fold and 1.6 to 2.3-fold, respectively, in patients with mild, moderate or severe renal impairment, relative to patients with normal renal function. Despite the higher plasma exposures, reductions in LDL-C were similar across all groups based on renal function.
Patients with Hepatic Impairment
Pharmacokinetic analysis of data from a dedicated hepatic impairment study reported increases in inclisiran Cmax and AUC of approximately 1.1- to 2.1-fold and 1.3- to 2.0-fold, respectively, in patients with mild and moderate hepatic impairment, relative to patients with normal hepatic function. Despite the higher plasma inclisiran exposures, reductions in LDL-C were similar between the groups of patients administered inclisiran with normal hepatic function and mild hepatic impairment. In patients with moderate hepatic impairment, baseline PCSK9 levels were lower and reductions in LDL-C were less than those observed in patients with normal hepatic function. LEQVIO has not been studied in patients with severe hepatic impairment.
Drug Interaction Studies
No formal clinical drug interaction studies have been performed. The components of LEQVIO are not substrates, inhibitors or inducers of cytochrome P450 enzymes or transporters. In a population pharmacokinetic analysis, concomitant use of inclisiran did not have a clinically significant impact on atorvastatin or rosuvastatin concentrations. LEQVIO is not expected to cause drug-drug interactions or to be affected by inhibitors or inducers of cytochrome P450 enzymes or transporters.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of inclisiran.
The immunogenicity of LEQVIO has been evaluated using screening and confirmatory immunoassays for the detection of binding anti-inclisiran antibodies.
Samples from 1,830 patients in the placebo-controlled clinical trials were tested for anti-drug antibodies [see Clinical Studies (14)]. Confirmed positivity was detected in 33 (2%) patients prior to receiving LEQVIO and in 90 (5%) patients during the 18 months of treatment with LEQVIO. Approximately 31 (2%) LEQVIO-treated patients with a negative sample at baseline had a persistent anti-inclisiran antibody response, defined as two confirmed positive samples separated by at least 16 weeks or a single confirmed positive final sample. There was no identified clinically significant effect of anti-inclisiran antibodies on pharmacodynamics, safety, or effectiveness of LEQVIO over the treatment duration of 18 months. However, the long-term consequences of continuing LEQVIO treatment in the presence of anti-inclisiran binding antibodies are unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year carcinogenicity study, Sprague-Dawley rats were administered subcutaneous doses of 40, 95, or 250 mg/kg inclisiran once every 28 days (1, 3, or 8 times the MRHD, based on BSA comparison/dose). Inclisiran was not carcinogenic up to the highest dose tested.
In a 26-week study in RasH2Tg mice, subcutaneous doses of 300, 600, or 1,500 mg/kg once every 28 days were administered. Inclisiran was not carcinogenic up to the highest dose tested.
Inclisiran was not mutagenic or clastogenic in a standard battery of genotoxicity tests, including a bacterial mutagenicity assay, an in vitro chromosome aberration assay using human peripheral lymphocytes, and an in vivo bone marrow micronucleus assay in rats.
Fertility and early embryonic-development studies were conducted in male and female rats. In male rats, inclisiran was administered subcutaneously at dose levels of 10, 50, and 250 mg/kg every 2 weeks for 4 weeks before cohabitation through mating, and until termination between Days 64 and 67. In female rats, inclisiran was administered subcutaneously at dose levels of 10, 50, and 250 mg/kg once every 4 days beginning 14 days prior to cohabitation and through mating, followed by 10, 50, or 150 mg/kg once daily during the gestation period up to Gestation Day 7. There were no adverse effects on fertility up to the highest dose examined, corresponding to 8 times the MRHD, based on BSA comparison/dose.
-
14 CLINICAL STUDIES
The efficacy of LEQVIO was investigated in three randomized, double-blind, placebo-controlled trials that enrolled 3,660 adults with HeFH, clinical ASCVD, or increased risk for ASCVD, who were taking maximally tolerated statin therapy and who required additional LDL-C lowering. Demographics and baseline disease characteristics were balanced between the treatment arms in all trials.
Primary Hyperlipidemia
Study 1 (ORION-10, NCT03399370) was a multicenter, double-blind, randomized, placebo-controlled 18-month trial in which 1,561 patients with ASCVD were randomized 1:1 to receive subcutaneous injections of either LEQVIO 284 mg (n = 781) or placebo (n = 780) on Day 1, Day 90, Day 270, and at Day 450. Patients were taking a maximally tolerated dose of statin with or without other lipid modifying therapy, and required additional LDL-C reduction. Patients were stratified by current use of statins or other lipid-modifying therapies. Patients taking PCSK9 inhibitors were excluded from the trial.
The mean age at baseline was 66 years (range: 35 to 90 years), 60% were ≥65 years old, 31% were women, 86% were White, 13% were Black or African American, 1% were Asian, and 14% identified as Hispanic or Latino ethnicity. Forty-five percent (45%) of patients had diabetes at baseline. The mean baseline LDL-C was 105 mg/dL. At the time of randomization, 89% of patients were receiving statin therapy and 69% were receiving high-intensity statin therapy.
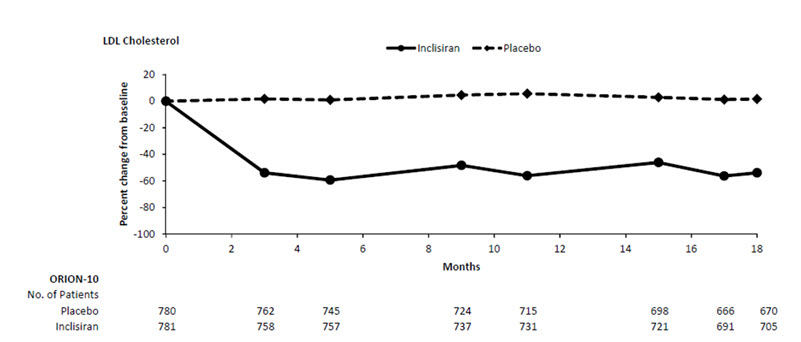
The primary efficacy outcome measure in Study 1 was the percent change from baseline to Day 510 in LDL-C. The difference between the LEQVIO and placebo groups in mean percentage change in LDL-C from baseline to Day 510 was -52% (95% CI: -56%, -49%; p < 0.0001). For additional results, see Table 2 and Figure 1.
Table 2: Changes in Lipid Parameters in Patients with Hyperlipidemia and ASCVD on Maximally Tolerated Statin Therapy (Mean % Change from Baseline to Day 510 in Study 1) Treatment Group LDL-C Total
CholesterolNon-HDL-C ApoB ApoB = apolipoprotein B; CI = confidence interval; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol
a11.5% of subjects on LEQVIO and 14.6% of subjects on placebo had missing LDL-C data at primary endpoint (Day 510). Missing data were imputed using a modified control-based multiple imputation to account for treatment adherence. Percent change from baseline in LDL-C was analyzed using analysis of covariance (ANCOVA) with fixed effect for treatment group and baseline LDL-C as a covariate. Other endpoints were analyzed using a mixed-effect model for repeated measure (MMRM) with fixed effects for treatment group, visit, interaction between treatment and visit, and baseline value. Missing data were imputed using a control-based pattern-mixture model approach.Day 510 (mean percentage change from baseline)a Placebo (n = 780) 1 0 0 -2 LEQVIO (n = 781) -51 -34 -47 -45 Difference from placebo (LS Mean) (95% CI) -52
(-56, -49)-33
(-35, -31)-47
(-50, -44)-43
(-46, -41)Figure 1: Mean Percent Change from Baseline in LDL-C Over 18 Months in Patients with Hyperlipidemia and ASCVD on Maximally Tolerated Statin Therapy (Study 1)
Study 2 (ORION-11, NCT03400800) was a multicenter, double-blind, randomized, placebo-controlled 18-month trial in which 1,617 adults with ASCVD or increased risk for ASCVD were randomized 1:1 to receive subcutaneous injections of either LEQVIO 284 mg (n = 810) or placebo (n = 807) on Day 1, Day 90, Day 270, and Day 450. Patients were taking a maximally tolerated dose of statin with or without other lipid modifying therapy and required additional LDL-C reduction. Patients were stratified by country and by current use of statins or other lipid-modifying therapies. Patients taking PCSK9 inhibitors were excluded from the trial.
The mean age at baseline was 65 years (range: 20 to 88 years), 55% were ≥65 years old, 28% were women, 98% were White, 1% were Black or African American, and <1% were Asian; <1% identified as Hispanic or Latino ethnicity. Thirty-five percent (35%) of patients had diabetes at baseline. The mean baseline LDL-C was 105 mg/dL. At the time of randomization, 95% of patients were receiving statin therapy and 78% were receiving high-intensity statin therapy.
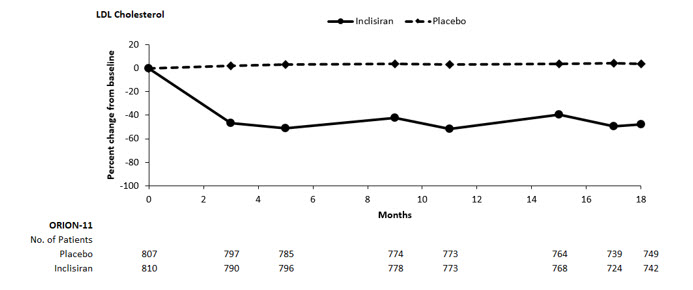
The primary efficacy outcome measure in Study 2 was the percent change from baseline to Day 510 in LDL-C. The difference between the LEQVIO and placebo groups in mean percentage change in LDL-C from baseline to Day 510 was -50% (95% CI: -53%, -47%; p < 0.0001). For additional results, see Table 3 and Figure 2.
Table 3: Changes in Lipid Parameters in Patients with Hyperlipidemia and ASCVD or Increased Risk for ASCVD on Maximally Tolerated Statin Therapy (Mean % Change from Baseline to Day 510 in Study 2) Treatment Group LDL-C Total
CholesterolNon-HDL-C ApoB ApoB = apolipoprotein B; CI = confidence interval; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol
a10.6% of subjects on LEQVIO and 8.4% of subjects on placebo had missing LDL-C data at primary endpoint (Day 510). Missing data were imputed using a modified control-based multiple imputation to account for treatment adherence. Percent change from baseline in LDL-C was analyzed using analysis of covariance (ANCOVA) with fixed effect for treatment group and baseline LDL-C as a covariate. Other endpoints were analyzed using mixed-effect model for repeated measure (MMRM) with fixed effects for treatment group, visit, interaction between treatment and visit, and baseline value. Missing data were imputed using a control-based pattern-mixture model approach.Day 510 (mean percentage change from baseline)a Placebo (n = 807) 4 2 2 1 LEQVIO (n = 810) -46 -28 -41 -38 Difference from placebo (LS Mean) (95% CI) -50
(-53, -47)-30
(-32, -28)-43
(-46, -41)-39
(-41, -37)Figure 2: Mean Percent Change from Baseline in LDL-C Over 18 Months in Patients with Hyperlipidemia and ASCVD or Increased Risk for ASCVD on Maximally Tolerated Statin Therapy (Study 2)
In a pooled analysis of Study 1 and Study 2, the observed treatment effect was similar across predefined subgroups, such as sex, age, race, disease characteristics, geographic regions, presence of diabetes, body mass index, baseline LDL-C levels, and intensity of statin treatment.
LDL-C Reduction in Patients with HeFH
Study 3 (ORION-9, NCT03397121) was a multicenter, double-blind, randomized, placebo-controlled 18-month trial in which 482 patients with HeFH were randomized 1:1 to receive subcutaneous injections of either LEQVIO 284 mg (n = 242) or placebo (n = 240) on Day 1, Day 90, Day 270, and at Day 450. Patients with HeFH were taking a maximally tolerated dose of statin with or without other lipid modifying therapy and required additional LDL-C reduction. The diagnosis of HeFH was made either by genotyping or clinical criteria using either the Simon Broome or WHO/Dutch Lipid Network criteria. Patients were stratified by country and by current use of statins or other lipid-modifying therapies. Patients taking PCSK9 inhibitors were excluded from the trial.
The mean age at baseline was 55 years (range: 21 to 80 years), 22% were ≥65 years old, 53% were women, 94% were White, 3% were Black or African American, and 3% were Asian; and 3% identified as Hispanic or Latino ethnicity. Ten percent (10%) of patients had diabetes at baseline. The mean baseline LDL-C was 153 mg/dL. At the time of randomization, 90% of patients were receiving statin therapy and 74% were receiving high-intensity statin therapy. Fifty-two percent (52%) of patients were treated with ezetimibe. The most commonly administered statins were atorvastatin and rosuvastatin.
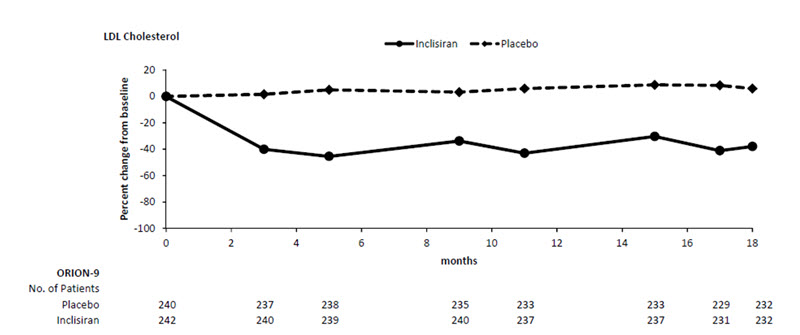
The primary efficacy outcome measure in Study 3 was the percent change from baseline to Day 510 in LDL-C. The difference between the LEQVIO and placebo groups in mean percentage change in LDL-C from baseline to Day 510 was -48% (95% CI: -54%, -42%; p < 0.0001). For additional results, see Table 4 and Figure 3.
Table 4: Changes in Lipid Parameters in Patients with HeFH on Maximally Tolerated Statin Therapy (Mean % Change from Baseline to Day 510 in Study 3) Treatment Group LDL-C Total
CholesterolNon-HDL-C ApoB ApoB = apolipoprotein B; CI = confidence interval; HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol
a4.5% of subjects on LEQVIO and 4.6% of subjects on placebo had missing LDL-C data at primary endpoint (Day 510). Missing data were imputed using a modified control-based multiple imputation to account for treatment adherence. Percent change from baseline in LDL-C was analyzed using analysis of covariance (ANCOVA) with fixed effect for treatment group and baseline LDL-C as a covariate. Other endpoints were analyzed using mixed-effect model for repeated measure (MMRM) with fixed effects for treatment group, visit, interaction between treatment and visit, and baseline value as a covariate. Missing data were imputed using a control-based pattern-mixture model approach.Day 510 (mean percentage change from baseline)a Placebo (n = 240) 8 7 7 3 LEQVIO (n = 242) -40 -25 -35 -33 Difference from placebo (LS Mean) (95% CI) -48
(-54, -42)-32
(-36, -28)-42
(-47, -37)-36
(-40, -32)Figure 3: Mean Percent Change from Baseline in LDL-C Over 18 Months in Patients with HeFH on Maximally Tolerated Statin Therapy (Study 3)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
LEQVIO injection is a clear, colorless to pale yellow solution, 284 mg/1.5 mL (189 mg/mL) of inclisiran supplied as:
Carton containing 1 single-dose prefilled syringe:
NDC 0078-1000-60
Store LEQVIO at controlled room temperature 20°C to 25°C (68°F to 77°F) with allowable excursions between 15°C and 30°C (59°F and 86°F) [see USP, Controlled Room Temperature (CRT)].
-
17 PATIENT COUNSELING INFORMATION
Pregnancy
Advise pregnant patients and patients who can become pregnant of the potential risk to a fetus. Advise patients to inform their healthcare provider of a known or suspected pregnancy to discuss if LEQVIO should be discontinued [see Use in Specific Populations (8.1)].
Injection Site Reactions
Advise patients that injection site reactions can occur with LEQVIO [see Adverse Reactions (6.1)].
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936For more information, visit www.leqvio.com or call 1-833-LEQVIO2 (1-833-537-8462).
© Novartis
T2024-47
-
INSTRUCTIONS FOR USE
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Issued: June 2024 INSTRUCTIONS FOR USE
LEQVIO® [leck' vee oh]
(inclisiran)
injection, for subcutaneous use
284 mg/1.5 mL single-dose Prefilled Syringe
The following information is intended for healthcare professionals only.
This Instructions for Use contains information on how to inject LEQVIO using the prefilled syringe.
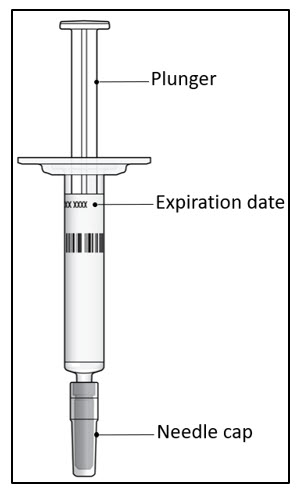
Important Information You Need to Know Before Injecting LEQVIO: - Do not use the prefilled syringe if any of the seals on the outer carton or the seal of the plastic tray are broken.
- Do not remove the needle cap until you are ready to inject.
- Do not use if the prefilled syringe has been dropped after removing the needle cap.
- Do not try to re-use or take apart the prefilled syringe.
Step 1. Inspect the prefilled syringe
It should appear clear and colorless to pale yellow. Do not use if particulate matter or discoloration is seen. You may see air bubbles in the liquid, which is normal. Do not try to remove the air.- Do not use the prefilled syringe if it looks damaged or if any of the solution for injection has leaked out of the prefilled syringe.
- Do not use the prefilled syringe after the expiration date (EXP), which is printed on the prefilled syringe label and carton.
Step 2. Select and prepare the injection site - Choose an injection site in the abdomen, upper arm, or thigh (see Figure A). Do not inject in areas of active skin disease or injury, such as sunburns, skin rashes, inflammation, or skin infection.
- Wipe the skin with an alcohol swab. Let the injection site dry before you inject the dose.
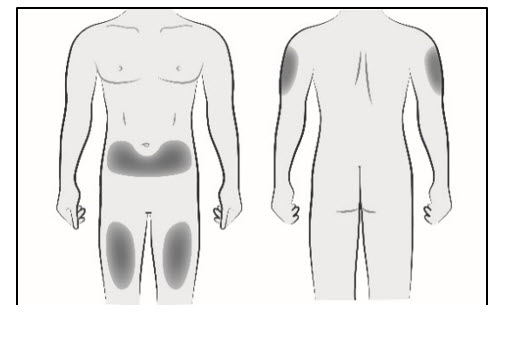
Figure AStep 3. Remove needle cap
Firmly pull straight to remove the needle cap from the prefilled syringe (see Figure B). You may see a drop of liquid at the end of the needle. This is normal.
Do not put the needle cap back on. Throw it away.
Note: Do not remove the needle cap until you are ready to inject. Early removal of the needle cap prior to injection can lead to drying of the drug product within the needle, which can result in needle clogging.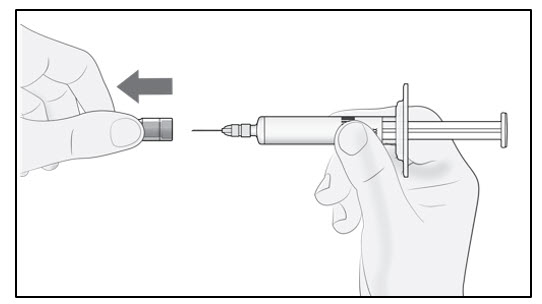
Figure BStep 4. Insert the needle
Gently pinch the skin at the injection site and hold the pinch throughout the injection. With the other hand insert the needle into the skin at an angle of approximately 45 degrees as shown (see Figure C).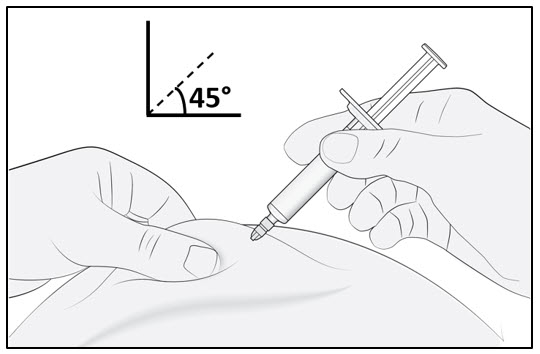
Figure CStep 5. Inject
Continue to pinch the skin. Slowly press the plunger as far as it will go (see Figure D). This will make sure that a full dose is injected.
Note: If you cannot depress the plunger following insertion of the needle, use a new prefilled syringe.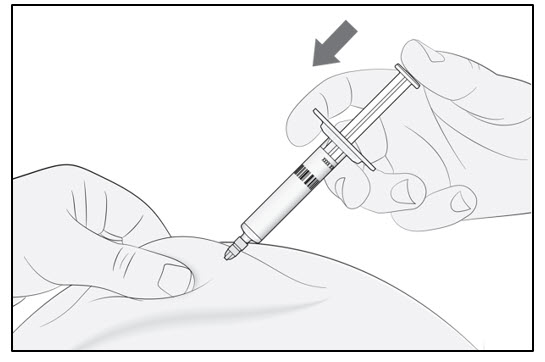
Figure DStep 6. Complete injection and dispose of the prefilled syringe
Remove the prefilled syringe from the injection site. Do not put the needle cap back on.
Dispose of the prefilled syringe in a FDA-cleared sharps disposal container right away after use.
For more information, visit www.leqvio.com or call 1-833-LEQVIO2 (1-833-537-8462).
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936© Novartis T2024-44
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LEQVIO
inclisiran injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0078-1000 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength INCLISIRAN SODIUM (UNII: UPC6BTX7PY) (INCLISIRAN - UNII:UOW2C71PG5) INCLISIRAN 284 mg in 1.5 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) PHOSPHORIC ACID (UNII: E4GA8884NN) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0078-1000-60 1 in 1 CARTON 12/22/2021 1 1.5 mL in 1 SYRINGE, GLASS; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 2 NDC:0078-1000-98 1 in 1 CARTON 02/17/2023 2 1.5 mL in 1 SYRINGE, GLASS; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214012 12/22/2021 Labeler - Novartis Pharmaceuticals Corporation (002147023)
