Label: SOLIRIS- eculizumab injection, solution, concentrate

- NDC Code(s): 25682-001-01
- Packager: Alexion Pharmaceuticals Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated July 2, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SOLIRIS safely and effectively. See full prescribing information for SOLIRIS.
SOLIRIS® (eculizumab) injection, for intravenous use
Initial U.S. Approval: 2007WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
See full prescribing information for complete boxed warning
SOLIRIS increases the risk of serious and life-threatening infections caused by Neisseria meningitidis.
- Complete or update meningococcal vaccination at least 2 weeks prior to the first dose of SOLIRIS, unless the risks of delaying SOLIRIS outweigh the risk of developing a serious infection. Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for meningococcal vaccination in patients receiving a complement inhibitor. (5.1)
- Patients receiving SOLIRIS are at increased risk for invasive disease caused by N. meningitidis, even if they develop antibodies following vaccination. Monitor patients for early signs and symptoms of meningococcal infections, and evaluate immediately if infection is suspected. (5.1)
SOLIRIS is available only through a restricted program called the ULTOMIRIS and SOLIRIS REMS. (5.2)
RECENT MAJOR CHANGES
Boxed Warning 02/2024 Boxed Warning 03/2024 Dosage and Administration (2.1) 02/2024 Dosage and Administration (2.1) 03/2024 Contraindications (4) 02/2024 Contraindications (4) 03/2024 Warnings and Precautions (5.1, 5.2) 02/2024 Warnings and Precautions (5.2) 03/2024 INDICATIONS AND USAGE
SOLIRIS is a complement inhibitor indicated for:
- the treatment of patients with paroxysmal nocturnal hemoglobinuria (PNH) to reduce hemolysis. (1.1)
- the treatment of patients with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy. (1.2)
Limitation of Use
SOLIRIS is not indicated for the treatment of patients with Shiga toxin E. coli related hemolytic uremic syndrome (STEC-HUS). - the treatment of generalized myasthenia gravis (gMG) in adult patients who are anti-acetylcholine receptor (AchR) antibody positive. (1.3)
- the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adult patients who are anti-aquaporin-4 (AQP4) antibody positive. (1.4)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 300 mg/30 mL (10 mg/mL) in a single-dose vial. (3)
CONTRAINDICATIONS
SOLIRIS is contraindicated for initiation in patients with unresolved serious Neisseria meningitidis infection. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
The most frequently reported adverse reactions in the PNH randomized trial (≥10% overall and greater than placebo) are: headache, nasopharyngitis, back pain, and nausea. (6.1)
The most frequently reported adverse reactions in aHUS single arm prospective trials (≥20%) are: headache, diarrhea, hypertension, upper respiratory infection, abdominal pain, vomiting, nasopharyngitis, anemia, cough, peripheral edema, nausea, urinary tract infections, pyrexia. (6.1)
The most frequently reported adverse reaction in the gMG placebo-controlled clinical trial (≥10%) is: musculoskeletal pain. (6.1)
The most frequently reported adverse reactions in the NMOSD placebo- controlled trial (≥10%) are: upper respiratory infection, nasopharyngitis, diarrhea, back pain, dizziness, influenza, arthralgia, pharyngitis, and contusion. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Alexion Pharmaceuticals, Inc. at 1-844-259-6783 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
1 INDICATIONS AND USAGE
1.1 Paroxysmal Nocturnal Hemoglobinuria (PNH)
1.2 Atypical Hemolytic Uremic Syndrome (aHUS)
1.3 Generalized Myasthenia Gravis (gMG)
1.4 Neuromyelitis Optica Spectrum Disorder (NMOSD)
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Vaccination and Prophylaxis for Meningococcal Infection
2.2 Recommended Dosage Regimen – PNH
2.3 Recommended Dosage Regimen – aHUS
2.4 Recommended Dosage Regimen – gMG and NMOSD
2.5 Dose Adjustment in Case of Plasmapheresis, Plasma Exchange, or Fresh Frozen Plasma Infusion
2.6 Preparation
2.7 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Meningococcal Infections
5.2 ULTOMIRIS and SOLIRIS REMS
5.3 Other Infections
5.4 Monitoring Disease Manifestations after SOLIRIS Discontinuation
5.5 Thrombosis Prevention and Management
5.6 Infusion-Related Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Plasmapheresis, Plasma Exchange, or Fresh Frozen Plasma Infusion
7.2 Neonatal Fc Receptor Blockers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Paroxysmal Nocturnal Hemoglobinuria (PNH)
14.2 Atypical Hemolytic Uremic Syndrome (aHUS)
14.3 Generalized Myasthenia Gravis (gMG)
14.4 Neuromyelitis Optica Spectrum Disorder (NMOSD)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS MENINGOCOCCAL INFECTIONS
SOLIRIS, a complement inhibitor, increases the risk of serious infections caused by Neisseria meningitidis [see Warnings and Precautions (5.1)]. Life-threatening and fatal meningococcal infections have occurred in patients treated with complement inhibitors. These infections may become rapidly life-threatening or fatal if not recognized and treated early.
- Complete or update vaccination for meningococcal bacteria (for serogroups A, C, W, Y, and B) at least 2 weeks prior to the first dose of SOLIRIS, unless the risks of delaying therapy with SOLIRIS outweigh the risk of developing a serious infection. Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for vaccinations against meningococcal bacteria in patients receiving a complement inhibitor. See Warnings and Precautions (5.1) for additional guidance on the management of the risk of serious infections caused by meningococcal bacteria.
- Patients receiving SOLIRIS are at increased risk for invasive disease caused by Neisseria meningitidis, even if they develop antibodies following vaccination. Monitor patients for early signs and symptoms of serious meningococcal infections and evaluate immediately if infection is suspected.
Because of the risk of serious meningococcal infections, SOLIRIS is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
-
1 INDICATIONS AND USAGE
1.1 Paroxysmal Nocturnal Hemoglobinuria (PNH)
SOLIRIS is indicated for the treatment of patients with paroxysmal nocturnal hemoglobinuria (PNH) to reduce hemolysis.
1.2 Atypical Hemolytic Uremic Syndrome (aHUS)
SOLIRIS is indicated for the treatment of patients with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiopathy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Vaccination and Prophylaxis for Meningococcal Infection
Vaccinate patients against meningococcal infection (serogroups A, C, W, Y and B) according to current ACIP recommendations at least 2 weeks prior to initiation of SOLIRIS [see Warnings and Precautions (5.1)].
If urgent SOLIRIS therapy is indicated in a patient who is not up to date with meningococcal vaccines according to ACIP recommendations, provide the patient with antibacterial drug prophylaxis and administer these vaccines as soon as possible.
Healthcare providers who prescribe SOLIRIS must enroll in the ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
2.2 Recommended Dosage Regimen – PNH
For patients 18 years of age and older, SOLIRIS therapy consists of:
- 600 mg weekly for the first 4 weeks, followed by
- 900 mg for the fifth dose 1 week later, then
- 900 mg every 2 weeks thereafter.
Administer SOLIRIS at the recommended dosage regimen time points, or within two days of these time points [see Warnings and Precautions (5.4)].
2.3 Recommended Dosage Regimen – aHUS
For patients 18 years of age and older, SOLIRIS therapy consists of:
- 900 mg weekly for the first 4 weeks, followed by
- 1200 mg for the fifth dose 1 week later, then
- 1200 mg every 2 weeks thereafter.
For patients less than 18 years of age, administer SOLIRIS based upon body weight, according to the following schedule (Table 1):
Table 1: Dosing Recommendations in aHUS Patients Less Than 18 Years of Age Patient Body Weight Induction Maintenance 40 kg and over 900 mg weekly × 4 doses 1200 mg at week 5;
then 1200 mg every 2 weeks30 kg to less than 40 kg 600 mg weekly × 2 doses 900 mg at week 3;
then 900 mg every 2 weeks20 kg to less than 30 kg 600 mg weekly × 2 doses 600 mg at week 3;
then 600 mg every 2 weeks10 kg to less than 20 kg 600 mg weekly × 1 dose 300 mg at week 2;
then 300 mg every 2 weeks5 kg to less than 10 kg 300 mg weekly × 1 dose 300 mg at week 2;
then 300 mg every 3 weeksAdminister SOLIRIS at the recommended dosage regimen time points, or within two days of these time points.
2.4 Recommended Dosage Regimen – gMG and NMOSD
For adult patients with generalized myasthenia gravis or neuromyelitis optica spectrum disorder, SOLIRIS therapy consists of:
- 900 mg weekly for the first 4 weeks, followed by
- 1200 mg for the fifth dose 1 week later, then
- 1200 mg every 2 weeks thereafter.
Administer SOLIRIS at the recommended dosage regimen time points, or within two days of these time points.
2.5 Dose Adjustment in Case of Plasmapheresis, Plasma Exchange, or Fresh Frozen Plasma Infusion
For adult and pediatric patients with aHUS, and adult patients with gMG or NMOSD, supplemental dosing of SOLIRIS is required in the setting of concomitant plasmapheresis or plasma exchange, or fresh frozen plasma infusion (PE/PI) (Table 2).
Table 2: Supplemental Dose of SOLIRIS after PE/PI Type of Plasma Intervention Most Recent SOLIRIS Dose Supplemental SOLIRIS Dose with Each Plasma Intervention Timing of Supplemental SOLIRIS Dose Plasmapheresis or plasma exchange 300 mg 300 mg per each plasmapheresis or plasma exchange session Within 60 minutes after each plasmapheresis or plasma exchange ≥600 mg 600 mg per each plasmapheresis or plasma exchange session Fresh frozen plasma infusion ≥300 mg 300 mg per infusion of fresh frozen plasma 60 minutes prior to each infusion of fresh frozen plasma 2.6 Preparation
Dilute SOLIRIS to a final admixture concentration of 5 mg/mL using the following steps:
- Withdraw the required amount of SOLIRIS from the vial into a sterile syringe.
- Transfer the recommended dose to an infusion bag.
- Dilute SOLIRIS to a final concentration of 5 mg/mL by adding the appropriate amount (equal volume of diluent to drug volume) of 0.9% Sodium Chloride Injection, USP; 0.45% Sodium Chloride Injection, USP; 5% Dextrose in Water Injection, USP; or Ringer's Injection, USP to the infusion bag.
The final admixed SOLIRIS 5 mg/mL infusion volume is 60 mL for 300 mg doses, 120 mL for 600 mg doses, 180 mL for 900 mg doses or 240 mL for 1200 mg doses (Table 3).
Table 3: Preparation and Reconstitution of SOLIRIS SOLIRIS Dose Diluent Volume Final Volume 300 mg 30 mL 60 mL 600 mg 60 mL 120 mL 900 mg 90 mL 180 mL 1200 mg 120 mL 240 mL Gently invert the infusion bag containing the diluted SOLIRIS solution to ensure thorough mixing of the product and diluent. Discard any unused portion left in a vial, as the product contains no preservatives.
Prior to administration, the admixture should be allowed to adjust to room temperature [18°-25° C, 64°-77° F]. The admixture must not be heated in a microwave or with any heat source other than ambient air temperature.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
2.7 Administration
Only administer as an intravenous infusion.
Do not administer as an intravenous push or bolus injection.
Administer the SOLIRIS admixture by intravenous infusion over 35 minutes in adults and 1 to 4 hours in pediatric patients via gravity feed, a syringe-type pump, or an infusion pump. Admixed solutions of SOLIRIS are stable for 24 h at 2°-8° C (36°-46° F) and at room temperature.
If an adverse reaction occurs during the administration of SOLIRIS, the infusion may be slowed or stopped at the discretion of the physician. If the infusion is slowed, the total infusion time should not exceed two hours in adults. Monitor the patient for at least one hour following completion of the infusion for signs or symptoms of an infusion-related reaction.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
SOLIRIS is contraindicated for initiation in patients with unresolved serious Neisseria meningitidis infection [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Meningococcal Infections
SOLIRIS, a complement inhibitor, increases a patient's susceptibility to serious, life-threatening, or fatal infections caused by meningococcal bacteria (septicemia and/or meningitis) in any serogroup, including non-groupable strains. Life-threatening and fatal meningococcal infections have occurred in both vaccinated and unvaccinated patients treated with complement inhibitors. The initiation of SOLIRIS treatment is contraindicated in patients with unresolved serious Neisseria meningitidis infection.
Complete or update meningococcal vaccination (for serogroups A, C, W, Y and B) at least 2 weeks prior to administration of the first dose of SOLIRIS, according to current ACIP recommendations for patients receiving a complement inhibitor. Revaccinate patients in accordance with ACIP recommendations, considering the duration of therapy with SOLIRIS. Note that ACIP recommends an administration schedule in patients receiving complement inhibitors that differs from the administration schedule in the vaccine prescribing information. If urgent SOLIRIS therapy is indicated in a patient who is not up to date with meningococcal vaccines according to ACIP recommendations, provide the patient with antibacterial drug prophylaxis and administer meningococcal vaccines as soon as possible. Various durations and regimens of antibacterial drug prophylaxis have been considered, but the optimal durations and drug regimens for prophylaxis and their efficacy have not been studied in unvaccinated or vaccinated patients receiving complement inhibitors, including SOLIRIS. The benefits and risks of treatment with SOLIRIS, as well as the benefits and risks of antibacterial drug prophylaxis in unvaccinated or vaccinated patients, must be considered against the known risks for serious infections caused by Neisseria meningitidis.
Vaccination does not eliminate the risk of serious meningococcal infections, despite development of antibodies following vaccination.
Closely monitor patients for early signs and symptoms of meningococcal infection and evaluate patients immediately if infection is suspected. Inform patients of these signs and symptoms and instruct patients to seek immediate medical care if these signs and symptoms occur. Promptly treat known infections. Meningococcal infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider interruption of SOLIRIS in patients who are undergoing treatment for serious meningococcal infection depending on the risks of interrupting treatment in the disease being treated.
SOLIRIS is available only through a restricted program under a REMS [see Warnings and Precautions (5.2)].
5.2 ULTOMIRIS and SOLIRIS REMS
SOLIRIS is available only through a restricted program under a REMS called ULTOMIRIS and SOLIRIS REMS, because of the risk of serious meningococcal infections [see Warnings and Precautions (5.1)].
Notable requirements of the ULTOMIRIS and SOLIRIS REMS include the following:
- Prescribers must enroll in the REMS.
- Prescribers must counsel patients about the risk of serious meningococcal infection.
- Prescribers must provide the patients with the REMS educational materials.
- Prescribers must assess patient vaccination status for meningococcal vaccines (against serogroups A, C, W, Y and B) and vaccinate if needed according to current ACIP recommendations two weeks prior to the first dose of SOLIRIS.
- Prescribers must provide a prescription for antibacterial drug prophylaxis if treatment must be started urgently and the patient is not up to date with meningococcal vaccines according to current ACIP recommendations at least two weeks prior to the first dose of SOLIRIS.
- Healthcare settings and pharmacies that dispense SOLIRIS must be certified in the REMS and must verify prescribers are certified.
- Patients must receive counseling from the prescriber about the need to receive meningococcal vaccines per ACIP recommendations, the need to take antibiotics as directed by the prescriber, and the signs and symptoms of meningococcal infection.
- Patients must be instructed to carry the Patient Safety Card with them at all times during and for 3 months following treatment with SOLIRIS.
Further information is available at www.UltSolREMS.com or 1-888-765-4747.
5.3 Other Infections
Serious infections with Neisseria species (other than Neisseria meningitidis), including disseminated gonococcal infections, have been reported.
SOLIRIS blocks terminal complement activation; therefore patients may have increased susceptibility to infections, especially with encapsulated bacteria such as infections with Neisseria meningitidis but also Streptococcus pneumoniae, Haemophilus influenzae, and to a lesser extent, Neisseria gonorrhoeae. Additionally, Aspergillus infections have occurred in immunocompromised and neutropenic patients. Children treated with SOLIRIS may be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenzae type b (Hib). Administer vaccinations for the prevention of Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) infections according to ACIP recommendations. Patients receiving SOLIRIS are at increased risk for infections due to these organisms, even if they develop antibodies following vaccination.
5.4 Monitoring Disease Manifestations after SOLIRIS Discontinuation
Treatment Discontinuation for PNH
Monitor patients after discontinuing SOLIRIS for at least 8 weeks to detect hemolysis.
Treatment Discontinuation for aHUS
After discontinuing SOLIRIS, monitor patients with aHUS for signs and symptoms of thrombotic microangiopathy (TMA) complications for at least 12 weeks. In aHUS clinical trials, 18 patients (5 in the prospective studies) discontinued SOLIRIS treatment. TMA complications occurred following a missed dose in 5 patients, and SOLIRIS was reinitiated in 4 of these 5 patients.
Clinical signs and symptoms of TMA include changes in mental status, seizures, angina, dyspnea, or thrombosis. In addition, the following changes in laboratory parameters may identify a TMA complication: occurrence of two, or repeated measurement of any one of the following: a decrease in platelet count by 25% or more compared to baseline or the peak platelet count during SOLIRIS treatment; an increase in serum creatinine by 25% or more compared to baseline or nadir during SOLIRIS treatment; or, an increase in serum LDH by 25% or more over baseline or nadir during SOLIRIS treatment.
If TMA complications occur after SOLIRIS discontinuation, consider reinstitution of SOLIRIS treatment, plasma therapy [plasmapheresis, plasma exchange, or fresh frozen plasma infusion (PE/PI)], or appropriate organ-specific supportive measures.
5.5 Thrombosis Prevention and Management
The effect of withdrawal of anticoagulant therapy during SOLIRIS treatment has not been established. Therefore, treatment with SOLIRIS should not alter anticoagulant management.
5.6 Infusion-Related Reactions
Administration of SOLIRIS may result in infusion-related reactions, including anaphylaxis or other hypersensitivity reactions. In clinical trials, no patients experienced an infusion-related reaction which required discontinuation of SOLIRIS. Interrupt SOLIRIS infusion and institute appropriate supportive measures if signs of cardiovascular instability or respiratory compromise occur.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the labeling:
- Serious Meningococcal Infections [see Warnings and Precautions (5.1)]
- Other Infections [see Warnings and Precautions (5.3)]
- Monitoring Disease Manifestations after SOLIRIS Discontinuation [see Warnings and Precautions (5.4)]
- Thrombosis Prevention and Management [see Warnings and Precautions (5.5)]
- Infusion-Related Reactions [see Warnings and Precautions (5.6)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Meningococcal infections are the most important adverse reactions experienced by patients receiving SOLIRIS. In PNH clinical studies, two patients experienced meningococcal sepsis. Both patients had previously received a meningococcal vaccine. In clinical studies among patients without PNH, meningococcal meningitis occurred in one unvaccinated patient. Meningococcal sepsis occurred in one previously vaccinated patient enrolled in the retrospective aHUS study during the post-study follow-up period [see Warnings and Precautions (5.1)].
PNH
The data described below reflect exposure to SOLIRIS in 196 adult patients with PNH, age 18-85, of whom 55% were female. All had signs or symptoms of intravascular hemolysis. SOLIRIS was studied in a placebo-controlled clinical study (PNH Study 1, in which 43 patients received SOLIRIS and 44, placebo); a single arm clinical study (PNH Study 2); and a long term extension study (E05-001). 182 patients were exposed for greater than one year. All patients received the recommended SOLIRIS dose regimen.
Table 4 summarizes the adverse reactions that occurred at a numerically higher rate in the SOLIRIS group than the placebo group and at a rate of 5% or more among patients treated with SOLIRIS.
Table 4: Adverse Reactions Reported in 5% or More of SOLIRIS Treated Patients with PNH and Greater than Placebo in the Controlled Clinical Study Reaction SOLIRIS
(N=43)
N (%)Placebo
(N=44)
N (%)Headache 19 (44) 12 (27) Nasopharyngitis 10 (23) 8 (18) Back pain 8 (19) 4 (9) Nausea 7 (16) 5 (11) Fatigue 5 (12) 1 (2) Cough 5 (12) 4 (9) Herpes simplex infections 3 (7) 0 Sinusitis 3 (7) 0 Respiratory tract infection 3 (7) 1 (2) Constipation 3 (7) 2 (5) Myalgia 3 (7) 1 (2) Pain in extremity 3 (7) 1 (2) Influenza-like illness 2 (5) 1 (2) In the placebo-controlled clinical study, serious adverse reactions occurred among 4 (9%) patients receiving SOLIRIS and 9 (21%) patients receiving placebo. The serious reactions included infections and progression of PNH. No deaths occurred in the study and no patients receiving SOLIRIS experienced a thrombotic event; one thrombotic event occurred in a patient receiving placebo.
Among 193 patients with PNH treated with SOLIRIS in the single arm, clinical study or the follow-up study, the adverse reactions were similar to those reported in the placebo-controlled clinical study. Serious adverse reactions occurred among 16% of the patients in these studies. The most common serious adverse reactions were: viral infection (2%), headache (2%), anemia (2%), and pyrexia (2%).
aHUS
The safety of SOLIRIS therapy in patients with aHUS was evaluated in four prospective, single-arm studies, three in adult and adolescent patients (Studies C08-002A/B, C08-003A/B, and C10-004), one in pediatric and adolescent patients (Study C10-003), and one retrospective study (Study C09-001r).
The data described below were derived from 78 adult and adolescent patients with aHUS in Studies C08-002A/B, C08-003A/B and C10-004. All patients received the recommended dosage of SOLIRIS. Median exposure was 67 weeks (range: 2-145 weeks). Table 5 summarizes all adverse events reported in at least 10% of patients in Studies C08-002A/B, C08-003A/B and C10-004 combined.
Table 5: Per Patient Incidence of Adverse Events in 10% or More Adult and Adolescent Patients Enrolled in Studies C08-002A/B, C08-003A/B and C10-004 Separately and in Total Number (%) of Patients C08-002A/B
(N=17)C08-003A/B
(N=20)C10-004
(N=41)Total
(N=78)- *
- includes the preferred terms hypertension, accelerated hypertension, and malignant hypertension.
Vascular Disorders Hypertension* 10 (59) 9 (45) 7 (17) 26 (33) Hypotension 2 (12) 4 (20) 7 (17) 13 (17) Infections and Infestations Bronchitis 3 (18) 2 (10) 4 (10) 9 (12) Nasopharyngitis 3 (18) 11 (55) 7 (17) 21 (27) Gastroenteritis 3 (18) 4 (20) 2 (5) 9 (12) Upper respiratory tract infection 5 (29) 8 (40) 2 (5) 15 (19) Urinary tract infection 6 (35) 3 (15) 8 (20) 17 (22) Gastrointestinal Disorders Diarrhea 8 (47) 8 (40) 12 (32) 29 (37) Vomiting 8 (47) 9 (45) 6 (15) 23 (30) Nausea 5 (29) 8 (40) 5 (12) 18 (23) Abdominal pain 3 (18) 6 (30) 6 (15) 15 (19) Nervous System Disorders Headache 7 (41) 10 (50) 15 (37) 32 (41) Blood and Lymphatic System Disorders Anemia 6 (35) 7 (35) 7 (17) 20 (26) Leukopenia 4 (24) 3 (15) 5 (12) 12 (15) Psychiatric Disorders Insomnia 4 (24) 2 (10) 5 (12) 11 (14) Renal and Urinary Disorders Renal Impairment 5 (29) 3 (15) 6 (15) 14 (18) Proteinuria 2 (12) 1 (5) 5 (12) 8 (10) Respiratory, Thoracic and Mediastinal Disorders Cough 4 (24) 6 (30) 8 (20) 18 (23) General Disorders and Administration Site Conditions Fatigue 3 (18) 4 (20) 3 (7) 10 (13) Peripheral edema 5 (29) 4 (20) 9 (22) 18 (23) Pyrexia 4 (24) 5 (25) 7 (17) 16 (21) Asthenia 3 (18) 4 (20) 6 (15) 13 (17) Eye Disorder 5 (29) 2 (10) 8 (20) 15 (19) Metabolism and Nutrition Disorders Hypokalemia 3 (18) 2 (10) 4 (10) 9 (12) Neoplasms benign, malignant, and unspecified (including cysts and polyps) 1 (6) 6 (30) 1 (20) 8 (10) Skin and Subcutaneous Tissue Disorders Rash 2 (12) 3 (15) 6 (15) 11 (14) Pruritus 1 (6) 3 (15) 4 (10) 8 (10) Musculoskeletal and Connective Tissue Disorders Arthralgia 1 (6) 2 (10) 7 (17) 10 (13) Back pain 3 (18) 3 (15) 2 (5) 8 (10) In Studies C08-002A/B, C08-003A/B and C10-004 combined, 60% (47/78) of patients experienced a serious adverse event (SAE). The most commonly reported SAEs were infections (24%), hypertension (5%), chronic renal failure (5%), and renal impairment (5%). Five patients discontinued SOLIRIS due to adverse events; three due to worsening renal function, one due to new diagnosis of Systemic Lupus Erythematosus, and one due to meningococcal meningitis.
Study C10-003 included 22 pediatric and adolescent patients, of which 18 patients were less than 12 years of age. All patients received the recommended dosage of SOLIRIS. Median exposure was 44 weeks (range: 1 dose-87 weeks).
Table 6 summarizes all adverse events reported in at least 10% of patients enrolled in Study C10-003.
Table 6: Per Patient Incidence of Adverse Reactions in 10% or More Patients Enrolled in Study C10-003 1 month to <12 yrs
(N=18)Total
(N=22)Eye Disorders 3 (17) 3 (14) Gastrointestinal Disorders Abdominal pain 6 (33) 7 (32) Diarrhea 5 (28) 7 (32) Vomiting 4 (22) 6 (27) Dyspepsia 0 3 (14) General Disorders and Administration Site Conditions Pyrexia 9 (50) 11 (50) Infections and Infestations Upper respiratory tract infection 5 (28) 7 (32) Nasopharyngitis 3 (17) 6 (27) Rhinitis 4 (22) 4 (18) Urinary Tract infection 3 (17) 4 (18) Catheter site infection 3 (17) 3 (14) Musculoskeletal and Connective Tissue Disorders Muscle spasms 2 (11) 3 (14) Nervous System Disorders Headache 3 (17) 4 (18) Renal and Urinary Disorders 3 (17) 4 (18) Respiratory, Thoracic and Mediastinal Disorders Cough 7 (39) 8 (36) Oropharyngeal pain 1 (6) 3 (14) Skin and Subcutaneous Tissue Disorders Rash 4 (22) 4 (18) Vascular Disorders Hypertension 4 (22) 4 (18) In Study C10-003, 59% (13/22) of patients experienced a serious adverse event (SAE). The most commonly reported SAEs were hypertension (9%), viral gastroenteritis (9%), pyrexia (9%), and upper respiratory infection (9%). One patient discontinued SOLIRIS due to an adverse event (severe agitation).
Analysis of retrospectively collected adverse event data from pediatric and adult patients enrolled in Study C09-001r (N=30) revealed a safety profile that was similar to that which was observed in the two prospective studies. Study C09-001r included 19 pediatric patients less than 18 years of age. Overall, the safety of SOLIRIS in pediatric patients with aHUS enrolled in Study C09-001r appeared similar to that observed in adult patients. The most common (≥15%) adverse events occurring in pediatric patients are presented in Table 7.
Table 7: Adverse Reactions Occurring in at Least 15% of Patients Less than 18 Years of Age Enrolled in Study C09-001r Number (%) of Patients < 2 yrs
(N=5)2 to < 12 yrs
(N=10)12 to <18 yrs
(N=4)Total
(N=19)- *
- includes the preferred terms upper respiratory tract infection and nasopharyngitis.
General Disorders and Administration Site Conditions Pyrexia 4 (80) 4 (40) 1 (25) 9 (47) Gastrointestinal Disorders Diarrhea 1 (20) 4 (40) 1 (25) 6 (32) Vomiting 2 (40) 1 (10) 1 (25) 4 (21) Infections and Infestations Upper respiratory tract infection* 2 (40) 3 (30) 1 (25) 6 (32) Respiratory, Thoracic and Mediastinal Disorders Cough 3 (60) 2 (20) 0 (0) 5 (26) Nasal congestion 2 (40) 2 (20) 0 (0) 4 (21) Cardiac Disorders Tachycardia 2 (40) 2 (20) 0 (0) 4 (21) Generalized Myasthenia Gravis (gMG)
In a 26-week placebo-controlled trial evaluating the effect of SOLIRIS for the treatment of gMG (gMG Study 1), 62 patients received SOLIRIS at the recommended dosage regimen and 63 patients received placebo [see Clinical Studies (14.3)]. Patients were 19 to 79 years of age, and 66% were female. Table 8 displays the most common adverse reactions from gMG Study 1 that occurred in ≥5% of SOLIRIS-treated patients and at a greater frequency than on placebo.
Table 8: Adverse Reactions Reported in 5% or More of SOLIRIS-Treated Patients in gMG Study 1 and at a Greater Frequency than in Placebo-Treated Patients SOLIRIS
(N=62)
N (%)Placebo
(N=63)
N (%)Gastrointestinal Disorders Abdominal pain 5 (8) 3 (5) General Disorders and Administration Site Conditions Peripheral edema 5 (8) 3 (5) Pyrexia 4 (7) 2 (3) Infections and Infestations Herpes simplex virus infections 5 (8) 1 (2) Injury, Poisoning, and Procedural Complications Contusion 5 (8) 2 (3) Musculoskeletal and Connective Tissue Disorders Musculoskeletal pain 9 (15) 5 (8) The most common adverse reactions (≥10%) that occurred in SOLIRIS-treated patients in the long-term extension to gMG Study 1, Study ECU-MG-302, and that are not included in Table 8 were headache (26%), nasopharyngitis (24%), diarrhea (15%), arthralgia (12%), upper respiratory tract infection (11%), and nausea (10%).
Neuromyelitis Optica Spectrum Disorder (NMOSD)
In a placebo-controlled trial evaluating the effect of SOLIRIS for the treatment of NMOSD (NMOSD Study 1), 96 patients received SOLIRIS at the recommended dosage regimen and 47 patients received placebo [see Clinical Studies (14.4)]. Patients were 19 to 75 years of age (mean 44 years of age), and 91% were female. Table 9 displays the most common adverse reactions from NMOSD Study 1 that occurred in ≥5% of SOLIRIS-treated patients and at a greater frequency than on placebo.
Table 9: Adverse Reactions Reported in 5% or More of SOLIRIS-Treated Patients in NMOSD Study 1 and at a Greater Frequency than in Placebo-Treated Patients SOLIRIS
(N=96)
N (%)Placebo
(N=47)
N (%)Events/Patients 1295/88 617/45 Blood and lymphatic system disorders Leukopenia 5 (5) 1 (2) Lymphopenia 5 (5) 0 (0) Eye disorders Cataract 6 (6) 2 (4) Gastrointestinal disorders Diarrhea 15 (16) 7 (15) Constipation 9 (9) 3 (6) General disorders and administration site conditions Asthenia 5 (5) 1 (2) Infections and infestations Upper respiratory tract infection 28 (29) 6 (13) Nasopharyngitis 20 (21) 9 (19) Influenza 11 (11) 2 (4) Pharyngitis 10 (10) 3 (6) Bronchitis 9 (9) 3 (6) Conjunctivitis 9 (9) 4 (9) Cystitis 8 (8) 1 (2) Hordeolum 7 (7) 0 (0) Sinusitis 6 (6) 0 (0) Cellulitis 5 (5) 1 (2) Injury, poisoning and procedural complications Contusion 10 (10) 2 (4) Metabolism and nutrition disorders Decreased appetite 5 (5) 1 (2) Musculoskeletal and connective tissue disorders Back pain 14 (15) 6 (13) Arthralgia 11 (11) 5 (11) Musculoskeletal pain 6 (6) 0 (0) Muscle spasms 5 (5) 2 (4) Nervous system disorders Dizziness 14 (15) 6 (13) Paraesthesia 8 (8) 3 (6) Respiratory, thoracic and mediastinal disorders Oropharyngeal pain 7 (7) 2 (4) Skin and subcutaneous tissue disorders Alopecia 5 (5) 2 (4) 6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of SOLIRIS. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to SOLIRIS exposure.
Fatal or serious infections: Neisseria gonorrhoeae, Neisseria meningitidis, Neisseria sicca/subflava, Neisseria spp unspecified.
-
7 DRUG INTERACTIONS
7.1 Plasmapheresis, Plasma Exchange, or Fresh Frozen Plasma Infusion
Concomitant use of SOLIRIS with plasma exchange (PE), plasmapheresis (PP) or fresh frozen plasma infusion (PE/PI) treatment can reduce serum eculizumab concentrations and requires a supplemental dose of SOLIRIS [see Dosage and Administration (2.5)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited data on outcomes of pregnancies that have occurred following SOLIRIS use in pregnant women have not identified a concern for specific adverse developmental outcomes (see Data). There are risks to the mother and fetus associated with untreated paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS) in pregnancy (see Clinical Considerations).
Animal studies using a mouse analogue of the SOLIRIS molecule (murine anti-C5 antibody) showed increased rates of developmental abnormalities and an increased rate of dead and moribund offspring at doses 2-8 times the human dose (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or fetal/neonatal risk
PNH in pregnancy is associated with adverse maternal outcomes, including worsening cytopenias, thrombotic events, infections, bleeding, miscarriages and increased maternal mortality, and adverse fetal outcomes, including fetal death and premature delivery.
aHUS in pregnancy is associated with adverse maternal outcomes, including pre-eclampsia and preterm delivery, and adverse fetal/neonatal outcomes, including intrauterine growth restriction (IUGR), fetal death and low birth weight.
Data
Human Data
A pooled analysis of prospectively (50.3%) and retrospectively (49.7%) collected data in more than 300 pregnant women with live births following exposure to SOLIRIS have not suggested safety concerns. However, these data cannot definitively exclude any drug-associated risk during pregnancy, because of the limited sample size.
Animal Data
Animal reproduction studies were conducted in mice using doses of a murine anti-C5 antibody that approximated 2-4 times (low dose) and 4-8 times (high dose) the recommended human SOLIRIS dose, based on a body weight comparison. When animal exposure to the antibody occurred in the time period from before mating until early gestation, no decrease in fertility or reproductive performance was observed. When maternal exposure to the antibody occurred during organogenesis, two cases of retinal dysplasia and one case of umbilical hernia were observed among 230 offspring born to mothers exposed to the higher antibody dose; however, the exposure did not increase fetal loss or neonatal death. When maternal exposure to the antibody occurred in the time period from implantation through weaning, a higher number of male offspring became moribund or died (1/25 controls, 2/25 low dose group, 5/25 high dose group). Surviving offspring had normal development and reproductive function.
8.2 Lactation
Risk Summary
Although limited published data does not report detectable levels of eculizumab in human milk, maternal IgG is known to be present in human milk. Available information is insufficient to inform the effect of eculizumab on the breastfed infant. There are no data on the effects of eculizumab on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SOLIRIS and any potential adverse effects on the breastfed child from eculizumab or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of SOLIRIS for the treatment of PNH, gMG, or NMOSD in pediatric patients have not been established.
The safety and effectiveness of SOLIRIS for the treatment of aHUS have been established in pediatric patients. Use of SOLIRIS in pediatric patients for this indication is supported by evidence from four adequate and well-controlled clinical studies assessing the safety and effectiveness of SOLIRIS for the treatment of aHUS. The studies included a total of 47 pediatric patients (ages 2 months to 17 years). The safety and effectiveness of SOLIRIS for the treatment of aHUS appear similar in pediatric and adult patients [see Adverse Reactions (6.1), and Clinical Studies (14.2)].
Administer vaccinations for the prevention of infection due to Neisseria meningitidis, Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) according to ACIP guidelines [see Warnings and Precautions (5.1, 5.3)].
8.5 Geriatric Use
Fifty-one patients 65 years of age or older (15 with PNH, 4 with aHUS, 26 with gMG, and 6 with NMOSD) were treated with SOLIRIS in clinical trials in the approved indications. Although there were no apparent age-related differences observed in these studies, the number of patients aged 65 and over is not sufficient to determine whether they respond differently from younger patients.
-
11 DESCRIPTION
Eculizumab, a complement inhibitor, is a recombinant humanized monoclonal IgG2/4κ antibody produced by murine myeloma cell culture and purified by standard bioprocess technology. Eculizumab contains human constant regions from human IgG2 sequences and human IgG4 sequences and murine complementarity-determining regions grafted onto the human framework light- and heavy-chain variable regions. Eculizumab is composed of two 448 amino acid heavy chains and two 214 amino acid light chains and has a molecular weight of approximately 148 kDa.
SOLIRIS (eculizumab) injection is a sterile, clear, colorless, preservative-free 10 mg/mL solution for intravenous infusion and is supplied in 30-mL single-dose vials. The product is formulated at pH 7 and each 30 mL vial contains 300 mg of eculizumab, polysorbate 80 (6.6 mg) (vegetable origin), sodium chloride (263.1 mg), sodium phosphate dibasic (53.4 mg), sodium phosphate monobasic (13.8 mg), and Water for Injection, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Eculizumab, the active ingredient in SOLIRIS, is a monoclonal antibody that specifically binds to the complement protein C5 with high affinity, thereby inhibiting its cleavage to C5a and C5b and preventing the generation of the terminal complement complex C5b-9.
SOLIRIS inhibits terminal complement-mediated intravascular hemolysis in PNH patients and complement-mediated thrombotic microangiopathy (TMA) in patients with aHUS.
The precise mechanism by which eculizumab exerts its therapeutic effect in gMG patients is unknown, but is presumed to involve reduction of terminal complement complex C5b-9 deposition at the neuromuscular junction.
The precise mechanism by which eculizumab exerts its therapeutic effect in NMOSD is unknown, but is presumed to involve inhibition of aquaporin-4-antibody induced terminal complement C5b-9 deposition.
12.2 Pharmacodynamics
In the placebo-controlled clinical study (PNH Study 1), SOLIRIS when administered as recommended reduced serum LDH levels from 2200 ± 1034 U/L (mean ± SD) at baseline to 700 ± 388 U/L by week one and maintained the effect through the end of the study at week 26 (327 ± 433 U/L) in patients with PNH. In the single arm clinical study (PNH Study 2), the effect was maintained through week 52 [see Clinical Studies (14)].
In patients with PNH, aHUS, gMG, and NMOSD, free C5 concentrations of < 0.5 mcg/mL was correlated with complete blockade of terminal complement activity.
12.3 Pharmacokinetics
Following intravenous maintenance doses of 900 mg once every 2 weeks in patients with PNH, the week 26 observed mean ± SD serum eculizumab maximum concentration (Cmax) was 194 ± 76 mcg/mL and the trough concentration (Ctrough) was 97 ± 60 mcg/mL. Following intravenous maintenance doses of 1200 mg once every 2 weeks in patients with aHUS, the week 26 observed mean ± SD Ctrough was 242 ± 101 mcg/mL. Following intravenous maintenance doses of 1200 mg once every 2 weeks in patients with gMG, the week 26 observed mean ± SD Cmax was 783 ± 288 mcg/mL and the Ctrough was 341 ± 172 mcg/mL. Following intravenous maintenance doses of 1200 mg once every 2 weeks in patients with NMOSD, at week 24, the observed mean±SD Cmax was 877±331 and the Ctrough was 429±188 mcg/mL.
Steady state was achieved 4 weeks after starting eculizumab treatment, with accumulation ratio of approximately 2-fold in all studied indications. Population pharmacokinetic analyses showed that eculizumab pharmacokinetics were dose-linear and time-independent over the 600 mg to 1200 mg dose range, with inter-individual variability of 21% to 38%.
Elimination
The half-life of eculizumab was approximately 270 h to 414 h.
Plasma exchange or infusion increased the clearance of eculizumab by approximately 250-fold and reduced the half-life to 1.26 h. Supplemental dosing is recommended when SOLIRIS is administered to patients receiving plasma exchange or infusion [see Dosage and Administration (2.5)].
Specific Populations
Age, Sex, and Race:
The pharmacokinetics of eculizumab were not affected by age (2 months to 85 years), sex, or race.
Renal Impairment:
Renal function did not affect the pharmacokinetics of eculizumab in PNH (creatinine clearance of 8 mL/min to 396 mL/min calculated using Cockcroft-Gault formula), aHUS (estimated glomerular filtration rate [eGFR] of 5 mL/min/1.73 m2 to105 mL/min/1.73 m2 using the Modification of Diet in Renal Disease [MDRD] formula), or gMG patients (eGFR of 44 mL/min/1.73 m2 to 168 mL/min/1.73 m2 using MDRD formula).
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of SOLIRIS or of other eculizumab products.
The immunogenicity of SOLIRIS has been evaluated using two different immunoassays for the detection of anti-eculizumab antibodies: a direct enzyme-linked immunosorbent assay (ELISA) using the Fab fragment of eculizumab as target was used for the PNH indication; and an electro-chemiluminescence (ECL) bridging assay using the eculizumab whole molecule as target was used for the aHUS, gMG, and NMOSD indications, as well as for additional patients with PNH. In the PNH population, antibodies to SOLIRIS were detected in 3/196 (2%) patients using the ELISA assay and in 5/161 (3%) patients using the ECL assay during the entire treatment period. In the aHUS population, antibodies to SOLIRIS were detected in 3/100 (3%) patients using the ECL assay during the entire treatment period. None of the 62 patients with gMG had antibodies to SOLIRIS detected following the 26-week active treatment. Two of the 96 (2%) SOLIRIS-treated patients with NMOSD had antibodies to SOLIRIS detected during the entire treatment period.
An ECL based neutralizing assay with a low sensitivity of 2 mcg/mL was performed to detect neutralizing antibodies for the 5 patients with PNH, the 3 patients with aHUS, and the 2 patients with NMOSD with anti-eculizumab antibody positive samples using the ECL assay. Two of 161 patients with PNH (1.2%) and 1 of 100 patients with aHUS (1%), and none of the 96 patients with NMOSD had low positive values for neutralizing antibodies.
No apparent correlation of antibody development to clinical response was observed.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal carcinogenicity studies of eculizumab have not been conducted.
Genotoxicity studies have not been conducted with eculizumab.
Effects of eculizumab upon fertility have not been studied in animals. Intravenous injections of male and female mice with a murine anti-C5 antibody at up to 4-8 times the equivalent of the clinical dose of SOLIRIS had no adverse effects on mating or fertility.
-
14 CLINICAL STUDIES
14.1 Paroxysmal Nocturnal Hemoglobinuria (PNH)
The safety and efficacy of SOLIRIS in PNH patients with hemolysis were assessed in a randomized, double-blind, placebo-controlled 26 week study (PNH Study 1, NCT00122330); PNH patients were also treated with SOLIRIS in a single arm 52 week study (PNH Study 2, NCT00122304) and in a long-term extension study (E05-001, NCT00122317). Patients received meningococcal vaccination prior to receipt of SOLIRIS. In all studies, the dose of SOLIRIS was 600 mg study drug every 7 ± 2 days for 4 weeks, followed by 900 mg 7 ± 2 days later, then 900 mg every 14 ± 2 days for the study duration. SOLIRIS was administered as an intravenous infusion over 25 - 45 minutes.
PNH Study 1:
PNH patients with at least four transfusions in the prior 12 months, flow cytometric confirmation of at least 10% PNH cells and platelet counts of at least 100,000/microliter were randomized to either SOLIRIS (n = 43) or placebo (n = 44). Prior to randomization, all patients underwent an initial observation period to confirm the need for RBC transfusion and to identify the hemoglobin concentration (the "set-point") which would define each patient's hemoglobin stabilization and transfusion outcomes. The hemoglobin set-point was less than or equal to 9 g/dL in patients with symptoms and was less than or equal to 7 g/dL in patients without symptoms. Endpoints related to hemolysis included the numbers of patients achieving hemoglobin stabilization, the number of RBC units transfused, fatigue, and health-related quality of life. To achieve a designation of hemoglobin stabilization, a patient had to maintain a hemoglobin concentration above the hemoglobin set-point and avoid any RBC transfusion for the entire 26 week period. Hemolysis was monitored mainly by the measurement of serum LDH levels, and the proportion of PNH RBCs was monitored by flow cytometry. Patients receiving anticoagulants and systemic corticosteroids at baseline continued these medications.
Major baseline characteristics were balanced (see Table 10).
Table 10: PNH Study 1 Patient Baseline Characteristics Study 1 Parameter Placebo
(N=44)SOLIRIS
(N=43)Mean age (SD) 38 (13) 42 (16) Gender - female (%) 29 (66) 23 (54) History of aplastic anemia or myelodysplastic syndrome (%) 12 (27) 8 (19) Patients with history of thrombosis (events) 8 (11) 9 (16) Concomitant anticoagulants (%) 20 (46) 24 (56) Concomitant steroids/immunosuppressant treatments (%) 16 (36) 14 (33) Packed RBC units transfused per patient in previous 12 months (median (Q1,Q3)) 17 (14, 25) 18 (12, 24) Mean Hgb level (g/dL) at setpoint (SD) 8 (1) 8 (1) Pre-treatment LDH levels (median, U/L) 2,234 2,032 Free hemoglobin at baseline (median, mg/dL) 46 41 Patients treated with SOLIRIS had significantly reduced (p< 0.001) hemolysis resulting in improvements in anemia as indicated by increased hemoglobin stabilization and reduced need for RBC transfusions compared to placebo treated patients (see Table 11). These effects were seen among patients within each of the three pre-study RBC transfusion strata (4 - 14 units; 15 - 25 units; > 25 units). After 3 weeks of SOLIRIS treatment, patients reported less fatigue and improved health-related quality of life. Because of the study sample size and duration, the effects of SOLIRIS on thrombotic events could not be determined.
Table 11: PNH Study 1 Results Placebo
(N=44)SOLIRIS
(N=43)Percentage of patients with stabilized hemoglobin levels 0 49 Packed RBC units transfused per patient (median) 10 0 (range) (2 - 21) (0 - 16) Transfusion avoidance (%) 0 51 LDH levels at end of study (median, U/L) 2,167 239 Free hemoglobin at end of study (median, mg/dL) 62 5 PNH Study 2 and Extension Study:
PNH patients with at least one transfusion in the prior 24 months and at least 30,000 platelets/microliter received SOLIRIS over a 52-week period. Concomitant medications included anti-thrombotic agents in 63% of the patients and systemic corticosteroids in 40% of the patients. Overall, 96 of the 97 enrolled patients completed the study (one patient died following a thrombotic event). A reduction in intravascular hemolysis as measured by serum LDH levels was sustained for the treatment period and resulted in a reduced need for RBC transfusion and less fatigue. 187 SOLIRIS-treated PNH patients were enrolled in a long term extension study. All patients sustained a reduction in intravascular hemolysis over a total SOLIRIS exposure time ranging from 10 to 54 months. There were fewer thrombotic events with SOLIRIS treatment than during the same period of time prior to treatment. However, the majority of patients received concomitant anticoagulants; the effects of anticoagulant withdrawal during SOLIRIS therapy was not studied [see Warnings and Precautions (5.5)].
14.2 Atypical Hemolytic Uremic Syndrome (aHUS)
Five single-arm studies [four prospective: C08-002A/B (NCT00844545 and NCT00844844), C08-003A/B (NCT00838513 and NCT00844428), C10-003 (NCT01193348), and C10-004 (NCT01194973); and one retrospective: C09-001r (NCT01770951)] evaluated the safety and efficacy of SOLIRIS for the treatment of aHUS. Patients with aHUS received meningococcal vaccination prior to receipt of SOLIRIS or received prophylactic treatment with antibiotics until 2 weeks after vaccination. In all studies, the dose of SOLIRIS in adult and adolescent patients was 900 mg every 7 ± 2 days for 4 weeks, followed by 1200 mg 7 ± 2 days later, then 1200 mg every 14 ± 2 days thereafter. The dosage regimen for pediatric patients weighing less than 40 kg enrolled in Study C09-001r and Study C10-003 was based on body weight [see Dosage and Administration (2.3)]. Efficacy evaluations were based on thrombotic microangiopathy (TMA) endpoints.
Endpoints related to TMA included the following:
- platelet count change from baseline
- hematologic normalization (maintenance of normal platelet counts and LDH levels for at least four weeks)
- complete TMA response (hematologic normalization plus at least a 25% reduction in serum creatinine for a minimum of four weeks)
- TMA-event free status (absence for at least 12 weeks of a decrease in platelet count of >25% from baseline, plasma exchange or plasma infusion, and new dialysis requirement)
- Daily TMA intervention rate (defined as the number of plasma exchange or plasma infusion interventions and the number of new dialyses required per patient per day).
aHUS Resistant to PE/PI (Study C08-002A/B)
Study C08-002A/B enrolled patients who displayed signs of thrombotic microangiopathy (TMA) despite receiving at least four PE/PI treatments the week prior to screening. One patient had no PE/PI the week prior to screening because of PE/PI intolerance. In order to qualify for enrollment, patients were required to have a platelet count ≤150 × 109/L, evidence of hemolysis such as an elevation in serum LDH, and serum creatinine above the upper limits of normal, without the need for chronic dialysis. The median patient age was 28 (range: 17 to 68 years). Patients enrolled in Study C08-002A/B were required to have ADAMTS13 activity level above 5%; observed range of values in the trial were 70%-121%. Seventy-six percent of patients had an identified complement regulatory factor mutation or auto-antibody. Table 12 summarizes the key baseline clinical and disease-related characteristics of patients enrolled in Study C08-002A/B.
Table 12: Baseline Characteristics of Patients Enrolled in Study C08-002A/B Parameter C08-002A/B
(N=17)Time from aHUS diagnosis until screening in months, median (min, max) 10 (0.26, 236) Time from current clinical TMA manifestation until screening in months, median (min, max) <1 (<1, 4) Baseline platelet count (× 109/L), median (range) 118 (62, 161) Baseline LDH (U/L), median (range) 269 (134, 634) Patients in Study C08-002A/B received SOLIRIS for a minimum of 26 weeks. In Study C08-002A/B, the median duration of SOLIRIS therapy was approximately 100 weeks (range: 2 weeks to 145 weeks).
Renal function, as measured by eGFR, was improved and maintained during SOLIRIS therapy. The mean eGFR (± SD) increased from 23 ± 15 mL/min/1.73m2 at baseline to 56 ± 40 mL/min/1.73m2 by 26 weeks; this effect was maintained through 2 years (56 ± 30 mL/min/1.73m2). Four of the five patients who required dialysis at baseline were able to discontinue dialysis.
Reduction in terminal complement activity and an increase in platelet count relative to baseline were observed after commencement of SOLIRIS. SOLIRIS reduced signs of complement-mediated TMA activity, as shown by an increase in mean platelet counts from baseline to 26 weeks. In Study C08-002A/B, mean platelet count (± SD) increased from 109 ± 32 ×109/L at baseline to 169 ± 72 ×109/L by one week; this effect was maintained through 26 weeks (210 ± 68 ×109/L), and 2 years (205 ± 46 ×109/L). When treatment was continued for more than 26 weeks, two additional patients achieved Hematologic Normalization as well as Complete TMA response. Hematologic Normalization and Complete TMA response were maintained by all responders. In Study C08-002A/B, responses to SOLIRIS were similar in patients with and without identified mutations in genes encoding complement regulatory factor proteins.
Table 13 summarizes the efficacy results for Study C08-002A/B.
Table 13: Efficacy Results for Study C08-002A/B Efficacy Parameter Study C08-002A/B at 26 wks*
(N=17)Study C08-002A/B at 2 yrs†
(N=17)Complete TMA response, n (%) 11 (65) 13 (77) Median Duration of complete TMA response, weeks (range) 38 (25, 56) 99 (25, 139) eGFR improvement ≥15 mL/min/1.73 m2, n (%) 9 (53) 10 (59) Median duration of eGFR improvement, days (range) 251 (70, 392) ND Hematologic normalization, n (%)
Median Duration of hematologic normalization, weeks (range)13 (76)
37 (25, 62)15 (88)
99 (25, 145)TMA event-free status, n (%) 15 (88) 15 (88) Daily TMA intervention rate, median (range) Before eculizumab 0.82 (0.04, 1.52) 0.82 (0.04, 1.52) On eculizumab treatment 0 (0, 0.31) 0 (0, 0.36) aHUS Sensitive to PE/PI (Study C08-003A/B)
Study C08-003A/B enrolled patients undergoing chronic PE/PI who generally did not display hematologic signs of ongoing thrombotic microangiopathy (TMA). All patients had received PT at least once every two weeks, but no more than three times per week, for a minimum of eight weeks prior to the first SOLIRIS dose. Patients on chronic dialysis were permitted to enroll in Study C08-003A/B. The median patient age was 28 years (range: 13 to 63 years). Patients enrolled in Study C08-003A/B were required to have ADAMTS13 activity level above 5%; observed range of values in the trial were 37%-118%. Seventy percent of patients had an identified complement regulatory factor mutation or auto-antibody. Table 14 summarizes the key baseline clinical and disease-related characteristics of patients enrolled in Study C08-003A/B.
Table 14: Baseline Characteristics of Patients Enrolled in Study C08-003A/B Parameter Study C08-003A/B
(N=20)Time from aHUS diagnosis until screening in months, median (min, max) 48 (0.66, 286) Time from current clinical TMA manifestation until screening in months, median (min, max) 9 (1, 45) Baseline platelet count (× 109/L), median (range) 218 (105, 421) Baseline LDH (U/L), median (range) 200 (151, 391) Patients in Study C08-003A/B received SOLIRIS for a minimum of 26 weeks. In Study C08-003A/B, the median duration of SOLIRIS therapy was approximately 114 weeks (range: 26 to 129 weeks).
Renal function, as measured by eGFR, was maintained during SOLIRIS therapy. The mean eGFR (± SD) was 31 ± 19 mL/min/1.73m2 at baseline, and was maintained through 26 weeks (37 ± 21 mL/min/1.73m2) and 2 years (40 ± 18 mL/min/1.73m2). No patient required new dialysis with SOLIRIS.
Reduction in terminal complement activity was observed in all patients after the commencement of SOLIRIS. SOLIRIS reduced signs of complement-mediated TMA activity, as shown by an increase in mean platelet counts from baseline to 26 weeks. Platelet counts were maintained at normal levels despite the elimination of PE/PI. The mean platelet count (± SD) was 228 ± 78 × 109/L at baseline, 233 ± 69 × 109/L at week 26, and 224 ± 52 × 109/L at 2 years. When treatment was continued for more than 26 weeks, six additional patients achieved Complete TMA response. Complete TMA Response and Hematologic Normalization were maintained by all responders. In Study C08-003A/B, responses to SOLIRIS were similar in patients with and without identified mutations in genes encoding complement regulatory factor proteins.
Table 15 summarizes the efficacy results for Study C08-003A/B.
Table 15: Efficacy Results for Study C08-003A/B Efficacy Parameter Study C08-003A/B at 26 wks*
(N=20)Study C08-003A/B at 2 yrs†
(N=20)- *
- At data cut-off (September 8, 2010).
- †
- At data cut-off (April 20, 2012).
- ‡
- In Study C08-003A/B, 85% of patients had normal platelet counts and 80% of patients had normal serum LDH levels at baseline, so hematologic normalization in this population reflects maintenance of normal parameters in the absence of PE/PI.
- §
- Calculated at each post-dose day of measurement (excluding Days 1 to 4) using a repeated measurement ANOVA model.
Complete TMA response, n (%) 5 (25) 11 (55) Median duration of complete TMA response, weeks (range) 32 (12, 38) 68 (38, 109) eGFR improvement ≥15 mL/min/1.73 m2, n (%) 1 (5) 8 (40) TMA Event-free status n (%) 16 (80) 19 (95) Daily TMA intervention rate, median (range) Before eculizumab 0.23 (0.05, 1.07) 0.23 (0.05, 1.07) On eculizumab treatment 0 0 (0, 0.01) Hematologic normalization‡, n (%)
Median duration of hematologic normalization, weeks (range)§18 (90)
38 (22, 52)18 (90)
114 (33, 125)Retrospective Study in Patients with aHUS (C09-001r)
The efficacy results for the aHUS retrospective study (Study C09-001r) were generally consistent with results of the two prospective studies. SOLIRIS reduced signs of complement-mediated TMA activity, as shown by an increase in mean platelet counts from baseline. Mean platelet count (± SD) increased from 171 ± 83 ×109/L at baseline to 233 ±109 ×109/L after one week of therapy; this effect was maintained through 26 weeks (mean platelet count (± SD) at week 26: 254 ± 79 ×109/L).
A total of 19 pediatric patients (ages 2 months to 17 years) received SOLIRIS in Study C09-001r. The median duration of SOLIRIS therapy was 16 weeks (range 4 to 70 weeks) for children <2 years of age (n=5), 31 weeks (range 19 to 63 weeks) for children 2 to <12 years of age (n=10), and 38 weeks (range 1 to 69 weeks) for patients 12 to <18 years of age (n=4). Fifty-three percent of pediatric patients had an identified complement regulatory factor mutation or auto-antibody.
Overall, the efficacy results for these pediatric patients appeared consistent with what was observed in patients enrolled in Studies C08-002A/B and C08-003A/B (Table 16). No pediatric patient required new dialysis during treatment with SOLIRIS.
Table 16: Efficacy Results in Pediatric Patients Enrolled in Study C09-001r Efficacy Parameter <2 yrs
(N=5)2 to <12 yrs
(N=10)12 to <18 yrs
(N=4)Total
(N=19)- *
- Of the 9 patients who experienced an eGFR improvement of at least 15 mL/min/1.73 m2, one received dialysis throughout the study period and another received SOLIRIS as prophylaxis following renal allograft transplantation.
- †
- Platelet count normalization was defined as a platelet count of at least 150,000 × 109/L on at least two consecutive measurements spanning a period of at least 4 weeks.
Complete TMA response, n (%) 2 (40) 5 (50) 1 (25) 8 (42) Patients with eGFR improvement ≥ 15 mL/min/1.73 m2, n (%)* 2 (40) 6 (60) 1 (25) 9 (47) Platelet count normalization, n (%)† 4 (80) 10 (100) 3 (75) 17 (89) Hematologic Normalization, n (%) 2 (40) 5 (50) 1 (25) 8 (42) Daily TMA intervention rate, median (range) Before eculizumab 1 (0, 2) <1 (0.07, 1.46) <1 (0, 1) 0.31 (0.00, 2.38) On eculizumab treatment <1 (0, <1) 0 (0, <1) 0 (0, <1) 0.00 (0.00, 0.08) Adult Patients with aHUS (Study C10-004)
Study C10-004 enrolled patients who displayed signs of thrombotic microangiopathy (TMA). In order to qualify for enrollment, patients were required to have a platelet count < lower limit of normal range (LLN), evidence of hemolysis such as an elevation in serum LDH, and serum creatinine above the upper limits of normal, without the need for chronic dialysis. The median patient age was 35 (range: 18 to 80 years). All patients enrolled in Study C10-004 were required to have ADAMTS13 activity level above 5%; observed range of values in the trial were 28%-116%. Fifty-one percent of patients had an identified complement regulatory factor mutation or auto-antibody. A total of 35 patients received PE/PI prior to eculizumab. Table 17 summarizes the key baseline clinical and disease-related characteristics of patients enrolled in Study C10-004.
Table 17: Baseline Characteristics of Patients Enrolled in Study C10-004 Parameter Study C10-004
(N=41)Time from aHUS diagnosis until start of study drug in months, median (range) 0.79 (0.03 – 311) Time from current clinical TMA manifestation until first study dose in months, median (range) 0.52 (0.03-19) Baseline platelet count (× 109/L), median (range) 125 (16 – 332) Baseline LDH (U/L), median (range) 375 (131 – 3318) Patients in Study C10-004 received SOLIRIS for a minimum of 26 weeks. In Study C10-004, the median duration of SOLIRIS therapy was approximately 50 weeks (range: 13 weeks to 86 weeks).
Renal function, as measured by eGFR, was improved during SOLIRIS therapy. The mean eGFR (± SD) increased from 17 ± 12 mL/min/1.73m2 at baseline to 47 ± 24 mL/min/1.73m2 by 26 weeks. Twenty of the 24 patients who required dialysis at study baseline were able to discontinue dialysis during SOLIRIS treatment.
Reduction in terminal complement activity and an increase in platelet count relative to baseline were observed after commencement of SOLIRIS. SOLIRIS reduced signs of complement-mediated TMA activity, as shown by an increase in mean platelet counts from baseline to 26 weeks. In Study C10-004, mean platelet count (± SD) increased from 119 ± 66 ×109/L at baseline to 200 ± 84 ×109/L by one week; this effect was maintained through 26 weeks (mean platelet count (± SD) at week 26: 252 ± 70 ×109/L). In Study C10-004, responses to SOLIRIS were similar in patients with and without identified mutations in genes encoding complement regulatory factor proteins or auto-antibodies to factor H.
Table 18 summarizes the efficacy results for Study C10-004.
Table 18: Efficacy Results for Study C10-004 Efficacy Parameter Study C10-004
(N=41)Complete TMA response, n (%), 23 (56) 95% CI 40,72 Median duration of complete TMA response, weeks (range) 42 (6, 75) Patients with eGFR improvement ≥ 15 mL/min/1.73m2, n (%) 22 (54) Hematologic Normalization, n (%) 36 (88) Median duration of hematologic normalization, weeks (range) 46 (10, 75) TMA Event-free Status, n (%) 37 (90) Daily TMA Intervention Rate, median (range) Before eculizumab 0.63 (0, 1.38) On eculizumab treatment 0 (0, 0.58) Pediatric and Adolescent Patients with aHUS (Study C10-003)
Study C10-003 enrolled patients who were required to have a platelet count < lower limit of normal range (LLN), evidence of hemolysis such as an elevation in serum LDH above the upper limits of normal, serum creatinine level ≥97 percentile for age without the need for chronic dialysis. The median patient age was 6.5 (range: 5 months to 17 years). Patients enrolled in Study C10-003 were required to have ADAMTS13 activity level above 5%; observed range of values in the trial were 38%-121%. Fifty percent of patients had an identified complement regulatory factor mutation or auto-antibody. A total of 10 patients received PE/PI prior to eculizumab. Table 19 summarizes the key baseline clinical and disease-related characteristics of patients enrolled in Study C10-003.
Table 19: Baseline Characteristics of Patients Enrolled in Study C10-003 Parameter Patients
1 month to <12 years
(N=18)All Patients
(N=22)Time from aHUS diagnosis until start of study drug in months, median (range) 0.51 (0.03 – 58) 0.56 (0.03-191) Time from current clinical TMA manifestation until first study dose in months, median (range) 0.23 (0.03 – 4) 0.2 (0.03-4) Baseline platelet count (× 109/L), median (range) 110 (19-146) 91 (19-146) Baseline LDH (U/L) median (range) 1510 (282-7164) 1244 (282-7164) Patients in Study C10-003 received SOLIRIS for a minimum of 26 weeks. In Study C10-003, the median duration of SOLIRIS therapy was approximately 44 weeks (range: 1 dose to 88 weeks).
Renal function, as measured by eGFR, was improved during SOLIRIS therapy. The mean eGFR (± SD) increased from 33 ± 30 mL/min/1.73m2 at baseline to 98 ± 44 mL/min/1.73m2 by 26 weeks. Among the 20 patients with a CKD stage ≥2 at baseline, 17 (85%) achieved a CKD improvement of ≥1 stage. Among the 16 patients ages 1 month to <12 years with a CKD stage ≥2 at baseline, 14 (88%) achieved a CKD improvement by ≥1 stage. Nine of the 11 patients who required dialysis at study baseline were able to discontinue dialysis during SOLIRIS treatment. Responses were observed across all ages from 5 months to 17 years of age.
Reduction in terminal complement activity was observed in all patients after commencement of SOLIRIS. SOLIRIS reduced signs of complement-mediated TMA activity, as shown by an increase in mean platelet counts from baseline to 26 weeks. The mean platelet count (± SD) increased from 88 ± 42 ×109/L at baseline to 281 ± 123 ×109/L by one week; this effect was maintained through 26 weeks (mean platelet count (±SD) at week 26: 293 ± 106 ×109/L). In Study C10-003, responses to SOLIRIS were similar in patients with and without identified mutations in genes encoding complement regulatory factor proteins or auto-antibodies to factor H.
Table 20 summarizes the efficacy results for Study C10-003.
Table 20: Efficacy Results for Study C10-003 Efficacy Parameter Patients
1 month to <12 years
(N=18)All Patients
(N=22)- *
- Through data cutoff (October 12, 2012).
Complete TMA response, n (%)
95% CI
Median Duration of complete TMA response, weeks (range)*11 (61)
36, 83
40 (14, 77)14 (64)
41, 83
37 (14, 77)eGFR improvement ≥15 mL/min/ 1.73∙m2∙n (%) 16 (89) 19 (86) Complete Hematologic Normalization, n (%)
Median Duration of complete hematologic normalization, weeks (range)14 (78)
38 (14, 77)18 (82)
38 (14, 77)TMA Event-Free Status, n (%) 17 (94) 21 (95) Daily TMA Intervention rate, median (range)
Before eculizumab treatment
On eculizumab treatment0.2 (0, 1.7)
0 (0, 0.01)0.4 (0, 1.7)
0 (0, 0.01)14.3 Generalized Myasthenia Gravis (gMG)
The efficacy of SOLIRIS for the treatment of gMG was established in gMG Study 1 (NCT01997229), a 26-week randomized, double-blind, parallel-group, placebo-controlled, multi-center trial that enrolled patients who met the following criteria at screening:
- Positive serologic test for anti-AChR antibodies,
- Myasthenia Gravis Foundation of America (MGFA) Clinical Classification Class II to IV,
- MG-Activities of Daily Living (MG-ADL) total score ≥6,
- Failed treatment over 1 year or more with 2 or more immunosuppressive therapies (ISTs) either in combination or as monotherapy, or failed at least 1 IST and required chronic plasmapheresis or plasma exchange (PE) or intravenous immunoglobulin (IVIg).
A total of 62 patients were randomized to receive SOLIRIS treatment and 63 were randomized to receive placebo. Baseline characteristics were similar between treatment groups, including age at diagnosis (38 years in each group), gender [66% female (eculizumab) versus 65% female (placebo)], and duration of gMG [9.9 (eculizumab) versus 9.2 (placebo) years]. Over 95% of patients in each group were receiving acetylcholinesterase (AchE) inhibitors, and 98% were receiving immunosuppressant therapies (ISTs). Approximately 50% of each group had been previously treated with at least 3 ISTs.
SOLIRIS was administered according to the recommended dosage regimen [see Dosage and Administration (2.4)].
The primary efficacy endpoint for gMG Study 1 was a comparison of the change from baseline between treatment groups in the Myasthenia Gravis-Specific Activities of Daily Living scale (MG-ADL) total score at Week 26. The MG-ADL is a categorical scale that assesses the impact on daily function of 8 signs or symptoms that are typically affected in gMG. Each item is assessed on a 4-point scale where a score of 0 represents normal function and a score of 3 represents loss of ability to perform that function (total score 0-24). A statistically significant difference favoring SOLIRIS was observed in the mean change from baseline to Week 26 in MG-ADL total scores [-4.2 points in the SOLIRIS-treated group compared with -2.3 points in the placebo-treated group (p=0.006)].
A key secondary endpoint in gMG Study 1 was the change from baseline in the Quantitative Myasthenia Gravis (QMG) total score at Week 26. The QMG is a 13-item categorical scale assessing muscle weakness. Each item is assessed on a 4-point scale where a score of 0 represents no weakness and a score of 3 represents severe weakness (total score 0-39). A statistically significant difference favoring SOLIRIS was observed in the mean change from baseline to Week 26 in QMG total scores [-4.6 points in the SOLIRIS-treated group compared with -1.6 points in the placebo-treated group (p=0.001)].
The results of the analysis of the MG-ADL and QMG from gMG Study 1 are shown in Table 21.
Table 21: Analysis of Change from Baseline to Week 26 in MG-ADL and QMG Total Scores in gMG Study 1 Efficacy Endpoints SOLIRIS-LS Mean
(N=62)
(SEM)Placebo-LS Mean
(N=63)
(SEM)SOLIRIS change relative to placebo – LS Mean Difference
(95% CI)p-values SEM= Standard Error of the Mean;
SOLIRIS-LSMean = least square mean for the treatment group;
Placebo-LSMean = least square mean for the placebo group;
LSMean-Difference (95% CI) = Difference in least square mean with 95% confidence interval;
p-values (testing the null hypothesis that there is no difference between the two treatment armsMG-ADL -4.2 (0.49) -2.3 (0.48) -1.9 (-3.3, -0.6) (0.006*; 0.014†) QMG -4.6 (0.60) -1.6 (0.59) -3.0 (-4.6, -1.3) (0.001 *; 0.005 †) In gMG Study 1, a clinical response was defined in the MG-ADL total score as at least a 3-point improvement and in QMG total score as at least a 5-point improvement. The proportion of clinical responders at Week 26 with no rescue therapy was statistically significantly higher for SOLIRIS compared to placebo for both measures. For both endpoints, and also at higher response thresholds (≥4-, 5-, 6-, 7-, or 8-point improvement on MG-ADL, and ≥6-, 7-, 8-, 9-, or 10-point improvement on QMG), the proportion of clinical responders was consistently greater for SOLIRIS compared to placebo. Available data suggest that clinical response is usually achieved by 12 weeks of SOLIRIS treatment.
14.4 Neuromyelitis Optica Spectrum Disorder (NMOSD)
The efficacy of SOLIRIS for the treatment of NMOSD was established in NMOSD Study 1 (NCT01892345), a randomized, double-blind, placebo-controlled trial that enrolled 143 patients with NMOSD who were anti-AQP4 antibody positive and met the following criteria at screening:
- History of at least 2 relapses in last 12 months or 3 relapses in the last 24 months, with at least 1 relapse in the 12 months prior to screening,
- Expanded Disability Status Scale (EDSS) score ≤ 7 (consistent with the presence of at least limited ambulation with aid),
- If on immunosuppressive therapy (IST), on a stable dose regimen,
- The use of concurrent corticosteroids was limited to 20 mg per day or less,
- Patients were excluded if they had been treated with rituximab or mitoxantrone within 3 months or with IVIg within 3 weeks prior to screening.
A total of 96 patients were randomized to receive SOLIRIS treatment and 47 were randomized to receive placebo.
The baseline demographic and disease characteristics were balanced between treatment groups. During the treatment phase of the trial, 76% percent of patients received concomitant IST, including chronic corticosteroids; 24% of patients did not receive concomitant IST or chronic corticosteroids during the treatment phase of the trial.
SOLIRIS was administered according to the recommended dosage regimen [see Dosage and Administration (2.4)].
The primary endpoint for NMOSD Study 1 was the time to the first adjudicated on-trial relapse. The time to the first adjudicated on-trial relapse was significantly longer in SOLIRIS-treated patients compared to placebo-treated patients (relative risk reduction 94%; hazard ratio 0.058; p < 0.0001) (Figure 1).
Figure 1: Kaplan-Meier Survival Estimates for Time to First Adjudicated On-Trial Relapse – Full Analysis Set
Note: Patients who did not experience an adjudicated on-trial relapse were censored at the end of the study period. Abbreviations: CI = confidence interval 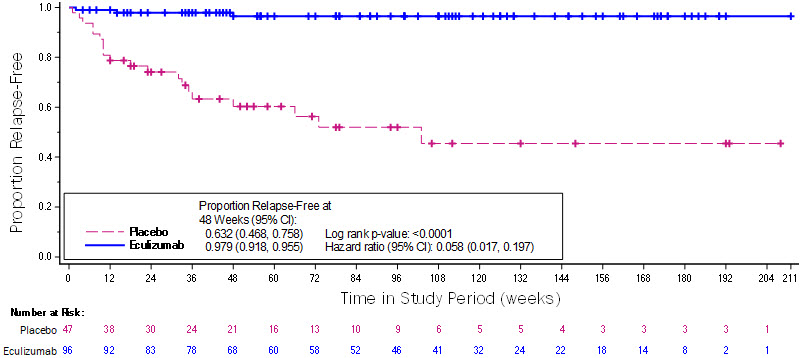
SOLIRIS-treated patients experienced similar improvement in time to first adjudicated on-trial relapse with or without concomitant treatment. SOLIRIS-treated patients had a 96% relative reduction in the adjudicated on-trial annualized relapse rate (ARR) compared to patients on placebo, as shown in Table 22.
Table 22: Adjudicated On-trial Annualized Relapse Rate – Full Analysis Set Variable Statistic Placebo
(N=47)SOLIRIS
(N=96)ARR = annualized relapse rate - *
- Based on a Poisson regression adjusted for randomization strata and historical ARR in 24 months prior to screening.
Total number of relapses Sum 21 3 Adjusted adjudicated ARR* Rate 0.350 0.016 Treatment effect* Rate ratio
(eculizumab/placebo)… 0.045 p-value … <0.0001 Compared to placebo-treated patients, SOLIRIS-treated patients had reduced annualized rates of hospitalizations (0.04 for SOLIRIS versus 0.31 for placebo), of corticosteroid administrations to treat acute relapses (0.07 for SOLIRIS versus 0.42 for placebo), and of plasma exchange treatments (0.02 for SOLIRIS versus 0.19 for placebo).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
SOLIRIS (eculizumab) injection is a sterile, preservative-free, clear, colorless solution supplied as one 300 mg/30 mL (10 mg/mL) single-dose vial per carton (NDC 25682-001-01).
Store SOLIRIS vials refrigerated at 2°-8° C (36°-46° F) in the original carton to protect from light until time of use. SOLIRIS vials may be stored in the original carton at controlled room temperature (not more than 25° C/77° F) for only a single period up to 3 days. Do not use beyond the expiration date stamped on the carton. Refer to Dosage and Administration (2) for information on the stability and storage of diluted solutions of SOLIRIS.
DO NOT FREEZE. DO NOT SHAKE.
-
17 PATIENT COUNSELING INFORMATION
Advise the patients and/or caregivers to read the FDA-approved patient labeling (Medication Guide).
Serious Meningococcal Infections
Advise patients of the risk of serious meningococcal infection. Inform patients of the need to complete or update their meningococcal vaccinations at least 2 weeks prior to receiving the first dose of SOLIRIS or receive antibacterial drug prophylaxis if SOLIRIS treatment must be initiated immediately and they have not been previously vaccinated. Inform patients of the requirement to be revaccinated according to current ACIP recommendations for meningococcal infection while on SOLIRIS therapy [see Warnings and Precautions (5.1)].
Inform patients that vaccination may not prevent serious meningococcal infection and to seek immediate medical attention if the following signs or symptoms occur[see Warnings and Precautions (5.1)]:
- fever
- fever and a rash
- fever with high heart rate
- headache with nausea or vomiting
- headache and a fever
- headache with a stiff neck or stiff back
- confusion
- muscle aches with flu-like symptoms
- eyes sensitive to light
Inform patients that they will be given a Patient Safety Card for SOLIRIS that they should carry with them at all times during and for 3 months following treatment with SOLIRIS. This card describes symptoms which, if experienced, should prompt the patient to immediately seek medical evaluation.
ULTOMIRIS and SOLIRIS REMS
SOLIRIS is available only through a restricted program called ULTOMIRIS and SOLIRIS REMS [see Warnings and Precautions (5.2)].
Inform the patient of the following notable requirements:
- Patients must receive counseling about the risk of serious meningococcal infections.
- Patients must receive written educational materials about this risk.
- Patients must be instructed to carry the Patient Safety Card with them at all times during and for 3 months following treatment with SOLIRIS.
- Patients must be instructed to complete or update meningococcal vaccines for serogroups A, C, W, Y and B per ACIP recommendations as directed by the prescriber prior to treatment with SOLIRIS.
- Patients must receive antibiotics as directed by the prescriber if they are not up to date with meningococcal vaccines and have to start SOLIRIS right away.
Other Infections
Counsel patients about gonorrhea prevention and advise regular testing for patients at-risk.
Inform patients that there may be an increased risk of other types of infections, particularly those due to encapsulated bacteria.
Aspergillus infections have occurred in immunocompromised and neutropenic patients.
Inform parents or caregivers of children receiving SOLIRIS for the treatment of aHUS that their child should be vaccinated against Streptococcus pneumoniae and Haemophilus influenzae type b (Hib) according to current medical guidelines.
Infusion-Related Reactions
Advise patients that administration of SOLIRIS may result in infusion-related reactions.
Discontinuation
Inform patients with PNH that they may develop serious hemolysis due to PNH when SOLIRIS is discontinued and that they will be monitored by their healthcare professional for at least 8 weeks following SOLIRIS discontinuation.
Inform patients with aHUS that there is a potential for TMA complications due to aHUS when SOLIRIS is discontinued and that they will be monitored by their healthcare professional for at least 12 weeks following SOLIRIS discontinuation. Inform patients who discontinue SOLIRIS to keep the Patient Safety Card with them for three months after the last SOLIRIS dose, because the increased risk of meningococcal infection persists for several weeks following discontinuation of SOLIRIS.
-
SPL UNCLASSIFIED SECTION
Manufactured by:
Alexion Pharmaceuticals, Inc.
121 Seaport Boulevard
Boston, MA 02210 USAUS License Number 1743
This product, or its use, may be covered by one or more US patents, including US Patent No. 6,355,245, US Patent No. 9,732,149 and US Patent No.9,718,880 in addition to others including patents pending.
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised 06/2024 MEDICATION GUIDE
SOLIRIS® (so-leer-is)
(eculizumab)
injection, for intravenous useWhat is the most important information I should know about SOLIRIS?
SOLIRIS is a medicine that affects your immune system. SOLIRIS may lower the ability of your immune system to fight infections.-
SOLIRIS increases your chance of getting serious meningococcal infections caused by Neisseria meningitidis bacteria. Meningococcal infections may quickly become life-threatening or cause death if not recognized and treated early.
- You must complete or update your meningococcal vaccine(s) at least 2 weeks before your first dose of SOLIRIS.
- If you have not completed your meningococcal vaccines and SOLIRIS must be started right away, you should receive the required vaccine(s) as soon as possible.
- If you have not been vaccinated and SOLIRIS must be started right away, you should also receive antibiotics to take for as long as your healthcare provider tells you.
- If you had a meningococcal vaccine in the past, you might need additional vaccines before starting SOLIRIS. Your healthcare provider will decide if you need additional meningococcal vaccines.
- Meningococcal vaccines do not prevent all meningococcal infections. Call your healthcare provider or get emergency medical care right away if you get any of these signs and symptoms of a serious meningococcal infection:
- fever
- fever with high heart rate
- headache and fever
- confusion
- muscle aches with flu-like symptoms
- fever and a rash
- headache with nausea or vomiting
- headache with a stiff neck or stiff back
- eyes sensitive to light
Your healthcare provider will give you a Patient Safety Card about the risk of serious meningococcal infection. Carry it with you at all times during treatment and for 3 months after your last dose of SOLIRIS. Your risk of meningococcal infection may continue for several weeks after your last dose of SOLIRIS. It is important to show this card to any healthcare provider who treats you. This will help them diagnose and treat you quickly.
SOLIRIS is only available through a program called the ULTOMIRIS and SOLIRIS Risk Evaluation and Mitigation Strategy (REMS).
Before you can receive SOLIRIS, your healthcare provider must:- enroll in the ULTOMIRIS and SOLIRIS REMS program
- counsel you about the risk of serious meningococcal infections
- give you information about the signs and symptoms of serious meningococcal infection
- make sure that you are vaccinated against serious infections caused by meningococcal bacteria and that you receive antibiotics if you need to start SOLIRIS right away and you are not up to date on your vaccines
- give you a Patient Safety Card about your risk of meningococcal infection, as discussed above
- If your child is treated with SOLIRIS, your child should receive vaccines against Streptococcus pneumoniae and Haemophilus influenzae type b (Hib).
- Certain people may be at risk of serious infections with gonorrhea. Talk to your healthcare provider about whether you are at risk for gonorrhea infection, about gonorrhea prevention, and regular testing.
- Certain fungal infections (aspergillus) may also happen if you take SOLIRIS and have a weak immune system or a low white blood cell count.
What is SOLIRIS?
SOLIRIS is a prescription medicine used to treat:- people with paroxysmal nocturnal hemoglobinuria (PNH).
- people with atypical hemolytic uremic syndrome (aHUS).
SOLIRIS is not for use in treating people with Shiga toxin E. coli related hemolytic uremic syndrome (STEC-HUS). - adults with generalized myasthenia gravis (gMG) who are anti-acetylcholine receptor (AchR) antibody positive.
- adults with neuromyelitis optica spectrum disorder (NMOSD) who are anti-aquaporin-4 (AQP4) antibody positive.
Who should not receive SOLIRIS?
Do not receive SOLIRIS if you have a serious meningococcal infection when you are starting SOLIRIS treatment.Before you receive SOLIRIS, tell your healthcare provider about all of your medical conditions, including if you: - have an infection or fever.
- are pregnant or plan to become pregnant. It is not known if SOLIRIS will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if SOLIRIS passes into your breast milk.
Know the medications you take and the vaccines you receive. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I receive SOLIRIS? - Your healthcare provider will give you SOLIRIS into your vein through an intravenous (IV) line usually over 35 minutes in adults and 1 to 4 hours in children.
- Adults will usually receive a SOLIRIS infusion:
- weekly for 5 weeks, then
- every 2 weeks.
- Children less than 18 years of age, your healthcare provider will decide how often you will receive SOLIRIS depending on your age and body weight.
- After each infusion, you should be monitored for at least 1 hour for infusion-related reactions. See "What are the possible side effects of SOLIRIS?" If you have an infusion-related reaction during your SOLIRIS infusion, your healthcare provider may decide to give SOLIRIS more slowly or stop your infusion.
- If you miss a SOLIRIS infusion, call your healthcare provider right away.
-
If you have PNH, your healthcare provider will need to monitor you closely for at least 8 weeks after stopping SOLIRIS. Stopping treatment with SOLIRIS may cause breakdown of your red blood cells due to PNH.
Symptoms or problems that can happen due to red blood cell breakdown include:
- drop in the number of your red blood cell count
- kidney problems
- drop in your platelet counts
- blood clots
- confusion
- difficulty breathing
- chest pain
-
If you have aHUS, your healthcare provider will need to monitor you closely for at least 12 weeks after stopping SOLIRIS for signs of worsening aHUS symptoms or problems related to abnormal clotting (thrombotic microangiopathy).
Symptoms or problems that can happen with abnormal clotting may include:
- stroke
- difficulty breathing
- confusion
- kidney problems
- seizure
- swelling in arms or legs
- chest pain (angina)
- a drop in your platelet count
What are the possible side effects of SOLIRIS?
SOLIRIS can cause serious side effects including:- See "What is the most important information I should know about SOLIRIS?"
-
Serious infusion-related reactions. Serious infusion-related reactions can happen during your SOLIRIS infusion. Tell your healthcare provider or nurse right away if you get any of these symptoms during your SOLIRIS infusion:
- chest pain
- trouble breathing or shortness of breath
- swelling of your face, tongue, or throat
- feel faint or pass out
The most common side effects in people with PNH treated with SOLIRIS include:- headache
- pain or swelling of your nose or throat (nasopharyngitis)
- back pain
- nausea
The most common side effects in people with aHUS treated with SOLIRIS include: - headache
- diarrhea
- high blood pressure (hypertension)
- common cold (upper respiratory infection
- stomach-area (abdominal pain)
- vomiting
- pain or swelling of your nose or throat (nasopharyngitis)
- low red blood cell count (anemia)
- cough
- swelling of legs or feet (peripheral edema
- nausea
- urinary tract infections
- fever
The most common side effects in people with gMG treated with SOLIRIS include: - muscle and joint (musculoskeletal) pain
The most common side effects in people with NMOSD treated with SOLIRIS include: - common cold (upper respiratory infection)
- pain or swelling of your nose or throat (nasopharyngitis)
- diarrhea
- back pain
- dizziness
- flu like symptoms (influenza) including fever, headache, tiredness, cough, sore throat, and body aches
- joint pain (arthralgia)
- throat irritation (pharyngitis)
- bruising (contusion)
Tell your healthcare provider about any side effect that bothers you or that does not go away. These are not all the possible side effects of SOLIRIS.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of SOLIRIS.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about SOLIRIS that is written for health professionals.What are the ingredients in SOLIRIS?
Active ingredient: eculizumab
Inactive ingredients: polysorbate 80 (vegetable origin), sodium chloride, sodium phosphate dibasic, sodium phosphate monobasic, and Water for Injection
Manufactured by: Alexion Pharmaceuticals, Inc., 121 Seaport Boulevard, Boston, MA 02210 USA. US License Number 1743 -
SOLIRIS increases your chance of getting serious meningococcal infections caused by Neisseria meningitidis bacteria. Meningococcal infections may quickly become life-threatening or cause death if not recognized and treated early.
- PRINCIPAL DISPLAY PANEL - 30 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
SOLIRIS
eculizumab injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:25682-001 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ECULIZUMAB (UNII: A3ULP0F556) (ECULIZUMAB - UNII:A3ULP0F556) ECULIZUMAB 300 mg in 30 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, MONOBASIC, UNSPECIFIED FORM (UNII: 3980JIH2SW) SODIUM PHOSPHATE, DIBASIC, UNSPECIFIED FORM (UNII: GR686LBA74) SODIUM CHLORIDE (UNII: 451W47IQ8X) POLYSORBATE 80 (UNII: 6OZP39ZG8H) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:25682-001-01 1 in 1 CARTON 04/02/2007 1 30 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125166 04/02/2007 Labeler - Alexion Pharmaceuticals Inc. (789359510)