Label: RAMIPRIL capsule

- NDC Code(s): 50090-5730-0, 50090-5730-1
- Packager: A-S Medication Solutions
- This is a repackaged label.
- Source NDC Code(s): 57237-225
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated December 3, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use RAMIPRIL CAPSULES safely and effectively. See full prescribing information for RAMIPRIL CAPSULES. RAMIPRIL capsules, for oral use ...
-
Table of ContentsTable of Contents
-
BOXED WARNING
(What is this?)
WARNING: FETAL TOXICITY
- When pregnancy is detected, discontinue ramipril as soon as possible [see Warnings and Precautions (5.6)].
- Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus [see Warnings and Precautions (5.6)].
-
1 INDICATIONS AND USAGE1.1 Hypertension - Ramipril capsules are indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular ...
-
2 DOSAGE AND ADMINISTRATION2.1 Hypertension - The recommended initial dose for patients not receiving a diuretic is 2.5 mg once a day. Adjust dose according to blood pressure response. The usual maintenance dosage range ...
-
3 DOSAGE FORMS AND STRENGTHSRamipril is supplied as hard gelatin capsules containing 1.25 mg, 2.5 mg, 5 mg, and 10 mg of ramipril. Ramipril Capsules USP, 1.25 mg are yellow/yellow size ‘4’ hard gelatin capsules imprinted ...
-
4 CONTRAINDICATIONSRamipril capsules are contraindicated in patients who are hypersensitive to this product or any other ACE inhibitor (e.g., a patient who has experienced angioedema during therapy with any other ...
-
5 WARNINGS AND PRECAUTIONS5.1 Anaphylactoid and Possibly Related Reactions - Presumably because drugs that act directly on the renin-angiotensin-aldosterone system (e.g., ACE inhibitors) affect the metabolism of ...
-
6 ADVERSE REACTIONS6.1 Clinical Trials Experience - Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly ...
-
7 DRUG INTERACTIONS7.1 Diuretics - Patients on diuretics, especially those in whom diuretic therapy was recently instituted, may occasionally experience an excessive reduction of blood pressure after initiation ...
-
8 USE IN SPECIFIC POPULATIONS8.1 Pregnancy - Pregnancy Category D - Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal ...
-
10 OVERDOSAGESingle oral doses of ramipril in rats and mice of 10 g/kg to 11 g/kg resulted in significant lethality. In dogs, oral doses as high as 1 g/kg induced only mild gastrointestinal distress. Limited ...
-
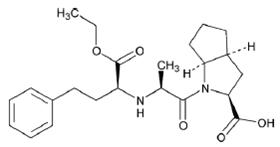
11 DESCRIPTIONRamipril is a 2-aza-bicyclo [3.3.0]-octane-3-carboxylic acid derivative. It is a white to almost white crystalline powder soluble in polar organic solvents and buffered aqueous solutions ...
-
12 CLINICAL PHARMACOLOGY12.1 Mechanism of Action - Ramipril and ramiprilat inhibit ACE in human subjects and animals. Angiotensin converting enzyme is a peptidyl dipeptidase that catalyzes the conversion of ...
-
13 NONCLINICAL TOXICOLOGY13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility - No evidence of a tumorigenic effect was found when ramipril was given by gavage to rats for up to 24 months at doses of up to 500 ...
-
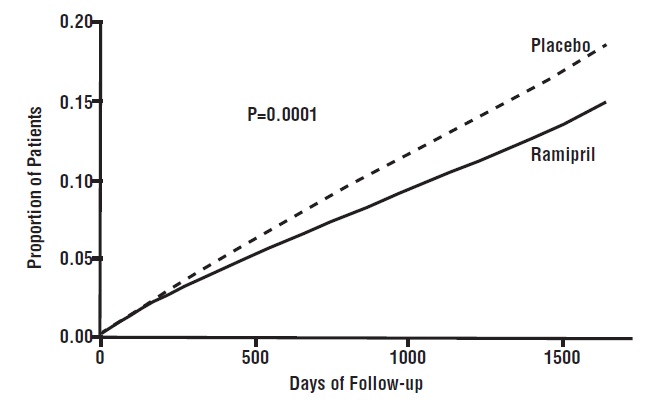
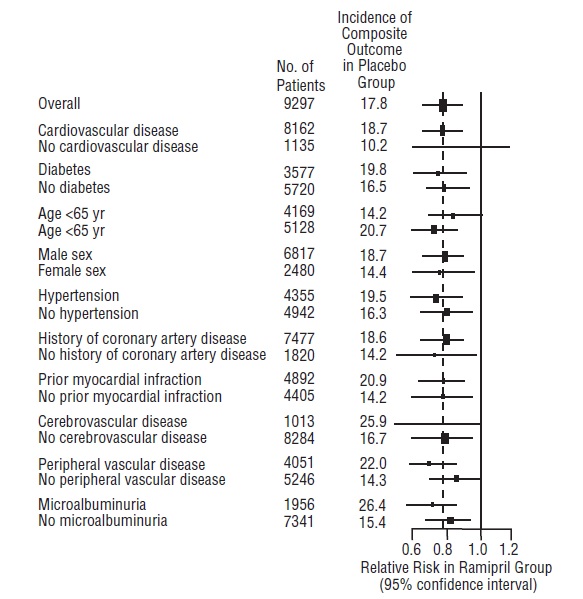
14 CLINICAL STUDIES14.1 Hypertension - Ramipril has been compared with other ACE inhibitors, beta-blockers, and thiazide diuretics as monotherapy for hypertension. It was approximately as effective as other ACE ...
-
16 HOW SUPPLIED/STORAGE AND HANDLINGProduct: 50090-5730 - NDC: 50090-5730-0 30 CAPSULE in a BOTTLE - NDC: 50090-5730-1 90 CAPSULE in a BOTTLE
-
17 PATIENT COUNSELING INFORMATIONAngioedema - Angioedema, including laryngeal edema, can occur with treatment with ACE inhibitors, especially following the first dose. Advise patients to immediately report any signs or ...
-
RAMIPRIL

-
INGREDIENTS AND APPEARANCEProduct Information