Label: CLONIDINE HYDROCHLORIDE tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 68071-2631-2, 68071-2631-3, 68071-2631-5, 68071-2631-6, view more - Packager: NuCare Pharmaceuticals,Inc.
- This is a repackaged label.
- Source NDC Code(s): 58657-647
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated February 1, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
Clonidine Hydrochloride Tablets, USP
Oral Antihypertensive - Tablets of 0.1 mg, 0.2 mg and 0.3 mg - Rx Only - Prescribing Information
-
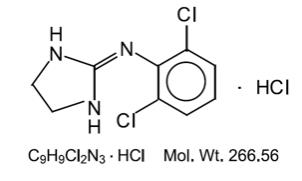
DESCRIPTION
Clonidine hydrochloride, USP is a centrally acting alpha-agonist hypotensive agent available as tablets for oral administration in three dosage strengths: 0.1 ...
-
CLINICAL PHARMACOLOGY
Clonidine stimulates alpha-adrenoreceptors in the brain stem. This action results in reduced sympathetic outflow from the central nervous system and in decreases in peripheral resistance, renal ...
-
INDICATIONS AND USAGE
Clonidine hydrochloride tablets, USP are indicated in the treatment of hypertension. Clonidine hydrochloride tablets, USP may be employed alone or concomitantly with other ...
-
CONTRAINDICATIONS
Clonidine hydrochloride tablets should not be used in patients with known hypersensitivity to clonidine (see - PRECAUTIONS).
-
WARNINGS
Withdrawal - Patients should be instructed not to discontinue therapy without consulting their physician. Sudden cessation of clonidine treatment has, in some cases, resulted in symptoms such ...
-
PRECAUTIONS
General - In patients who have developed localized contact sensitization to transdermal clonidine, continuation of transdermal clonidine or substitution of oral clonidine hydrochloride therapy ...
-
ADVERSE REACTIONS
Most adverse effects are mild and tend to diminish with continued therapy. The most frequent (which appear to be dose-related) are dry mouth, occurring in about 40 of 100 patients; drowsiness ...
-
OVERDOSAGE
Hypertension may develop early and may be followed by hypotension, bradycardia, respiratory depression, hypothermia, drowsiness, decreased or absent reflexes, weakness, irritability and miosis ...
-
DOSAGE AND ADMINISTRATION
Adults - The dose of clonidine hydrochloride tablets must be adjusted according to the patient's individual blood pressure response. The following is a general guide to ...
-
HOW SUPPLIED
Clonidine Hydrochloride Tablets, USP are supplied as follows: 0.1 mg: A pink tablet, in the shape of “ ". NDC 68071-2631-5 BOTTLES OF 15 - NDC 68071-2631-3 BOTTLES OF 30 - NDC 68071-2631-6 ...
-
PRINCIPAL DISPLAY PANEL

-
INGREDIENTS AND APPEARANCEProduct Information
 ".
".