Label: TERBUTALINE SULFATE injection, solution


- NDC Code(s): 63323-665-00, 63323-665-01
- Packager: Fresenius Kabi USA, LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated July 12, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
WARNING: PROLONGED TOCOLYSIS
Terbutaline sulfate has not been approved and should not be used for prolonged tocolysis (beyond 48 to 72 hours). In particular, terbutaline sulfate should not be used for maintenance tocolysis in the outpatient or home setting. Serious adverse reactions, including death, have been reported after administration of terbutaline sulfate to pregnant women. In the mother, these adverse reactions include increased heart rate, transient hyperglycemia, hypokalemia, cardiac arrhythmias, pulmonary edema and myocardial ischemia. Increased fetal heart rate and neonatal hypoglycemia may occur as a result of maternal administration (see CONTRAINDICATIONS, Prolonged Tocolysis).
-
DESCRIPTION
Terbutaline Sulfate Injection, USP, is a beta-adrenergic agonist bronchodilator available as a sterile, nonpyrogenic, aqueous solution in vials, for subcutaneous administration. Each mL of solution contains: 1 mg of terbutaline sulfate USP (0.82 mg of the free base), and Water for Injection, USP. Sodium chloride is used for isotonicity, and hydrochloric acid for adjustment to a pH of 3.0 to 5.0. Terbutaline sulfate is (±)-α-[( tert-butyl-amino) methyl]-3,5-dihydroxybenzyl alcohol sulfate (2:1) (salt). The structural formula is:
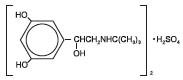
(C12H19NO3)2 • H2SO4 M.W. 548.65
Terbutaline sulfate USP is a white to gray-white crystalline powder. It is odorless or has a faint odor of acetic acid. It is soluble in water and in 0.1N hydrochloric acid, slightly soluble in methanol, and insoluble in chloroform.
-
CLINICAL PHARMACOLOGY
Terbutaline is a beta-adrenergic receptor agonist. In vitro and in vivo pharmacologic studies have demonstrated that terbutaline exerts a preferential effect on beta 2-adrenergic receptors. While it is recognized that beta 2-adrenergic receptors are the predominant receptors in bronchial smooth muscle, data indicate that there is a population of beta 2-receptors in the human heart, existing in a concentration between 10% to 50%. The precise function of these receptors has not been established (see WARNINGS). Controlled clinical studies in patients given terbutaline subcutaneously have not revealed a preferential beta 2-adrenergic effect.
The pharmacologic effects of beta-adrenergic agonists, including terbutaline, are at least in part attributable to stimulation through beta-adrenergic receptors of intracellular adenyl cyclase, the enzyme which catalyzes the conversion of adenosine triphosphate (ATP) to cyclic 3’,5’-adenosine monophosphate (cAMP). Increased cAMP levels are associated with relaxation of bronchial smooth muscle and inhibition of release of mediators of immediate hypersensitivity from cells, especially from mast cells.
Controlled clinical studies have shown that terbutaline relieves bronchospasm in acute and chronic obstructive pulmonary disease by significantly increasing pulmonary flow rates (e.g., an increase of 15% or more in FEV 1). After subcutaneous administration of 0.25 mg of terbutaline, a measurable change in expiratory flow rate usually occurs within 5 minutes, and a clinically significant increase in FEV 1 occurs within 15 minutes. The maximum effect usually occurs within 30 to 60 minutes, and clinically significant bronchodilator activity may continue for 1.5 to 4 hours. The duration of clinically significant improvement is comparable to that observed with equimilligram doses of epinephrine.
Preclinical
Studies in laboratory animals (minipigs, rodents, and dogs) have demonstrated the occurrence of cardiac arrhythmias and sudden death (with histological evidence of myocardial necrosis) when beta-agonists and methylxanthines are administered concurrently. The clinical significance of these findings is unknown.
Pharmacokinetics
Subcutaneous administration of 0.5 mg of terbutaline to 17 healthy, adult, male subjects resulted in mean (SD) peak plasma terbutaline concentration of 9.6 (3.6) ng/mL, which was observed at a median (range) time of 0.5 (0.08 to 1) hours after dosing. The mean (SD) AUC (0 to 48) and total body clearance values were 29.4 (14.2) hr • ng/mL, and 311 (112) mL/min respectively. The terminal half-life was determined in 9 of the 17 subjects and had a mean (SD) of 5.7 (2) hours.
After subcutaneous administration of 0.25 mg of terbutaline to two male subjects, peak terbutaline serum concentrations of 5.2 and 5.3 ng/mL were observed at about 20 minutes after dosing.
Elimination half-life of the drug in 10 of 14 patients was approximately 2.9 hours after subcutaneous administration, but longer elimination half-lives (between 6 to 14 hours) were found in the other 4 patients. About 90% of the drug was excreted in the urine at 96 hours after subcutaneous administration, with about 60% of this being unchanged drug. It appears that the sulfate conjugate is a major metabolite of terbutaline and urinary excretion is the primary route of elimination.
- INDICATIONS AND USAGE
-
CONTRAINDICATIONS
Prolonged Tocolysis
Terbutaline sulfate has not been approved and should not be used for prolonged tocolysis (beyond 48 to 72 hours). In particular, terbutaline sulfate should not be used for maintenance tocolysis in the outpatient or home setting (see BOXED WARNING: PROLONGED TOCOLYSIS).
-
WARNINGS
Deterioration of Asthma
Asthma may deteriorate acutely over a period of hours or chronically over several days or longer. If the patient needs more doses of terbutaline than usual, this may be a marker of destabilization of asthma and requires reevaluation of the patient and treatment regimen, giving special consideration to the possible need for anti-inflammatory treatment, e.g., corticosteroids.
Use of Anti-Inflammatory Agents
The use of beta-adrenergic agonist bronchodilators alone may not be adequate to control asthma in many patients. Early consideration should be given to adding anti-inflammatory agents, e.g., corticosteroids.
Cardiovascular Effects
Terbutaline, like all other beta-adrenergic agonists, can produce a clinically significant cardiovascular effect in some patients as measured by pulse rate, blood pressure, and/or symptoms. Although such effects are uncommon after administration of terbutaline at recommended doses, if they occur, the drug may need to be discontinued. In addition, beta-agonists have been reported to produce electrocardiogram (ECG) changes, such as flattening of the T wave, prolongation of the QTc interval, and ST segment depression. The clinical significance of these findings is unknown. Therefore, terbutaline, like all sympathomimetic amines, should be used with caution in patients with cardiovascular disorders, especially coronary insufficiency, cardiac arrhythmias, and hypertension.
-
PRECAUTIONS
General
Terbutaline, as with all sympathomimetic amines, should be used with caution in patients with cardiovascular disorders, including ischemic heart disease, hypertension, and cardiac arrhythmias; in patients with hyperthyroidism or diabetes mellitus; and in patients who are unusually responsive to sympathomimetic amines or who have convulsive disorders. Significant changes in systolic and diastolic blood pressure have been seen and could be expected to occur in some patients after use of any beta-adrenergic bronchodilator.
Immediate hypersensitivity reactions and exacerbations of bronchospasm have been reported after terbutaline administration.
Beta-adrenergic agonist medications may produce significant hypokalemia in some patients, possibly through intracellular shunting, which has the potential to produce adverse cardiovascular effects. The decrease is usually transient, not requiring supplementation.
Large doses of intravenous terbutaline have been reported to aggravate pre-existing diabetes mellitus and ketoacidosis.
Drug Interactions
The concomitant use of terbutaline with other sympathomimetic agents is not recommended, since the combined effect on the cardiovascular system may be deleterious to the patient.
Monoamine Oxidase Inhibitors or Tricyclic Antidepressants
Terbutaline should be administered with extreme caution to patients being treated with monoamine oxidase inhibitors or tricyclic antidepressants, or within 2 weeks of discontinuation of such agents, since the action of terbutaline on the vascular system may be potentiated.
Beta-Blockers
Beta-adrenergic receptor blocking agents not only block the pulmonary effect of beta-agonists, such as terbutaline, but may produce severe bronchospasm in asthmatic patients. Therefore, patients with asthma should not normally be treated with beta-blockers. However, under certain circumstances, e.g., as prophylaxis after myocardial infarction, there may be no acceptable alternatives to the use of beta-adrenergic blocking agents in patients with asthma. In this setting, cardioselective beta-blockers could be considered, although they should be administered with caution.
Diuretics
The ECG changes and/or hypokalemia that may result from the administration of non-potassium-sparing diuretics (such as loop or thiazide diuretics) can be acutely worsened by beta-agonists, especially when the recommended dose of the beta-agonist is exceeded. Although the clinical significance of these effects is not known, caution is advised in the coadministration of beta-agonists with non-potassium-sparing diuretics.
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year study in Sprague-Dawley rats, terbutaline sulfate caused a significant and dose-related increase in the incidence of benign leiomyomas of the mesovarium at dietary doses of 50 mg/kg and above (approximately 810 times the maximum recommended daily subcutaneous (sc) dose for adults on a mg/m 2 basis). In a 21-month study in CD-1 mice, terbutaline showed no evidence of tumorigenicity at dietary doses up to 200 mg/kg (approximately 1,600 times the maximum recommended daily sc dose for adults on a mg/m 2 basis). The mutagenicity potential of terbutaline has not been determined.
Reproduction studies in rats using terbutaline demonstrated no impairment of fertility at oral doses up to 50 mg/kg (approximately 810 times the maximum recommended daily sc dose for adults on a mg/m 2 basis).
Pregnancy: Teratogenic Effects
Pregnancy Category C
There are no adequate and well-controlled studies of terbutaline sulfate in pregnant women. Published animal studies show that rat offspring exhibit alterations in behavior and brain development, including decreased cellular proliferation and differentiation when dams were treated subcutaneously with terbutaline during the late stage of pregnancy and lactation period. Terbutaline exposures in rat dams were approximately 24 to 48 times the common human dose in adults of 2 to 4 mg/day, on a mg/m 2 basis.
Terbutaline sulfate has not been approved and should not be used for prolonged tocolysis (beyond 48 to 72 hours). In particular, terbutaline sulfate should not be used for maintenance tocolysis in the outpatient or home setting. Serious adverse reactions, including death, have been reported after administration of terbutaline sulfate to pregnant women. In the mother, these adverse reactions include increased heart rate, transient hyperglycemia, hypokalemia, cardiac arrhythmias, pulmonary edema and myocardial ischemia. Increased fetal heart rate and neonatal hypoglycemia may occur as a result of maternal administration (see BOXED WARNING: PROLONGED TOCOLYSIS and CONTRAINDICATIONS, Prolonged Tocolysis).
In animal embryofetal developmental studies, no teratogenic effects were observed in offspring when pregnant rats and rabbits received terbutaline sulfate at oral doses up to 50 mg/kg/day, approximately 810 and 1600 times, respectively, the maximum recommended daily subcutaneous dose for adults, on a mg/m 2 basis.
Terbutaline sulfate should be used during pregnancy only if the potential benefits justify the potential risk to the fetus.
Use in Labor and Delivery
Because of the potential for beta-agonist interference with uterine contractility, use of terbutaline for relief of bronchospasm during labor should be restricted to those patients in whom the benefits clearly outweigh the risk.
Terbutaline crosses the placenta. After single dose IV administration of terbutaline to 22 women in late pregnancy who were delivered by elective Cesarean section due to clinical reasons, umbilical blood levels of terbutaline were found to range from 11% to 48% of the maternal blood levels.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Therefore, terbutaline should be used during nursing only if the potential benefit justifies the possible risk to the newborn.
Pediatric Use
Terbutaline is not recommended for patients under the age of 12 years because of insufficient clinical data to establish safety and effectiveness.
Geriatric Use
Clinical studies of Terbutaline Sulfate Injection, USP did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Fresenius Kabi USA, LLC at 1-800-551-7176 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Adverse reactions observed with terbutaline are similar to those commonly seen with other sympathomimetic agents. All these reactions are transient in nature and usually do not require treatment.
The following table compares adverse reactions seen in patients treated with terbutaline (0.25 mg and 0.5 mg), with those seen in patients treated with epinephrine injection (0.25 mg and 0.5 mg), during eight double-blind crossover studies involving a total of 214 patients.
Incidence (%) of Adverse Reactions
Terbutaline (%)
0.25 mg
N=77
0.5 mg
N=205
Reaction
Central Nervous System
Tremor
7.8
38
Nervousness
16.9
30.7
Dizziness
1.3
10.2
Headache
7.8
8.8
Drowsiness
11.7
9.8
Cardiovascular
Palpitations
7.8
22.9
Tachycardia
1.3
1.5
Respiratory
Dyspnea
0
2
Chest discomfort
1.3
1.5
Gastrointestinal
Nausea/vomiting
1.3
3.9
Systemic
Weakness
1.3
0.5
Flushed feeling
0
2.4
Sweating
0
2.5
Pain at injection site
2.6
0.5
Epinephrine (%)
0.25 mg
N=153
0.5 mg
N=61
Reaction
Central Nervous System
Tremor
16.3
18
Nervousness
8.5
31.1
Dizziness
7.8
3.3
Headache
3.3
9.8
Drowsiness
14.4
8.2
Cardiovascular
Palpitations
7.8
29.5
Tachycardia
2.6
0
Respiratory
Dyspnea
2
0
Chest discomfort
2.6
0
Gastrointestinal
Nausea/vomiting
1.3
11.5
Systemic
Weakness
2.6
1.6
Flushed feeling
1.3
0
Sweating
0
0
Pain at injection site
2.6
1.6
Note: Some patients received more than one dosage strength of terbutaline and epinephrine. In addition, there were reports of anxiety, muscle cramps, and dry mouth (<0.5%). There have been rare reports of elevations in liver enzymes and of hypersensitivity vasculitis with terbutaline administration.
-
OVERDOSAGE
The median sc lethal dose of terbutaline in mature rats was approximately 165 mg/kg (approximately 2,700 times the maximum recommended daily sc dose for adults on a mg/m 2 basis). The median sc lethal dose of terbutaline in young rats was approximately 2,000 mg/kg (approximately 32,000 times the maximum recommended daily sc dose for adults on a mg/m 2 basis).
The expected symptoms with overdosage are those of excessive beta-adrenergic stimulation and/or occurrence or exaggeration of any of the symptoms listed under ADVERSE REACTIONS, e.g., seizures, angina, hypertension or hypotension, tachycardia with rates up to 200 beats per minute, arrhythmias, nervousness, headache, tremor, dry mouth, palpitation, nausea, dizziness, fatigue, malaise, and insomnia. Hypokalemia may also occur. There is no specific antidote. Treatment consists of discontinuation of terbutaline together with appropriate symptomatic therapy. The judicious use of a cardioselective beta-receptor blocker may be considered, bearing in mind that such medication can produce bronchospasm. There is insufficient evidence to determine if dialysis is beneficial for overdosage of terbutaline.
-
DOSAGE AND ADMINISTRATION
Terbutaline Sulfate Injection, USP should be used only for subcutaneous administration and not intravenous infusion. Sterility and accurate dosing cannot be assured if the vials are not used in accordance with DOSAGE AND ADMINISTRATION.
Discard unused portion after single patient use.
The usual subcutaneous dose of Terbutaline Sulfate Injection, USP is 0.25 mg injected into the lateral deltoid area. If significant clinical improvement does not occur within 15 to 30 minutes, a second dose of 0.25 mg may be administered. If the patient then fails to respond within another 15 to 30 minutes, other therapeutic measures should be considered. The total dose within 4 hours should not exceed 0.5 mg.
Note: Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
-
HOW SUPPLIED
Terbutaline Sulfate Injection, USP
Product
CodeUnit of Sale Strength Each 660501 NDC 63323-665-01
Unit of 251 mg per mL
NDC 63323-665-00
2 mL Amber, Type 1 Glass
Single Dose VialDiscard unused portion after single patient use.
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature].
Protect from light. Do not use if solution is discolored.
Vial stoppers do not contain natural rubber latex.
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TERBUTALINE SULFATE
terbutaline sulfate injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63323-665 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TERBUTALINE SULFATE (UNII: 576PU70Y8E) (TERBUTALINE - UNII:N8ONU3L3PG) TERBUTALINE SULFATE 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 8.9 mg in 1 mL HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63323-665-01 25 in 1 TRAY 03/10/2011 1 NDC:63323-665-00 1 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA076887 03/10/2011 Labeler - Fresenius Kabi USA, LLC (608775388) Establishment Name Address ID/FEI Business Operations Fresenius Kabi USA, LLC 023648251 MANUFACTURE(63323-665)


