Label: AGRYLIN- anagrelide hydrochloride capsule

- NDC Code(s): 54092-063-01
- Packager: Takeda Pharmaceuticals America, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated December 29, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AGRYLIN safely and effectively. See full prescribing information for AGRYLIN.
AGRYLIN® (anagrelide) capsules, for oral use
Initial U.S. Approval: 1997INDICATIONS AND USAGE
AGRYLIN is a platelet reducing agent indicated for the treatment of thrombocythemia, secondary to myeloproliferative neoplasms, to reduce the elevated platelet count and the risk of thrombosis and to ameliorate associated symptoms including thrombo-hemorrhagic events. (1)
DOSAGE AND ADMINISTRATION
- The starting dose for adults is 0.5 mg four times a day or 1 mg twice a day. (2.1)
- The starting dose for pediatric patients is 0.5 mg per day. (2.1)
- Maintain the starting dose for at least one week and then titrate to maintain target platelet counts. (2.2)
- Do not exceed a dose increment of 0.5 mg/day in any one week. Do not exceed 10 mg/day or 2.5 mg in a single dose. (2.2)
- Moderate hepatic impairment: Start with 0.5 mg per day. (2.3)
DOSAGE FORMS AND STRENGTHS
Capsules: 0.5 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Cardiovascular Toxicity: QT prolongation and ventricular tachycardia have been reported with AGRYLIN. Obtain a pre-treatment cardiovascular examination including an ECG in all patients. Monitor patients for cardiovascular effects. (5.1)
- Pulmonary Hypertension: Assess underlying cardiopulmonary disease prior to initiating therapy. (5.2)
- Bleeding Risk: Monitor patients for bleeding, including those receiving concomitant therapy with other drugs known to cause bleeding. (5.3)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥ 5%) are headache, palpitations, diarrhea, asthenia, edema, nausea, abdominal pain, dizziness, pain, dyspnea, cough, flatulence, vomiting, fever, peripheral edema, rash, chest pain, anorexia, tachycardia, malaise, paresthesia, back pain, pruritus, and dyspepsia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1-800-828-2088 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 10/2021
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Starting Dosage
2.2 Dose Titration Based Upon Platelet Response
2.3 Dose Modifications for Hepatic Impairment
2.4 Clinical Monitoring
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Toxicity
5.2 Pulmonary Hypertension
5.3 Bleeding Risk
5.4 Pulmonary Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drugs that Prolong QT
7.2 PDE3 Inhibitors
7.3 Aspirin and Drugs that Increase Bleeding Risk
7.4 CYP450 Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.2 Dose Titration Based Upon Platelet Response
Continue the starting dose for at least one week and then titrate to reduce and maintain the platelet count below 600,000/µL, and ideally between 150,000/µL and 400,000/µL. The dose increment should not exceed 0.5 mg/day in any one week. Dosage should not exceed 10 mg/day or 2.5 mg in a single dose. Most patients will experience an adequate response at a dose of 1.5 to 3.0 mg/day. Monitor platelet counts weekly during titration then monthly or as necessary.
2.3 Dose Modifications for Hepatic Impairment
In patients with moderate hepatic impairment (Child Pugh score 7-9) start AGRYLIN therapy at a dose of 0.5 mg/day and monitor frequently for cardiovascular events [see Warnings and Precautions (5.1), Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. Patients with moderate hepatic impairment who have tolerated AGRYLIN therapy for one week may have their dose increased. The dose increase increment should not exceed 0.5 mg/day in any one week. Avoid use of AGRYLIN in patients with severe hepatic impairment.
2.4 Clinical Monitoring
AGRYLIN therapy requires clinical monitoring, including complete blood counts, assessment of hepatic and renal function, and electrolytes.
To prevent the occurrence of thrombocytopenia, monitor platelet counts every two days during the first week of treatment and at least weekly thereafter until the maintenance dosage is reached. Typically, platelet counts begin to respond within 7 to 14 days at the proper dosage. In the clinical trials, the time to complete response, defined as platelet count ≤600,000/µL, ranged from 4 to 12 weeks. In the event of dosage interruption or treatment withdrawal, the rebound in platelet count is variable, but platelet counts typically will start to rise within 4 days and return to baseline levels in one to two weeks, possibly rebounding above baseline values. Monitor platelet counts frequently.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Toxicity
Torsades de pointes and ventricular tachycardia have been reported with AGRYLIN. Obtain a pre-treatment cardiovascular examination including an ECG in all patients. During treatment with AGRYLIN monitor patients for cardiovascular effects and evaluate as necessary.
AGRYLIN increases the QTc interval of the electrocardiogram and increases the heart rate in healthy volunteers [see Clinical Pharmacology (12.2)].
Do not use AGRYLIN in patients with known risk factors for QT interval prolongation, such as congenital long QT syndrome, a known history of acquired QTc prolongation, medicinal products that can prolong QTc interval and hypokalemia [see Drug Interactions (7.1)].
Hepatic impairment increases anagrelide exposure and could increase the risk of QTc prolongation. Monitor patients with hepatic impairment for QTc prolongation and other cardiovascular adverse reactions. The potential risks and benefits of AGRYLIN therapy in a patient with mild and moderate hepatic impairment should be assessed before treatment is commenced. Reduce AGRYLIN dose in patients with moderate hepatic impairment. Avoid use of AGRYLIN in patients with severe hepatic impairment.
In patients with heart failure, bradyarrhythmias, or electrolyte abnormalities, consider periodic monitoring with electrocardiograms [see Clinical Pharmacology (12.2)].
AGRYLIN is a phosphodiesterase 3 (PDE3) inhibitor and may cause vasodilation, tachycardia, palpitations, and congestive heart failure. Other drugs that inhibit PDE3 have caused decreased survival when compared with placebo in patients with Class III-IV congestive heart failure [see Drug Interactions (7.2)].
In patients with cardiac disease, use AGRYLIN only when the benefits outweigh the risks.
5.2 Pulmonary Hypertension
Cases of pulmonary hypertension have been reported in patients treated with AGRYLIN. Evaluate patients for signs and symptoms of underlying cardiopulmonary disease prior to initiating and during AGRYLIN therapy [see Adverse Reactions (6.1)].
5.3 Bleeding Risk
Use of concomitant AGRYLIN and aspirin increased major hemorrhagic events in a postmarketing study. Assess the potential risks and benefits for concomitant use of AGRYLIN with aspirin, since bleeding risks may be increased. Monitor patients for bleeding, including those receiving concomitant therapy with other drugs known to cause bleeding (e.g., anticoagulants, PDE3 inhibitors, NSAIDs, antiplatelet agents, selective serotonin reuptake inhibitors) [see Drug Interactions (7.3), Clinical Pharmacology (12.3)].
5.4 Pulmonary Toxicity
Interstitial lung diseases (including allergic alveolitis, eosinophilic pneumonia and interstitial pneumonitis) have been reported to be associated with the use of AGRYLIN in post-marketing reports. Most cases presented with progressive dyspnea with lung infiltrations. The time of onset ranged from 1 week to several years after initiating AGRYLIN. If suspected, discontinue AGRYLIN and evaluate. Symptoms may improve after discontinuation [see Adverse Reactions (6)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Cardiovascular Toxicity [see Warnings and Precautions (5.1)]
- Pulmonary Hypertension [see Warnings and Precautions (5.2)]
- Bleeding Risk [see Warnings and Precautions (5.3)]
- Pulmonary Toxicity [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Studies in Adult Patients
In three single-arm clinical studies, 942 patients [see Clinical Trials (14)] diagnosed with myeloproliferative neoplasms of varying etiology (ET: 551; PV: 117; OMPN: 274) were exposed to AGRYLIN with a mean duration of approximately 65 weeks. Serious adverse reactions reported in these patients included the following: congestive heart failure, myocardial infarction, cardiomyopathy, cardiomegaly, complete heart block, atrial fibrillation, cerebrovascular accident, pericardial effusion [see Warnings and Precautions (5.1)], pleural effusion, pulmonary infiltrates, pulmonary fibrosis, pulmonary hypertension, and pancreatitis. Of the 942 patients treated with AGRYLIN, 161 (17%) were discontinued from the study because of adverse reactions or abnormal laboratory test results. The most common adverse reactions resulting in treatment discontinuation were headache, diarrhea, edema, palpitations, and abdominal pain.
The most frequently reported adverse reactions to AGRYLIN (in 5% or greater of 942 patients with myeloproliferative neoplasms) in clinical trials were listed in Table 1.
Table 1 Adverse Reactions Reported in Clinical Studies of AGRYLIN in at least 5% of Patients Adverse Reactions AGRYLIN
(N=942)
(%)Cardiac disorders Palpitations 26% Tachycardia 8% Chest pain 8% General disorders and administration site conditions Asthenia 23% Edema 21% Pain 15% Fever 9% Peripheral edema 9% Malaise 6% Gastrointestinal disorders Diarrhea 26% Nausea 17% Abdominal pain 16% Vomiting 10% Flatulence 10% Anorexia 8% Dyspepsia 5% Respiratory, thoracic and mediastinal disorders Dyspnea 12% Cough 6% Skin and subcutaneous tissue disorders Rash 8% Pruritus 6% Musculoskeletal and connective tissue disorders Back pain 6% Nervous system disorders Headache 44% Dizziness 15% Paresthesia 6% Adverse Reactions (frequency 1% to < 5%) included:
General disorders and administration site conditions: Flu symptoms, chills.
Cardiac disorders: Arrhythmia, angina pectoris, heart failure, syncope.
Vascular disorders: Hemorrhage, hypertension, postural hypotension, vasodilatation.
Gastrointestinal disorders: Constipation, gastrointestinal hemorrhage, gastritis.
Blood and lymphatic system disorders: Anemia, thrombocytopenia, ecchymosis.
Hepatobiliary disorders: Elevated liver enzymes.
Musculoskeletal and connective tissue disorders: Arthralgia, myalgia.
Psychiatric disorders: Depression, confusion, nervousness.
Nervous system disorders: Somnolence, insomnia, amnesia, migraine headache.
Respiratory, thoracic and mediastinal disorders: Epistaxis, pneumonia.
Skin and subcutaneous tissue disorders: Alopecia.
Eye disorders: Abnormal vision, diplopia.
Ear and labyrinth disorders: Tinnitus.
Renal and urinary disorders: Hematuria, renal failure.
Other less frequent adverse reactions (<1%) were:
Cardiac disorders: Ventricular tachycardia, supraventricular tachycardia.
Nervous system disorders: Hypoesthesia.
Clinical Study in Pediatric Patients
The frequency of adverse reactions observed in pediatric patients was similar to adult patients. The most common adverse reactions observed in pediatric patients were fever, epistaxis, headache, and fatigue during the 3-month AGRYLIN treatment in the study. Episodes of increased pulse and decreased systolic or diastolic blood pressure beyond the normal ranges in the absence of clinical symptoms were observed. Other adverse reactions reported in these pediatric patients receiving AGRYLIN treatment were palpitations, headache, nausea, vomiting, abdominal pain, back pain, anorexia, fatigue, and muscle cramps.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-marketing use of AGRYLIN. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac disorders: Prinzmetal angina, Torsades de pointes.
Respiratory, thoracic and mediastinal disorders: Interstitial lung diseases (including allergic alveolitis, eosinophilic pneumonia, and interstitial pneumonitis) [see Warnings and Precautions (5.4)].
Renal and urinary disorders: Tubulointerstitial nephritis.
Hepatobiliary disorders: Clinically significant hepatotoxicity (including symptomatic ALT and AST elevations and elevations greater than three times the ULN).
Nervous system disorders: Cerebral infarction
Other adverse reactions in pediatric patients reported in spontaneous reports and literature reviews include:
Blood and lymphatic system disorders: Anemia.
Skin and subcutaneous tissue disorders: Cutaneous photosensitivity.
Investigations: Elevated leukocyte count.
-
7 DRUG INTERACTIONS
7.1 Drugs that Prolong QT
Avoid use of AGRYLIN in patients taking medications that may prolong QT interval (including, but not limited to, chloroquine, clarithromycin, haloperidol, methadone, moxifloxacin, amiodarone, disopyramide, procainamide, and pimozide) [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
7.2 PDE3 Inhibitors
AGRYLIN is a phosphodiesterase 3 (PDE3) inhibitor. Avoid use of drug products with similar properties such as inotropes and other PDE3 inhibitors (e.g., cilostazol, milrinone) [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
7.3 Aspirin and Drugs that Increase Bleeding Risk
Co-administration of single-dose or repeat-dose AGRYLIN and aspirin showed greater ex vivo anti-platelet aggregation effects than administration of aspirin alone [see Clinical Pharmacology (12.3)]. Results from an observational study in patients with essential thrombocythemia suggest the rate of major hemorrhagic events (MHEs) in patients treated with AGRYLIN is higher than in those subjects treated with another cytoreductive treatment. The majority of the major hemorrhagic events occurred in patients who were also receiving concomitant anti-aggregatory treatment (primarily, aspirin). Therefore, the potential risks of the concomitant use of AGRYLIN with aspirin should be assessed, particularly in patients with a high-risk profile for hemorrhage, before treatment is initiated [see Warnings and Precautions (5.3)].
Monitor patients for bleeding, particularly those receiving concomitant therapy with other drugs known to cause bleeding (e.g., anticoagulants, PDE3 inhibitors, NSAIDs, antiplatelet agents, selective serotonin reuptake inhibitors).
7.4 CYP450 Interactions
CYP1A2 inhibitors: AGRYLIN and its active metabolite are primarily metabolized by CYP1A2. Drugs that inhibit CYP1A2 (e.g., fluvoxamine, ciprofloxacin) could increase the exposure of AGRYLIN. Monitor patients for cardiovascular events and titrate doses accordingly when CYP1A2 inhibitors are co-administered.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from case reports with AGRYLIN use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal embryo-fetal studies, delayed fetal development (delayed skeletal ossification and reduced body weight) was observed in rats administered anagrelide hydrochloride during organogenesis at doses approximately 97 times the maximum clinical dose (10 mg/day) based on body surface area (see Data). There are adverse effects on maternal and fetal outcomes associated with thrombocythemia in pregnancy (see Clinical Considerations).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Thrombotic events, such as stroke, deep vein thrombosis, or myocardial infarction, can be complications of thrombocythemia. Thrombocythemia in pregnancy is associated with an increased risk for miscarriage, stillbirth, and other maternal outcomes, such as preeclampsia.
Data
Animal Data
Anagrelide hydrochloride was administered orally to pregnant rats and rabbits during the period of organogenesis at doses up to 900 mg/kg/day in rats and up to 20 mg/kg/day in rabbits (875 and 39 times, respectively, the maximum clinical dose of 10 mg/day based on body surface area). In rats, developmental delays were observed including reductions in fetal weight at 300 and 900 mg/kg/day and delays in skeletal ossification at doses of 100 mg/kg/day and higher. The dose of 100 mg/kg/day (600 mg/m2/day) in rats is approximately 97 times the maximum clinical dose based on body surface area. No adverse embryo-fetal effects were detected in rabbits at the highest dose of 20 mg/kg/day (39 times the maximal clinical dose based on body surface area).
In a pre- and post-natal study conducted in female rats, anagrelide hydrochloride administered at oral doses of 60 mg/kg/day (58 times the maximum clinical dose based on body surface area) or higher during organogenesis through lactation produced delay or blockage of parturition, deaths of non-delivering pregnant dams and their fully developed fetuses, and increased mortality in the pups born.
In a placental transfer study, a single oral dose of [14C]-anagrelide hydrochloride (3 mg/kg) was administered to pregnant rats on gestation Day 17. Drug-related radioactivity was detected in maternal and fetal tissue.
8.2 Lactation
Risk Summary
There is no information regarding the presence of anagrelide in human milk, the effect on the breastfed child, or the effects on milk production. Anagrelide or its metabolites have been detected in the milk of lactating rats (see Data). Because of the potential for serious adverse reactions, including thrombocytopenia, in a breastfed child, advise patients that breastfeeding is not recommended during treatment with AGRYLIN, and for one week following the last dose.
8.3 Females and Males of Reproductive Potential
Infertility
Females
Based on findings from animal studies, AGRYLIN may impair female fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of AGRYLIN have been established in pediatric patients 7 years of age and older. There are no data for pediatric patients less than 7 years of age. Use of AGRYLIN in these pediatric patients is supported by evidence from adequate and well controlled studies of AGRYLIN in adults with additional pharmacokinetic, pharmacodynamic, and safety data in 18 pediatric patients aged 7 through 16 years with thrombocythemia secondary to ET [see Dosage and Administration (2.1), Clinical Pharmacology (12.3), and Clinical Studies (14)].
There were no apparent trends or differences in the types of adverse events observed between the pediatric patients compared with those of the adult patients [see Adverse Reactions (6.1)].
8.5 Geriatric Use
Of the 942 subjects in clinical studies of AGRYLIN, 42.1% were 65 years and over, while 14.9% were 75 years and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in response between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
Hepatic metabolism is the major route of anagrelide clearance. Exposure to anagrelide is increased 8-fold in patients with moderate hepatic impairment [see Clinical Pharmacology (12.3)] and dose reduction is required [see Dosage and Administration (2.3)]. Use of AGRYLIN in patients with severe hepatic impairment has not been studied. Avoid use of AGRYLIN in patients with severe hepatic impairment. The potential risks and benefits of AGRYLIN therapy in a patient with mild and moderate hepatic impairment should be assessed before treatment is commenced. Assess hepatic function before and during AGRYLIN treatment [see Warnings and Precautions (5.1)].
-
10 OVERDOSAGE
At higher than recommended doses, AGRYLIN has been shown to cause hypotension. There have been postmarketing case reports of intentional overdose with AGRYLIN. Reported symptoms include sinus tachycardia and vomiting. Symptoms resolved with supportive management. Platelet reduction from AGRYLIN therapy is dose-related; therefore, thrombocytopenia, which can potentially cause bleeding, is expected from overdosage.
In case of overdosage, stop AGRYLIN dosing and monitor platelet counts for thrombocytopenia and observe for possible complications such as bleeding. Consider resumption of AGRYLIN dosing once the platelet count returns to the normal range.
-
11 DESCRIPTION
AGRYLIN (anagrelide hydrochloride) is a platelet-reducing agent. Its chemical name is 6,7-dichloro-1,5-dihydroimidazo[2,1-b]quinazolin-2(3H)-one monohydrochloride monohydrate. The molecular formula is C10H7Cl2N3O∙HCl∙H2O which corresponds to a molecular weight of 310.55. The structural formula is:
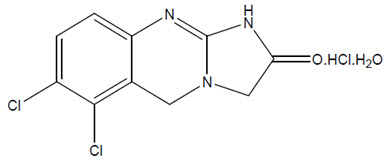
Anagrelide hydrochloride is an off-white powder. It is very slightly soluble in water and sparingly soluble in dimethyl sulfoxide and dimethylformamide.
AGRYLIN is supplied as capsules for oral administration, containing 0.5 mg of anagrelide (equivalent to 0.61 mg of anagrelide hydrochloride USP). The capsules also contain anhydrous lactose NF, crospovidone NF, lactose monohydrate NF, magnesium stearate NF, microcrystalline cellulose NF, and povidone NF as inactive ingredients. The capsule shell contains gelatin, titanium dioxide, and black iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism by which anagrelide reduces blood platelet count is unknown. In cell culture studies, anagrelide suppressed expression of transcription factors including GATA-1 and FOG-1 required for megakaryocytopoiesis, ultimately leading to reduced platelet production.
12.2 Pharmacodynamics
In blood withdrawn from normal volunteers treated with anagrelide, a disruption was found in the postmitotic phase of megakaryocyte development and a reduction in megakaryocyte size and ploidy. At therapeutic doses, anagrelide does not produce significant changes in white cell counts or coagulation parameters, and may have a small, but clinically insignificant effect on red cell parameters. The active metabolite, 3-hydroxy anagrelide, has similar potency and efficacy to that of anagrelide in the platelet lowering effect; however, exposure (measured by plasma AUC) to 3-hydroxy anagrelide is approximately 2-fold higher compared to anagrelide. Anagrelide and 3-hydroxy anagrelide inhibit cyclic AMP phosphodiesterase 3 (PDE3) and 3-hydroxy anagrelide is approximately forty times more potent than anagrelide (IC50s = 0.9 and 36 nM, respectively). PDE3 inhibition does not alter platelet production. PDE3 inhibitors, as a class can inhibit platelet aggregation. However, significant inhibition of platelet aggregation is observed only at doses of anagrelide higher than those typically required to reduce platelet count. PDE3 inhibitors have cardiovascular (CV) effects including vasodilation, positive inotropy and chronotropy.
Cardiac Electrophysiology
The effect of anagrelide dose (0.5 mg and 2.5 mg single doses) on the heart rate and QTc interval prolongation potential was evaluated in a double-blind, randomized, placebo- and active-controlled, cross-over study in 60 healthy adult men and women.
A dose-related increase in heart rate was observed, with the maximum increase occurring around the time of maximal drug concentration (0.5 – 4 hours). The maximum change in mean heart rate occurred at 2 hours after administration and was +7.8 beats per minute (bpm) for 0.5 mg and +29.1 bpm for 2.5 mg.
Dose-related increase in mean QTc was observed. The maximum mean (95% upper confidence bound) change in QTcI (individual subject correction) from placebo after baseline-correction was 7.0 (9.8) ms and 13.0 (15.7) ms following anagrelide doses of 0.5 mg and 2.5 mg, respectively.
12.3 Pharmacokinetics
Dose proportionality has been found in the dose range 0.5 mg to 2.5 mg.
Absorption
Following oral administration of AGRYLIN, at least 70% is absorbed from the gastrointestinal tract. In fasted subjects, anagrelide peak plasma concentrations occur within about 1 hour after administration.
Pharmacokinetic data obtained from healthy volunteers comparing the pharmacokinetics of anagrelide in the fed and fasted states showed that administration of a 1 mg dose of anagrelide with food decreased the Cmax by 14%, but increased the AUC by 20%. Food decreased the Cmax of the active metabolite 3-hydroxy anagrelide by 29%, although it had no effect on the AUC.
Elimination
Anagrelide and 3-hydroxy anagrelide are eliminated with plasma half-lives of approximately 1.5 and 2.5 hours, respectively. Anagrelide and 3-hydroxy anagrelide do not accumulate in plasma when the clinical dose regimens are administered.
Metabolism: Anagrelide is primarily metabolized by CYP1A2 to the active metabolite, 3-hydroxy anagrelide, which is subsequently metabolized by CYP1A2 to the inactive metabolite, RL603. Less than 1% of the administered dose is recovered in the urine as anagrelide, and approximately 3% and 16-20% of the administered dose is recovered as 3-hydroxy anagrelide and RL603, respectively.
Drug Interactions
Aspirin: In two pharmacodynamic interaction studies in healthy subjects, co-administration of single-dose anagrelide 1 mg and aspirin 900 mg or repeat-dose anagrelide 1 mg once daily and aspirin 75 mg once daily showed greater ex vivo anti-platelet aggregation effects than administration of aspirin alone. Co-administered anagrelide 1 mg and aspirin 900 mg single-doses had no effect on bleeding time, prothrombin time (PT) or activated partial thromboplastin time (aPTT).
Specific Populations
Pediatric: Dose-normalized Cmax and AUC of anagrelide were higher in children and adolescents (age range 7 through 16 years) with essential thrombocythemia, by 17% and 56%, respectively, than in adult patients (19 through 57 years).
Geriatric: Cmax and AUC of anagrelide were 36% and 61% higher, respectively, in elderly patients (age range 65 through 75 years), than in younger adults (age range 22 through 50 years), but Cmax and AUC of the active metabolite, 3-hydroxy anagrelide, were 42% and 37% lower, respectively, in the elderly patients.
Renal Impairment: Pharmacokinetic study at a single dose of 1 mg anagrelide in subjects with severe renal impairment (creatinine clearance <30 mL/min) showed no significant effects on the pharmacokinetics of anagrelide.
Hepatic Impairment: A pharmacokinetic study at a single dose of 1 mg anagrelide in subjects with moderate hepatic impairment (Child Pugh score 7-9) showed a 2-fold increase in mean anagrelide Cmax and an 8-fold increase in total exposure (AUC) to anagrelide compared with healthy subjects. Additionally, subjects with moderate hepatic impairment showed 24% lower mean 3-hydroxy anagrelide Cmax and 77% higher mean 3-hydroxy anagrelide AUC compared to healthy subjects.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a two-year rat carcinogenicity study a higher incidence of uterine adenocarcinoma, relative to controls, was observed in females receiving 30 mg/kg/day (at least 174 times human AUC exposure after a 1 mg twice daily dose). Adrenal pheochromocytomas were increased relative to controls in males receiving 3 mg/kg/day and above, and in females receiving 10 mg/kg/day and above (at least 10 and 18 times respectively human AUC exposure after a 1 mg twice daily dose).
Anagrelide hydrochloride was not mutagenic in the bacterial mutagenesis (Ames) assay or the mouse lymphoma cell (L5178Y, TK+/-) forward mutation assay, and was not clastogenic in the in vitro chromosome aberration assay using human lymphocytes or the in vivo mouse micronucleus test.
Anagrelide hydrochloride at oral doses up to 240 mg/kg/day (233 times the recommended human dose of 10 mg/day based on body surface area) had no effect on fertility and reproductive function of male rats. However, in fertility studies in female rats, oral doses of 30 mg/kg/day (29 times the recommended maximum human dose based on body surface area) or higher resulted in increased pre- and post-implantation loss and a decrease in the number of live embryos.
13.2 Animal Toxicology and/or Pharmacology
In the 2-year rat study, a significant increase in non-neoplastic lesions was observed in anagrelide treated males and females in the adrenal (medullary hyperplasia), heart (myocardial hypertrophy and chamber distension), kidney (hydronephrosis, tubular dilation and urothelial hyperplasia), and bone (femur enostosis). Vascular effects were observed in tissues of the pancreas (arteritis/periarteritis, intimal proliferation, and medial hypertrophy), kidney (arteritis/periarteritis, intimal proliferation, and medial hypertrophy), sciatic nerve (vascular mineralization), and testes (tubular atrophy and vascular infarct) in anagrelide-treated males.
-
14 CLINICAL STUDIES
Clinical Studies in Adult Patients:
A total of 942 patients with myeloproliferative neoplasms including 551 patients with Essential Thrombocythemia (ET), 117 patients with Polycythemia Vera (PV), 178 patients with Chronic Myelogenous Leukemia (CML), and 96 patients with other myeloproliferative neoplasms (OMPN), were treated with AGRYLIN in three clinical trials. Patients with OMPN included 87 patients who had Myeloid Metaplasia with Myelofibrosis (MMM), and 9 patients who had unclassified myeloproliferative neoplasms.
Patients were enrolled in clinical trials if their platelet count was ≥900,000/µL on two occasions or ≥650,000/µL on two occasions with documentation of symptoms associated with thrombocythemia. The mean duration of AGRYLIN therapy for ET, PV, CML, and OMPN patients was 65, 67, 40, and 44 weeks, respectively; 23% of patients received treatment for 2 years. Patients were treated with AGRYLIN starting at doses of 0.5-2.0 mg every 6 hours. The dose was increased if the platelet count was still high, but to no more than 12 mg each day. Efficacy was defined as reduction of platelet count to or near physiologic levels (150,000-400,000/µL). The criteria for defining subjects as "responders" were reduction in platelets for at least 4 weeks to ≤600,000/µL, or by at least 50% from baseline value. Subjects treated for less than 4 weeks were not considered evaluable. The results are depicted graphically below:
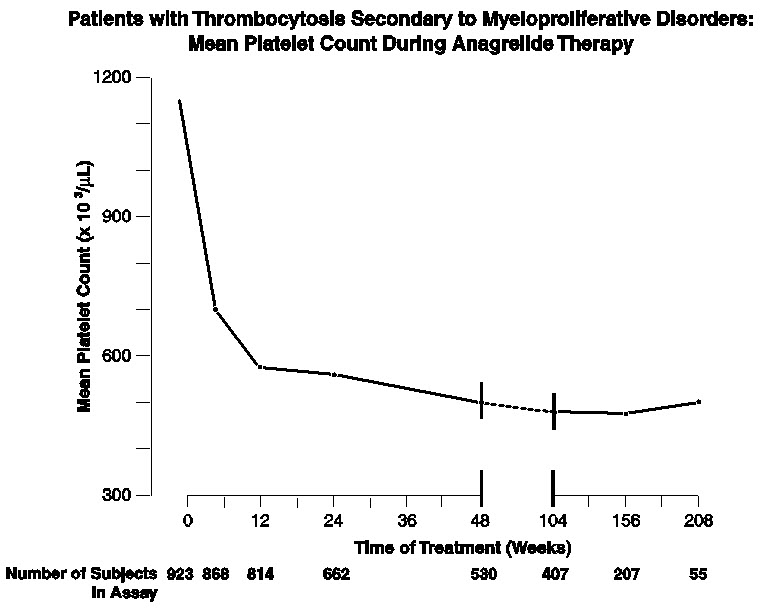
Time on Treatment Weeks Years Baseline 4 12 24 48 2 3 4 Mean* 1131 683 575 526 484 460 437 457 N 923† 868 814 662 530 407 207 55 AGRYLIN was effective in phlebotomized patients as well as in patients treated with other concomitant therapies including hydroxyurea, aspirin, interferon, radioactive phosphorus, and alkylating agents.
Clinical Study in Pediatric Patients:
An open-label safety and PK/PD study was conducted in 18 pediatric patients 7 through 16 years of age (8 children 7 through 11 years of age and 10 adolescents 12 through 16 years of age, mean age of 12 years; 8 males and 10 females) with thrombocythemia secondary to ET as compared to 17 adult patients (mean age of 66 years, 9 males and 8 females). Prior to entry on to the study, 17 of 18 pediatric patients and 12 of 17 adult patients had received AGRYLIN treatment for an average of 2 years. The median starting total daily dose, determined by retrospective chart review, for pediatric and adult patients with ET who had received AGRYLIN prior to study entry was 1 mg for each of the three age groups (7 through 11 and 12 through 16 years of age and adults). The starting dose for 6 AGRYLIN-naive patients at study entry was 0.5 mg once daily. At study completion, the median total daily maintenance doses were similar across age groups, median of 1.75 mg for children of 7 through 11 years of age, 2.25 mg in adolescents 12 through 16 years of age, and 1.5 mg for adults.
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
- Dose: Tell the patient that their dose will be adjusted on a weekly basis until they are on a dose that lowers their platelets to an appropriate level. This will also help the patient to adjust to common side effects. Tell the patient to contact their doctor if they experience tolerability issues, so the dose or dosing frequency can be adjusted [see Dosage and Administration (2)].
- Cardiovascular effects: Tell the patient to contact a doctor immediately if they experience chest pain, palpitations, or feel their heartbeat is irregular [see Warnings and Precautions (5.1)].
- Risk of Pulmonary Hypertension: Tell the patient to contact a doctor immediately if they experience shortness of breath, swelling in legs or ankles, or lips and skin turn a bluish color [see Warnings and Precautions (5.2)].
- Risk of bleeding: Warn the patient that concomitant aspirin (or other medicines that affect blood clotting) may increase the risk of bleeding. Tell the patient to contact a doctor immediately if they experience signs or symptoms of bleeding (e.g., vomit blood, pass bloody or black stools) or experience unexplained bruising/bruise more easily than usual [see Warnings and Precautions (5.3), Drug Interactions (7.1)].
- Lactation: Advise patients not to breastfeed during treatment with AGRYLIN, and for one week following the last dose [see Use in Specific Populations (8.2)].
- Infertility: Advise females of reproductive potential treatment with AGRYLIN may impair fertility [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 0.5 mg Capsule Bottle Label
-
INGREDIENTS AND APPEARANCE
AGRYLIN
anagrelide hydrochloride capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54092-063 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANAGRELIDE HYDROCHLORIDE ANHYDROUS (UNII: VNS4435G39) (ANAGRELIDE - UNII:K9X45X0051) ANAGRELIDE 0.5 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) CROSPOVIDONE (UNII: 2S7830E561) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POVIDONE K30 (UNII: U725QWY32X) Product Characteristics Color WHITE (white) Score no score Shape CAPSULE (CAPSULE) Size 14mm Flavor Imprint Code S063 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54092-063-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 03/14/1997 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020333 03/14/1997 Labeler - Takeda Pharmaceuticals America, Inc. (039997266) Establishment Name Address ID/FEI Business Operations Sterling Wisconsin, LLC 054452136 ANALYSIS(54092-063) , API MANUFACTURE(54092-063) Establishment Name Address ID/FEI Business Operations Catalent Pharma Solutions, LLC 014167995 ANALYSIS(54092-063) Establishment Name Address ID/FEI Business Operations Patheon Manufacturing Services LLC 079415560 ANALYSIS(54092-063) , LABEL(54092-063) , MANUFACTURE(54092-063) , PACK(54092-063) Establishment Name Address ID/FEI Business Operations Powdersize, LLC 080120056 PARTICLE SIZE REDUCTION(54092-063)
 063" in black ink. Each capsule contains 0.5 mg anagrelide equivalent to 0.61 mg anagrelide hydrochloride USP.
063" in black ink. Each capsule contains 0.5 mg anagrelide equivalent to 0.61 mg anagrelide hydrochloride USP.