Label: DROXIDOPA- droxidopa capsule



- NDC Code(s): 31722-010-90, 31722-014-90, 31722-015-90
- Packager: Camber Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated August 17, 2021
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DROXIDOPA CAPSULES safely and effectively. See full prescribing information for DROXIDOPA CAPSULES.
DROXIDOPA capsules, for oral use
Initial U.S. Approval: 2014
WARNING: SUPINE HYPERTENSION
See full prescribing information for complete boxed warning.Monitor supine blood pressure prior to and during treatment and more frequently when increasing doses. Elevating the head of the bed lessens the risk of supine hypertension, and blood pressure should be measured in this position. If supine hypertension cannot be managed by elevation of the head of the bed, reduce to discontinue droxidopa [ see Warnings and Precautions ( 5.1)].
INDICATIONS AND USAGE
Droxidopa capsules are indicated for the treatment of orthostatic dizziness, lightheadedness, or the “feeling that you are about blank out” in adult patients with symptomatic neurogenic orthostatic hypotension (nOH) caused by primary autonomic failure (Parkinson's disease [PD], multiple system atrophy, and pure autonomic failure), dopamine beta-hydroxylase deficiency, and non-diabetic autonomic neuropathy. Effectiveness beyond 2 weeks of treatment has not been established. The continued effectiveness of droxidopa capsules should be assessed periodically ( 1).
DOSAGE AND ADMINISTRATION
• Starting dose is 100 mg three times during the day ( 2.1)
• Titrate by 100 mg three times daily, up to a maximum dose of 600 mg three times daily ( 2.1)
• Take consistently with or without food (2.1)
• To reduce the potential for supine hypertension, elevate the head of the bed and give the last dose at least 3 hours prior to bedtime ( 2.1)
• Take droxidopa capsules whole ( 2.1)DOSAGE FORMS AND STRENGTHS
100 mg, 200 mg, and 300 mg capsule ( 3)
CONTRAINDICATIONS
History of hypersensitivity to the drug or its ingredients (4)
WARNINGS AND PRECAUTIONS
• Droxidopa capsules may cause supine hypertension and may increase cardiovascular risk if supine hypertension is not well-managed ( 5.1)
• Hyperpyrexia and confusion ( 5.2)
• May exacerbate symptoms in patients with existing ischemic heart disease, arrhythmias, and congestive heart failure ( 5.3)
• Allergic reactions ( 5.4)
ADVERSE REACTIONS
The most common adverse reactions (>5% and ≥3% compared to placebo) are headache, dizziness, nausea, and hypertension ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Annora Pharma Private Limited at 1-866-495-1995 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
Use of DOPA decarboxylase inhibitors may require dose adjustments for droxidopa capsules ( 7.2)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2021
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SUPINE HYPERTENSION
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Supine Hypertension
5.2 Hyperpyrexia and Confusion
5.3 Ischemic Heart Disease, Arrhythmias, and Congestive Heart Failure
5.4 Allergic Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drugs that Increase Blood Pressure
7.2 Parkinson's Medications
7.3 Non-selective MAO Inhibitors
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
10.1 Symptoms
10.2 Treatment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Studies in Neurogenic Orthostatic Hypotension
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SUPINE HYPERTENSION
Monitor supine blood pressure prior to and during treatment and more frequently when increasing doses. Elevating the head of the bed lessens the risk of supine hypertension, and blood pressure should be measured in this position. If supine hypertension cannot be managed by elevation of the head of the bed, reduce or discontinue droxidopa capsules [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
Droxidopa capsules are indicated for the treatment of orthostatic dizziness, lightheadedness, or the “feeling that you are about to black out” in adult patients with symptomatic neurogenic orthostatic hypotension (nOH) caused by primary autonomic failure (Parkinson's disease [PD], multiple system atrophy, and pure autonomic failure), dopamine beta-hydroxylase deficiency, and non-diabetic autonomic neuropathy. Effectiveness beyond 2 weeks of treatment has not been established. The continued effectiveness of droxidopa capsules should be assessed periodically.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended starting dose of droxidopa capsules are 100 mg, taken orally three times daily: upon arising in the morning, at midday, and in the late afternoon at least 3 hours prior to bedtime (to reduce the potential for supine hypertension during sleep). Administer droxidopa capsules consistently, either with food or without food. Take droxidopa capsule whole. Titrate to symptomatic response, in increments of 100 mg three times daily every 24 to 48 hours up to a maximum dose of 600 mg three times daily (i.e., a maximum total daily dose of 1,800 mg).
Monitor supine blood pressure prior to initiating droxidopa capsules and after increasing the dose.
Patients who miss a dose of droxidopa capsules should take their next scheduled dose.
-
3 DOSAGE FORMS AND STRENGTHS
Droxidopa capsules are available in 100 mg, 200 mg, and 300 mg strengths as specified below.
• 100 mg: Pink opaque size '3' hard gelatin capsule imprinted with 'V' on cap and '26' on body filled with white to light brown color powder.
• 200 mg: Light blue opaque size '2' hard gelatin capsule imprinted with 'V' on cap and '28' on body filled with white to light brown color powder.
• 300 mg: White opaque size '1el' hard gelatin capsule imprinted with 'V' on cap and '32' on body filled with white to light brown color powder. -
4 CONTRAINDICATIONS
Droxidopa capsules are contraindicated in patients who have a history of hypersensitivity to the drug or its ingredients [see Warnings and
Precautions ( 5.4)]. -
5 WARNINGS AND PRECAUTIONS
5.1 Supine Hypertension
Droxidopa capsules are therapy may cause or exacerbate supine hypertension in patients with nOH. Patients should be advised to elevate the head of the bed when resting or sleeping. Monitor blood pressure, both in the supine position and in the recommended head-elevated sleeping position. Reduce or discontinue droxidopa if supine hypertension persists. If supine hypertension is not well-managed, droxidopa may increase the risk of cardiovascular events, particularly stroke.
5.2 Hyperpyrexia and Confusion
Postmarketing cases of a symptom complex resembling neuroleptic malignant syndrome (NMS) have been reported with droxidopa use during postmarketing surveillance. Observe patients carefully when the dosage of droxidopa is changed or when concomitant levodopa is reduced abruptly or discontinued, especially if the patient is receiving neuroleptics.
NMS is an uncommon but life-threatening syndrome characterized by fever or hyperthermia, muscle rigidity, involuntary movements, altered consciousness, and mental status changes. The early diagnosis of this condition is important for the appropriate management of these patients.
5.3 Ischemic Heart Disease, Arrhythmias, and Congestive Heart Failure
Droxidopa capsules may exacerbate existing ischemic heart disease, arrhythmias, and congestive heart failure. Careful consideration should be given to this potential risk prior to initiating therapy in patients with these conditions.
5.4 Allergic Reactions
Hypersensitivity reactions including anaphylaxis, angioedema, bronchospasm, urticaria and rash have been reported in postmarketing experience. Some of these reactions resulted in emergency treatment. If a hypersensitivity reaction occurs, discontinue the drug and initiate appropriate therapy.
-
6 ADVERSE REACTIONS
The following adverse reactions with droxidopa are included in more detail in the Warnings and Precautions section of the label:
• Supine Hypertension [ see Warnings and Precautions ( 5.1)]
• Hyperpyrexia and Confusion [ see Warnings and Precautions ( 5.2)]
• May exacerbate existing ischemic heart disease, arrhythmias, and congestive heart failure [ see Warnings and Precautions ( 5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety evaluation of droxidopa is based on two placebo-controlled studies 1 to 2 weeks in duration (Studies 301 and 302), one 8-week placebo-controlled study (Study 306), and two long-term, open-label extension studies (Studies 303 and 304). In the placebo-controlled studies, a total of 485 patients with Parkinson's disease, multiple system atrophy, pure autonomic failure, dopamine beta-hydroxylase deficiency, or non-diabetic autonomic neuropathy were randomized and treated, 245 with droxidopa and 240 with placebo [see Clinical Studies (14)].
Placebo-Controlled Experience
The most commonly observed adverse reactions (those occurring at an incidence of greater than 5% in the droxidopa group and with at least a 3% greater incidence in the droxidopa group than in the placebo group) in droxidopa -treated patients during the three placebo-controlled trials were headache, dizziness, nausea, and hypertension. The most common adverse reactions leading to discontinuation from droxidopa were hypertension or increased blood pressure and nausea.
Table 1. Most Common Adverse Reactions Occurring More Frequently in the Droxidopa Group
Study 301 and Study 302
(1 to 2 Weeks Randomized Treatment)
Study 306
(8 to 10 Weeks Randomized Treatment)
Placebo
(N=132)
n (%)
Droxidopa
(N=131)
n (%)
Placebo
(N=108)
n (%)
Droxidopa
(N=114)
n (%)
Headache
4 (3.0)
8 (6.1)
8 (7.4)
15 (13.2)
Dizziness
2 (1.5)
5 (3.8)
5 (4.6)
11 (9.6)
Nausea
2 (1.5)
2 (1.5)
5 (4.6)
10 (8.8)
Hypertension
0
2 (1.5)
1 (0.9)
8 (7.0)
Note: n=number of patients. Adverse reactions that were reported in greater than 5% of patients in the droxidopa group and with at least a 3% greater incidence in the droxidopa group than in the placebo group were from Study 306.
Long-Term, Open-Label Trials with Droxidopa
In the long-term, open-label extension studies, a total of 422 patients, mean age 65 years, were treated with droxidopa for a mean total exposure of approximately one year. The commonly reported adverse events were falls (24%), urinary tract infections (15%), headache (13%), syncope (13%), and dizziness (10%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of droxidopa. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac Disorders: Chest pain
Eye Disorders: Blurred vision
Gastrointestinal Disorders: Pancreatitis, abdominal pain, vomiting, diarrhea
General Disorders and Administration Site Conditions: Fatigue
Nervous System Disorders: Cerebrovascular accident
Psychiatric Disorders: Psychosis, hallucination, delirium, agitation, memory disorder
-
7 DRUG INTERACTIONS
7.1 Drugs that Increase Blood Pressure
Administering droxidopa in combination with other agents that increase blood pressure (e.g., norepinephrine, ephedrine, midodrine, and triptans) would be expected to increase the risk for supine hypertension.
7.2 Parkinson's Medications
Dopa-decarboxylase inhibitors may require dose adjustments for droxidopa.
7.3 Non-selective MAO Inhibitors
The concomitant use of selective MAO-B inhibitors, such as rasagiline or selegiline, was permitted in the droxidopa clinical trials. However, based on mechanism of action, the use of non-selective MAO inhibitors and linezolid should be avoided as there is a potential for increased blood pressure when taken with droxidopa.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on use of droxidopa in pregnant women and risk of major birth defects or miscarriage. Droxidopa did not produce significant reproductive toxicity in pregnant female rats or rabbits or in their fetuses. However, when pregnant female rats were dosed during days 7 to 17 of gestation (the period of fetal organogenesis) with doses of droxidopa corresponding to 0.3, 1 and 3 times the maximum recommended daily dose of 1,800 mg in a 60 kg patient, based on body surface area, and when their male and female offspring (who were exposed only during fetal life) were subsequently bred, the female offspring exhibited a dose-dependent reduction in the number of live fetuses across all three doses and an increased number of embryonic/fetal deaths at the two higher doses ( see Data).
The estimated background risk of major birth defects and miscarriage in the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Anima Data
During a multigenerational reproductive toxicity study in rats, pregnant females were dosed during days 7 to 17 of gestation (the period of fetal organogenesis) with doses of droxidopa corresponding to 0.3, 1 and 3 times the maximum recommended daily dose of 1,800 mg in a 60 kg patient. Reduced weight gain, renal lesions, and a small number of deaths were observed in females treated with the two higher doses. When their male and female offspring (who were exposed to droxidopa only during fetal life) were subsequently bred, the female offspring exhibited a dose-dependent reduction in the number of live fetuses across all three doses and an increased number of embryonic/fetal deaths at the two higher doses.8.2 Lactation
Risk Summary
There is no information regarding the presence of droxidopa or its active metabolite(s) in human milk, the effects of droxidopa on the breastfed child, nor the effects of droxidopa on milk production/excretion. Droxidopa is present in rat milk with peak concentrations seen 4 hours after oral drug administration and drug excretion into milk still occurring 48 hours after administration ( see Data). However, due to species-specific differences in lactation physiology, animal lactation data typically do not reliably predict levels in humans. Because of the potential for serious adverse reactions, including reduced weight gain in breastfed infants, advise a woman not to breastfeed during treatment with droxidopa.
Data
Anima Data
In rats, oral administration of droxidopa resulted in excretion into breast milk with peak concentrations seen 4 hours after administration, and excretion still occurring 48 hours after administration. When the drug was administered to nursing dams during the period of lactation at a dose corresponding to 3 times the maximum recommended daily dose of 1,800 mg in a 60 kg patient when based on body surface area, reduced weight gain and reduced survival were observed in the offspring. Despite the observed decreased weight gain, physical development was normal (with respect to timing and organ morphology).
8.4 Pediatric Use
The safety and effectiveness of droxidopa in pediatric patients have not been established.
8.5 Geriatric Use
A total of 197 patients with symptomatic nOH aged 75 years or above were included in the droxidopa clinical program. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
Droxidopa and its metabolites are primarily cleared renally. Patients with mild or moderate renal impairment (GFR greater than 30 mL/min) were included in clinical trials and did not have a higher frequency of adverse reactions. Clinical experience with droxidopa in patients with severe renal function impairment (GFR less than 30 mL/min) is limited.
-
10 OVERDOSAGE
10.1 Symptoms
There have been cases of overdose reported during postmarketing surveillance. A patient ingested 7,700 mg of droxidopa and experienced a hypertensive crisis that resolved promptly with treatment. Another patient treated with a total daily dose of 2,700 mg of droxidopa experienced hypertension and an intracranial hemorrhage.
10.2 Treatment
There is no known antidote for droxidopa overdosage. In case of an overdose that may result in an excessively high blood pressure, discontinue droxidopa and treat with appropriate symptomatic and supportive therapy. Counsel patients to remain in a standing or seated position until their blood pressure drops below an acceptable limit.
-
11 DESCRIPTION
Droxidopa capsules contain droxidopa, which is a synthetic amino acid precursor of norepinephrine, for oral administration. Chemically, droxidopa is (2S,3R)-2-amino-3-(3,4-dihdroxyphenyl)-3- hydroxypropanoic acid (or) L-threo-3,4- dihydroxy phenyl serine. It has the following structural formula:
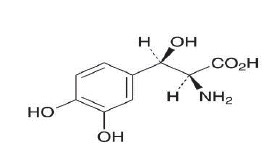
Droxidopa is a white to light brown color powder. It is soluble in 1N hydrochloric acid, slightly soluble in water and practically insoluble in ethanol (99.5%). It has a molecular weight of 213.19 and a molecular formula of C 9H 11NO 5.
Droxidopa capsules also contain the following inactive ingredients: corn starch, mannitol, and magnesium stearate. The capsule shell is printed with black ink. The black ink contain black iron oxide, potassium hydroxide, propylene glycol, shellac and strong ammonia solution. The capsule shell contains gelatin and titanium dioxide, in addition 100 mg and 200 mg capsule shells contain FD&C Blue 1 and FD&C Red 3.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The exact mechanism of action of droxidopa in the treatment of neurogenic orthostatic hypotension is unknown. Droxidopa is a synthetic amino acid analog that is directly metabolized to norepinephrine by dopa-decarboxylase, which is extensively distributed throughout the body. Droxidopa is believed to exert its pharmacological effects through norepinephrine and not through the parent molecule or other metabolites. Norepinephrine increases blood pressure by inducing peripheral arterial and venous vasoconstriction. Droxidopa in humans induces small and transient rises in plasma norepinephrine.
12.2 Pharmacodynamics
Peak droxidopa plasma concentrations are associated with increases in systolic and diastolic blood pressures. Droxidopa has no clinically significant effect on standing or supine heart rates in patients with autonomic failure.
Cardiac Electrophysiology
No prolongation of the QTc interval was observed with droxidopa at single oral doses up to 2,000 mg, as shown in a dedicated thorough QT study.
12.3 Pharmacokinetics
Absorption
Peak plasma concentrations (C max) of droxidopa were reached by 1 to 4 hours post-dose (mean of approximately 2 hours) in healthy volunteers. High-fat meals have a moderate impact on droxidopa exposure with C max and area under the plasma concentration-time curve (AUC) decreasing by 35% and 20%, respectively. The C max was delayed by approximately 2 hours with a high-fat meal.
Distribution
Pre-clinical studies suggest that droxidopa can cross the blood-brain barrier. Droxidopa exhibits plasma protein binding of 75% at 100 ng/mL and 26% at 10,000 ng/mL. The estimated apparent volume of distribution of droxidopa is about 200 L in humans.
Elimination
The total clearance of droxidopa after oral administration (CL/F) was approximately 400 mL/hr following administration of a single 300 mg dose.
Metabolism
The metabolism of droxidopa is mediated by catecholamine pathway and not through the cytochrome P450 system. Droxidopa is initially converted to methoxylated dihydroxyphenylserine (3-OM-DOPS), a major metabolite, by catechol-O-methyltransferase (COMT), to norepinephrine by DOPA decarboxylase (DDC), or to protocatechualdehyde by DOPS aldolase. After oral dosing in humans, plasma norepinephrine levels peak within 3 to 4 hours but are generally very low (less than 1 ng/mL) and variable with no consistent relationship with dose. The contribution of the metabolites of droxidopa other than norepinephrine to its pharmacological effects is not well understood.
Excretion
The mean elimination half-life of droxidopa is approximately 2.5 hours in humans. The major route of elimination of droxidopa and its metabolites is via the kidneys in both animals and in humans. Studies in animals with radiolabeled drug showed that ~75% of the administered radioactivity was excreted in urine within 24 hours of oral dosing.
Specific Populations
There are no clinically relevant effects of age, body mass index, or sex on the pharmacokinetics of droxidopa. A population pharmacokinetic analysis suggests that hepatic function, assessed by aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and total bilirubin, did not influence the exposure to droxidopa. The controlled clinical trials included patients with mild to moderate renal impairment. No dose adjustments are required in patients with mild to moderate renal impairment.
Drug Interaction Studies
No dedicated drug-drug interaction studies were performed for droxidopa. Patients in the Phase 3 trials with droxidopa received concomitant levodopa/carbidopa, dopamine agonists, MAO-B inhibitors, COMT inhibitors and other medications used to treat Parkinson’s disease. Carbidopa, a peripheral dopa-decarboxylase inhibitor, could prevent the conversion of droxidopa to norepinephrine outside of the central nervous system (CNS). Patients taking droxidopa with L-DOPA/dopa-decarboxylase inhibitor combination drugs had decreased clearance of droxidopa, an increase in overall exposure (AUC) to droxidopa of approximately 100%, and an increase in overall exposure to 3-OM-DOPS of approximately 50%. However, in clinical trials, it was found that the decreased clearance was not associated with a significant need for a different treatment dose or increases in associated adverse events. Dopamine agonists, amantadine derivatives, and MAO-B inhibitors do not appear to affect droxidopa clearance, and no dose adjustments are required. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies have been conducted at dosages up to 1,000 mg/kg/day in mice and up to 100 mg/kg/day in rats with no indication of carcinogenic effects. Based on dose per unit body surface area, these two doses correspond to approximately 3 and 0.5 times, respectively, the maximum recommended total daily dose of 1,800 mg in a 60 kg patient. Droxidopa was clastogenic in Chinese hamster ovary cells (chromosome aberration assay), but was not mutagenic in bacteria (Ames assay), and was not clastogenic in a mouse micronucleus assay.
Studies in rats show that droxidopa has no effect on fertility.
13.2 Animal Toxicology and/or Pharmacology
In long-term chronic toxicity studies, rats and mice treated for 52 and 80 weeks, respectively, at doses up to 300 mg/kg/day in rats and 1,000 mg/kg/day in mice had increased incidences of renal and cardiac lesions (rats and mice) and deaths (rats only). The doses at which these effects were not seen represented 0.2 and 0.3 times, in rats and mice, respectively, the maximum recommended total daily dose of 1,800 mg in a 60 kg patient, when based on body surface area.
No signs of toxicity were observed in monkeys or dogs given droxidopa for 13 weeks at doses 32 times (3,000 mg/kg/day) and 37 times (2,000 mg/kg/day), respectively, the maximum human dose.
-
14 CLINICAL STUDIES
14.1 Studies in Neurogenic Orthostatic Hypotension
Clinical studies (described below) examined the efficacy of droxidopa in the short-term (1 to 2 weeks) and over longer-term periods (8 weeks; 3 months). Studies 301 and 306B showed a treatment effect of droxidopa at Week 1, but none of the studies demonstrated continued efficacy beyond 2 weeks of treatment.
Study 306B was a multi-center, double-blind, randomized, placebo-controlled, parallel-group study in patients with symptomatic nOH and Parkinson’s disease. Patients entering the study were required to have a decrease of at least 20 mm Hg or 10 mm Hg, respectively, in systolic or diastolic blood pressure, within 3 minutes after standing, as well as symptoms associated with neurogenic orthostatic hypotension. The study had an initial dose titration period that lasted up to 2 weeks in which patients received placebo or 100 to 600 mg of droxidopa three times daily, followed by an 8-week treatment period.
Efficacy was measured using the OHSA Item #1 score (“dizziness, lightheadedness, feeling faint, and feeling like you might black out”) at Week 1, in patients who had completed titration and 1 week of maintenance therapy.
A total of 171 patients were enrolled, and 147 patients were included in the efficacy analysis. The mean age was 72 years, and patients were mostly Caucasian. During the study, 94% of placebo-treated patients and 88% on droxidopa were taking dopa-decarboxylase inhibitors; 17% of placebo-treated patients and 26% on droxidopa were taking fludrocortisone. There were more premature discontinuations in the droxidopa group (28%) than in the placebo group (20%).
In both groups, the mean baseline dizziness score was 5.1 on an 11-point scale. At Week 1, patients showed a statistically significant mean 0.9 unit decrease in dizziness with droxidopa versus placebo ( P=0.028), but the effect did not persist beyond Week 1. The data at all time points are shown in Figure 1.
Patients receiving droxidopa also had a greater increase, compared to placebo, in the Week 1 lowest standing systolic blood pressure within 3 minutes after standing (5.6 mm Hg; P=0.032).
Figure 1. Mean Change in OHSA Item 1 Score by Week in Study 306B
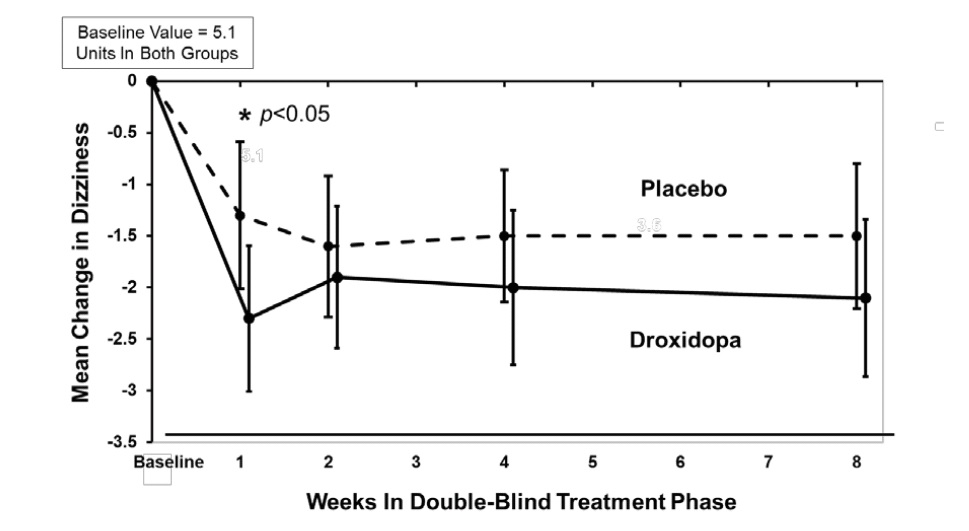
Note: The graph is based on observed data only. The error bars are the 95% confidence interval of the mean change from baseline in OHSA Item 1 scores.
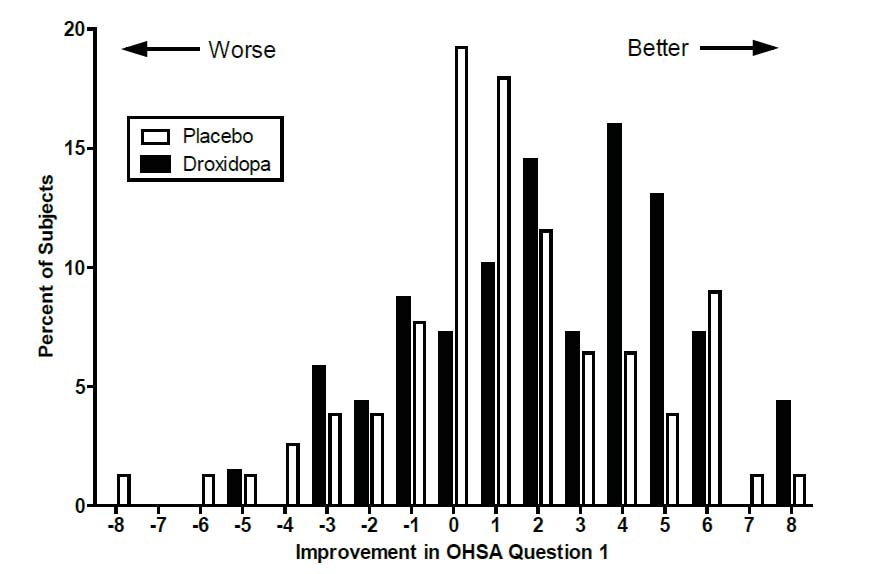
Figure 2. Distribution of Patients by Change in OHSA Item 1, Baseline to Week 1, in Study 306B
Figure 2 shows the distribution of changes from Baseline to Week 1 in the OHSA Item #1 score. Overall, the figure shows that patients treated with droxidopa improved more than those treated with placebo.
Study 301 was a multicenter, multinational, double-blind, randomized, placebo-controlled, parallel-group study in patients with symptomatic neurogenic orthostatic hypotension. The study included an initial open-label dose titration period, a 7-day washout period, and a randomized double-blind 7-day treatment period. To be eligible for enrollment, patients were required to have a decrease in systolic or diastolic blood pressure of at least 20 or 10 mm Hg, respectively, within 3 minutes after standing. The study was enriched, such that only patients who had been identified as “responders” during the titration period were randomized to droxidopa or placebo. To be considered a responder, a patient had to demonstrate improvement on the OHSA Item #1 score by at least 1 point, as well as an increase in systolic blood pressure of at least 10 mm Hg post-standing, during the open-label dose titration period. Patients who dropped out during the titration period because of side effects or other reasons were also not included in the double-blind portion of the study.
Patients had a primary diagnosis of Parkinson’s disease (n=60), pure autonomic failure (n=36), or multiple system atrophy (n=26). The mean age was 60 years, and most were Caucasian. 45% of patients were taking dopa-decarboxylase inhibitors, and 29% were taking fludrocortisone.
Efficacy was measured using the Orthostatic Hypotension Questionnaire (OHQ), a patient-reported outcome that measures symptoms of nOH and their impact on the patient’s ability to perform daily activities that require standing and walking. The OHQ includes OHSA Item #1 as one of several components. A statistically significant treatment effect was not demonstrated on OHQ (treatment effect of 0.4 unit, P=0.19).
The mean baseline dizziness score on OHSA Item #1 (“dizziness, lightheadedness, feeling faint, and feeling like you might black out”) was 5.2 units on an 11-point scale. At Week 1 of treatment, patients showed a mean 0.7 unit decrease in dizziness with droxidopa versus placebo ( P=0.06).
Study 302 (n=101) was a placebo-controlled, 2-week randomized withdrawal study of droxidopa in patients with symptomatic nOH. Study 303 (n=75) was an extension of Studies 301 and 302, where patients received their titrated dose of droxidopa for 3 months and then entered a 2-week randomized withdrawal phase. Neither study showed a statistically significant difference between treatment arms on its primary endpoint. Considering these data, the effectiveness of droxidopa beyond 2 weeks is uncertain, and patients should be evaluated periodically to determine whether droxidopa is continuing to provide a benefit.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Droxidopa capsules are supplied in the following dosage strengths:
100 mg: Pink opaque size '3' hard gelatin capsule imprinted with 'V' on cap and '26' on body filled with white to light brown color powder.
90-count bottle NDC 31722-014-90200 mg: Light blue opaque size '2' hard gelatin capsule imprinted with 'V' on cap and '28' on body filled with white to light brown color powder.
90-count bottle NDC 31722-015-90
300 mg: White opaque size '1el' hard gelatin capsule imprinted with 'V' on cap and '32' on body filled with white to light brown color powder.
90-count bottle NDC 31722-010-90
Dispense in a tight, light resistant container with a child resistant closure as defined in the USP.
-
17 PATIENT COUNSELING INFORMATION
Elevations in Blood Pressure
Counsel patients that droxidopa causes elevations in blood pressure and increases the risk of supine hypertension, which could lead to strokes, heart attacks, and death. Instruct patients to rest and sleep in an upper-body elevated position and monitor blood pressure. Instruct patients how to manage observed blood pressure elevations. To reduce the risk of supine hypertension, in addition to raising the upper body, the late afternoon dose of droxidopa should be taken at least three hours before bedtime [see Warnings and Precautions ( 5.1)].
Concomitant Treatments
Counsel patients about the concomitant use of drugs to treat other conditions that may have an additive effect with droxidopa [see Drug Interactions ( 7)].
Allergic Reactions
Counsel patients to discontinue droxidopa and seek immediate medical attention if any signs or symptoms of a hypersensitivity reaction such as anaphylaxis, angioedema, bronchospasm, urticaria or rash occur [see Warnings and Precautions ( 5.4)].
Lactation
Advise women not to breastfeed during treatment with droxidopa [see Use in Specific Populations ( 8.2)].Food
Patients should take droxidopa the same way each time, either with food or without food [see Dosage and Administration ( 2.1)].Missed Dose
If a dose is missed, patients should take the next dose at the regularly scheduled time and should not double the dose.

Manufactured for:
Camber Pharmaceuticals, Inc.
Piscataway, NJ 08854
By: Annora Pharma Pvt. Ltd.
Sangareddy - 502313, Telangana, India.
Revised: 07/2021
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
DROXIDOPA
droxidopa capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:31722-014 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DROXIDOPA (UNII: J7A92W69L7) (DROXIDOPA - UNII:J7A92W69L7) DROXIDOPA 100 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) MANNITOL (UNII: 3OWL53L36A) MAGNESIUM STEARATE (UNII: 70097M6I30) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: MB5IUD6JUA) AMMONIA (UNII: 5138Q19F1X) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C RED NO. 3 (UNII: PN2ZH5LOQY) Product Characteristics Color pink Score no score Shape CAPSULE Size 16mm Flavor Imprint Code V;26 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:31722-014-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 08/09/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA211726 08/09/2021 DROXIDOPA
droxidopa capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:31722-015 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DROXIDOPA (UNII: J7A92W69L7) (DROXIDOPA - UNII:J7A92W69L7) DROXIDOPA 200 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) MANNITOL (UNII: 3OWL53L36A) MAGNESIUM STEARATE (UNII: 70097M6I30) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: MB5IUD6JUA) AMMONIA (UNII: 5138Q19F1X) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C RED NO. 3 (UNII: PN2ZH5LOQY) Product Characteristics Color blue (light blue) Score no score Shape CAPSULE Size 18mm Flavor Imprint Code V;28 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:31722-015-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 08/09/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA211726 08/09/2021 DROXIDOPA
droxidopa capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:31722-010 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DROXIDOPA (UNII: J7A92W69L7) (DROXIDOPA - UNII:J7A92W69L7) DROXIDOPA 300 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) MANNITOL (UNII: 3OWL53L36A) MAGNESIUM STEARATE (UNII: 70097M6I30) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: MB5IUD6JUA) AMMONIA (UNII: 5138Q19F1X) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color white Score no score Shape CAPSULE Size 20mm Flavor Imprint Code V;32 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:31722-010-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 02/18/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA211726 02/18/2021 Labeler - Camber Pharmaceuticals, Inc. (826774775) Establishment Name Address ID/FEI Business Operations Annora Pharma Private Limited 650980746 analysis(31722-010, 31722-014, 31722-015) , manufacture(31722-010, 31722-014, 31722-015)


