Label: HYQVIA (immune globulin infusion 10%- human with recombinant human hyaluronidase kit















-
NDC Code(s):
0944-2510-02,
0944-2511-02,
0944-2512-02,
0944-2513-02, view more0944-2514-02, 0944-2715-25, 0944-2716-05, 0944-2717-10, 0944-2718-20, 0944-2719-30, 0944-2720-03, 0944-2721-03, 0944-2722-03, 0944-2723-03, 0944-2724-03
- Packager: Takeda Pharmaceuticals America, Inc.
- Category: PLASMA DERIVATIVE
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated November 12, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use HYQVIA® safely and effectively. See full prescribing information for HYQVIA.
HYQVIA® [Immune Globulin Infusion 10% (Human) with Recombinant Human Hyaluronidase] Solution, for subcutaneous administration
Initial U.S. Approval: 2014WARNING: THROMBOSIS
See full prescribing information for complete boxed warning.
- Thrombosis may occur with immune globulin products, including HYQVIA. Risk factors may include advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling vascular catheters, hyperviscosity and cardiovascular risk factors. (5.2)
- For patients at risk of thrombosis, administer HYQVIA at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. (5.2)
- Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk of hyperviscosity. (5.2)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
HYQVIA is an Immune Globulin Infusion 10% (Human) with a Recombinant Human Hyaluronidase (rHuPH20) indicated for the treatment of
DOSAGE AND ADMINISTRATION
For subcutaneous use only
The recommended rHuPH20 dose is 80 U/g Immune globulin G (IgG), which corresponds to 0.5 mL rHuPH20 solution per 10 mL Immune Globulin Infusion 10% (Human) solution for both indications. (2.1)
PI (2.1)
Initiation of Treatment with HYQVIA
- Administer the first dose approximately one week after the last infusion of their previous treatment. (2.1)
- Increase the dose and frequency from a 1-week dose to a 3-or 4-week dose. (2.1)
Switching from Immune Globulin Intravenous (Human) [IGIV] treatment:
- Use same dose and frequency as previous intravenous treatment after the initial ramp-up. (2.1)
Naïve to Immune Globulin Subcutaneous (Human) [IGSC] treatment or switching from IGSC treatment:
- 300 to 600 mg/kg at 3 to 4-week intervals, after initial ramp-up. (2.1)
CIDP (2.1)
Switching from Immune Globulin Intravenous (Human) [IGIV] treatment:
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- History of anaphylactic or severe systemic hypersensitivity reactions to Immune Globulin (Human). (4)
- IgA deficient patients with antibodies against IgA and a history of hypersensitivity. (4)
- Known systemic hypersensitivity to hyaluronidase including Recombinant Human Hyaluronidase of HYQVIA. (4)
- Known systemic hypersensitivity to human albumin (in the hyaluronidase solution). (4)
WARNINGS AND PRECAUTIONS
- IgA-deficient patients with anti-IgA antibodies are at greater risk of severe hypersensitivity and anaphylactic reactions. (5.1)
- Thrombosis may occur following treatment with immune globulin products including HYQVIA. (5.2)
- Antibodies to rHuPH20 can develop. The potential exists for such antibodies to cross-react with endogenous Hyaluronidase (PH20) which is known to be expressed in the adult male testes, epididymis, and sperm. It is unknown whether these antibodies may interfere with fertilization in humans. The clinical significance of these antibodies is not known. (5.3)
- Aseptic Meningitis Syndrome (AMS) may occur. Discontinue treatment if AMS symptoms appear. (5.4)
- Acute intravascular hemolysis may occur. Monitor for clinical signs and symptoms of hemolysis and hemolytic anemia. (5.5)
- Infusion into or around an infected area can spread a localized infection. (5.7)
- Monitor for pulmonary adverse reactions (transfusion-related acute lung injury [TRALI]). (5.8)
- May carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. (5.9)
ADVERSE REACTIONS
PI: The most common adverse reactions observed in clinical trials in >5% of subjects were local reactions, headache, antibody formation against Recombinant Human Hyaluronidase (rHuPH20), fatigue, nausea, pyrexia, and vomiting. (6)
CIDP: The most common adverse reactions observed in >5% of study subjects in clinical studies (Study 1, Study 2) of HYQVIA for CIDP were local reactions, headache, pyrexia, nausea, fatigue, erythema, pruritus, increased lipase, abdominal pain, back pain, and pain in extremity. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals U.S.A., Inc. at 1-877-TAKEDA-7 (1-877-825-3327) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Passive transfer of antibodies may transiently interfere with the immune responses to live virus vaccines, such as measles, mumps, and rubella. (7)
USE IN SPECIFIC POPULATIONS
Geriatric: In patients over age 65 or in any patient at risk of developing renal insufficiency, do not exceed the recommended dose, and consider infusing HYQVIA at lower, more frequent doses. (8.5)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: THROMBOSIS
1 INDICATIONS AND USAGE
1.1 Primary Immunodeficiency (PI)
1.2 Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Administration
2.3 Preparation and Handling
2.4 Instructions for Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
5.2 Thrombosis
5.3 Immunogenicity of rHuPH20
5.4 Aseptic Meningitis Syndrome (AMS)
5.5 Hemolysis
5.6 Renal Dysfunction/Failure
5.7 Spread of Localized Infection
5.8 Transfusion-Related Acute Lung Injury (TRALI)
5.9 Transmissible Infectious Agents
5.10 Interference with Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Primary Immunodeficiency (PI)
14.2 Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: THROMBOSIS
- Thrombosis may occur with immune globulin products, including HYQVIA. Risk factors may include advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling vascular catheters, hyperviscosity and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors. (5.2)
- For patients at risk of thrombosis, administer HYQVIA at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. (5.2)
- Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk of hyperviscosity. (5.2)
-
1 INDICATIONS AND USAGE
1.1 Primary Immunodeficiency (PI)
HYQVIA is an Immune Globulin Infusion 10% (Human) with a Recombinant Human Hyaluronidase (rHuPH20) indicated for the treatment of Primary Immunodeficiency (PI) in adults and pediatric patients two years of age and older. This includes, but is not limited to, common variable immunodeficiency (CVID), X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies.1,2
-
2 DOSAGE AND ADMINISTRATION
For subcutaneous use only
2.1 Dose
The recommended rHuPH20 dose is 80 U/g Immune Globulin (IgG), which corresponds to 0.5 mL rHuPH20 solution per 10 mL Immune Globulin Infusion 10% (Human) solution for both indications.
The recommended Immune Globulin Infusion 10% (Human) dose is given below.
Primary Immunodeficiency (PI)
Initiation of Treatment with HYQVIA
- For patients previously on another IgG treatment, administer the first dose approximately one week after the last infusion of their previous treatment.
- Gradually increase the dose and frequency from a 1-week dose to a 3-or 4-week dose (see ramp-up schedule in Table 1).
- Initiating treatment at a full monthly dose was not evaluated in the clinical trial.
Table 1: Initial Treatment Interval/Dosage Ramp-Up Schedule Week Infusion Number Dose Interval Example for 30 grams per 4 weeks 1 1st infusion 1-week-dose 7.5 grams 2 2nd infusion 2-week-dose 15 grams 3 No infusion Not Applicable (NA) NA 4 3rd infusion 3-week-dose 22.5 grams 5 No infusion NA NA 6 No infusion NA NA 7 4th infusion (if required) 4-week-dose 30 grams For patients switching from Immune Globulin Intravenous (Human) [IGIV] treatment:
- Administer HYQVIA at the same dose and frequency as the previous intravenous treatment, after the initial dose ramp-up.
For patients at risk for measles exposure:
- If a patient has been exposed to measles, it may be prudent to administer a dose of HYQVIA as soon as possible and within 6 days of exposure. A dose of 400 mg/kg should provide a serum level >240 mIU/mL of measles antibodies for at least two weeks.
- If a patient is at risk of future measles exposure and receives a dose of less than 530 mg/kg every 3 to 4 weeks, the dose should be increased to at least 530 mg/kg. This should provide a serum level of 240 mIU/mL of measles antibodies for at least 22 days after infusion.
Individualization of Dose
- If HYQVIA is administered at the same dose and frequency, the serum IgG levels from HYQVIA should be comparable to serum IgG levels from intravenous treatment.
For dose adjustment:
- Calculate the difference between the patient's serum IgG trough level during HYQVIA treatment and the IgG trough level during the previous intravenous treatment.
- Find this difference (in mg/dL) in the columns of Table 2 and the corresponding amount (in mL) by which to increase or decrease the dose based on the patient's body weight and desired change in IgG trough level.
Table 2: Individualization in Volume Administered per Dosing Interval for Intended Change in IgG Trough Level* (Difference in IgG Trough Levels) Body Weight 100 mg/dL 200 mg/dL 300 mg/dL 400 mg/dL - *
- Derived using a slope of 3.3 kg/dL
10 kg 3 mL 6 mL 9 mL 12 mL 20 kg 6 mL 12 mL 18 mL 24 mL 30 kg 9 mL 18 mL 27 mL 36 mL 40 kg 12 mL 24 mL 36 mL 48 mL 50 kg 15 mL 30 mL 45 mL 61 mL 60 kg 18 mL 36 mL 55 mL 73 mL 70 kg 21 mL 42 mL 64 mL 85 mL 80 kg 24 mL 48 mL 73 mL 97 mL 90 kg 27 mL 55 mL 82 mL 109 mL 100 kg 30 mL 61 mL 91 mL 121 mL 110 kg 33 mL 67 mL 100 mL 133 mL 120 kg 36 mL 73 mL 109 mL 145 mL 130 kg 39 mL 79 mL 118 mL 158 mL 140 kg 42 mL 85 mL 127 mL 170 mL Example 1: A patient with a body weight of 80 kg has a measured IgG trough level of 800 mg/dL and the reference trough level is 1000 mg/dL. The trough level difference is 200 mg/dL (1000 mg/dL minus 800 mg/dL). The dose of HYQVIA would be increased by 48 mL (4.8 grams) per dosing interval.
Example 2: A patient with a body weight of 60 kg has a measured IgG trough level of 1000 mg/dL and the reference trough level is 900 mg/dL. The trough level difference is -100 mg/dL (900 mg/dL minus 1000 mg/dL). The dose of HYQVIA would be decreased by 18 mL (1.8 grams) per dosing interval.
HYQVIA can be used to administer a full therapeutic dose in one site up to every four weeks. Adjust the frequency and number of infusion sites taking into consideration volume, total infusion time, and tolerability. Adjust the frequency as needed so that the patient receives the same weekly equivalent dose.
Example 3: When adjusting a dose of 30 grams administered every 3 weeks, administer 40 grams of HYQVIA every 4 weeks. If a higher trough level is required relative to intravenous treatment at 3- or 4-week intervals, increase the dose or decrease the dosing interval. Evaluate the use of a second site or infusing at shorter intervals when the volume of HYQVIA is greater than 600 mL.
If a patient misses a dose, administer the missed dose as soon as possible and then resume scheduled treatments as applicable.
If HYQVIA is administered at a different interval than the previous treatment, either intravenously or subcutaneously, then Table 2 should not be used and the dose of HYQVIA should be adjusted, if necessary, based on clinical response.
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
General instruction:
- Before initiating therapy with HYQVIA, calculate the weekly equivalent dose to plan for the ramp-up schedule. Dose and dosing frequency can be adjusted based on the individual clinical response.
- A dose ramp-up schedule is recommended by gradually increasing the SC infusion volume until the full dose is reached to ensure the patients’ tolerability.
- Depending on the treating physician's discretion, in patients who tolerate the first two infusions well, subsequent infusions may be administered by gradually increasing doses and decreasing dose intervals, considering the volume and total infusion time.
- Doses less than or equal to 0.4 g/kg may be administered without a ramp-up provided acceptable patient tolerance.
For patients switching from Immune Globulin Intravenous (Human) [IGIV] treatment:
- Patients switching from intravenous administration of immune globulin must be on stable1 doses of IGIV.
- Before initiating therapy with HYQVIA, calculate the weekly equivalent dose by dividing the last IGIV dose by the IGIV dose interval in weeks.
- The starting dose and dosing frequency of HYQVIA is the same as the patient’s previous IGIV treatment. The typical dosing interval range in the clinical trial for HYQVIA was 4 weeks. For patients with less frequent IGIV dosing (greater than 4 weeks), the dosing interval can be converted to 3 or 4 weeks while maintaining the same monthly equivalent IgG dose.
- Administer the calculated one-week dose (1st infusion) two weeks after the last IGIV infusion as directed in section 2.3. One week after the first HYQVIA dose, administer another weekly equivalent dose (2nd infusion).
- A ramp-up period can take up to 9 weeks (Table 3), depending on the dosing interval and tolerability.
- 1
- Variations in the dosing interval of up to ±7 days or monthly equivalent dose amount of up to ±20% between the subject’s IgG infusions are considered a stable dose.
Table 3: IGIV to HYQVIA Infusion Dose Ramp-up Schedule in Study 1 Week* Infusion Number Dose Interval Example for 100 g every 4 weeks - *
- Clock starts one week after the last IGIV dose is administered. Week 1 is the week that starts one week after the last IGIV dose.
1 No infusion Not applicable (NA) NA 2 1st infusion 1-week-dose 25g 3 2nd infusion 1-week-dose 25g 4 3rd infusion 2-week-dose 50g 5 No infusion NA NA 6 4th infusion 3-week-dose 75g 7 No infusion NA NA 8 No infusion NA NA 9 5th infusion 4-week-dose 100g (Full dose reached) 2.2 Administration
HYQVIA should be administered by a healthcare professional, caregiver or self-administered by the patient after appropriate training.
- Infusion of Immune Globulin Infusion 10% (Human) requires an infusion pump capable of infusing a patient's therapeutic dose at infusion rates up to 300 mL/hr/site. The pump must have the ability to titrate the flow rate up or down if required to improve tolerability. To ensure maximum flow rates, use a subcutaneous needle set that is 24 gauge and labeled for high flow rates.
- Infusion site leakage can occur during or after subcutaneous administration of immunoglobulin, including HYQVIA. Consider using longer needles (14 or 12 mm rather than 9 mm) and/or more than one infusion site.
- Infuse the two components of HYQVIA sequentially, beginning with the rHuPH20. If using two or three infusion sites, divide the rHuPH20 evenly between all sites.
- Initiate infusion of the full dose of the Immune Globulin Infusion 10% (Human) through the same subcutaneous needle set within approximately 10 minutes of the rHuPH20 infusion. For each full or partial vial of Immune Globulin Infusion 10% (Human) used, administer the entire contents of the rHuPH20 vial.
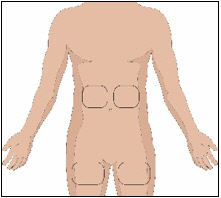
Selection of Infusion Site(s)
The suggested site(s) for the infusion of HYQVIA are the abdomen and thighs. If two sites are used, the two infusion sites should be on opposite sides of the body. If using three infusion sites, the sites should be at least 10 cm apart. Avoid bony prominences, or areas that are scarred, inflamed, or infected.
Volume per Site
Primary immunodeficiency (PI)
Administer up to 600 mL per site for patients whose body weight is greater than or equal to 40 kg and up to 300 mL per site for patients whose body weight is less than 40 kg.
A second site can be used at the discretion of the physician and patient based on tolerability and total volume. If a second site is used, administer half the total volume of rHuPH20 of HYQVIA in each site.
Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP)
On a given infusion day, the maximum infusion volume should not exceed 1200 mL for subjects weighing ≥40 kg or 600 mL for subjects weighing <40 kg. If the maximum daily dose limit is exceeded or the patient cannot tolerate the infusion volume, the dose may be administered over multiple days in divided doses with 48 to 72 hours between doses to allow absorption of infusion fluid at the infusion site(s). The dose can be administered at 1, 2, or 3 infusion sites with a maximum infusion volume of 600 mL per site (or as tolerated). If using three sites, the maximum is 400 mL per site.
Rate of Infusion
Primary immunodeficiency (PI)
Administer the rHuPH20 of HYQVIA at an initial rate per site of approximately 1 to 2 mL per minute, or as tolerated.
Administer Immune Globulin Infusion 10% (Human) of HYQVIA at rates as shown in Table 4 for the initial infusions. If the patient tolerates these infusions at the full dose and maximum rate, adjust both the time intervals and number of rate changes of the ramp-up used for successive infusions at the discretion of the physician and patient.
Table 4: Recommended Immune Globulin Infusion 10% (Human) Infusion Rates First 2 Infusions Subsequent 2 or 3 Infusions Subjects <40 kg
(<88lbs)Subjects ≥40 kg
(≥88lbs)Subjects <40 kg
(<88lbs)Subjects ≥40 kg
(≥88lbs)Intervals Rate per site Rate per site Rate per site Rate per site Minutes mL per hour mL per hour mL per hour mL per hour 5 - 15 5 10 10 10 5 - 15 10 30 20 30 5 - 15 20 60 40 120 5 - 15 40 120 80 240 Remainder of infusion 80 240 160 300 Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP)
The full dose of rHuPH20 solution is infused at a rate of 1 to 2 mL per minute (60 mL to 120 mL/hr) per infusion site or as tolerated. Immune Globulin Infusion, 10% (Human) can be administered through the same subcutaneous needle set within approximately 10 minutes after the rHuPH20 infusion is completed.
Patients with body weight of 40 kg or above
The Immune Globulin Infusion 10% (Human) should be infused at an initial rate of 10 mL per hour per infusion site. If tolerated, the rate of the administration may be increased at intervals of 5 to 15 minutes and to a maximum infusion rate of 240 mL per hour per infusion site for the initial one or two infusions. For subsequent infusions the rate can be adjusted to a maximum of 300 mL per hour per infusion site.
Patients with body weight under 40 kg
The Immune Globulin Infusion, 10% (Human) should be infused at an initial rate of 5 mL per hour per infusion site. If well tolerated, the rate of the administration may be increased at intervals of 5 to 15 minutes and to a maximum infusion rate of 80 mL per hour per infusion site for the initial one or two infusions. For subsequent infusions the rate can be adjusted to a maximum of 160 mL per hour per infusion site.
2.3 Preparation and Handling
- Visually inspect both vials of HYQVIA for discoloration and particulate matter prior to administration.
- The appearance of the Immune Globulin Infusion 10% (Human) of HYQVIA can vary from clear or slightly opalescent and colorless or pale yellow.
- The appearance of the rHuPH20 of HYQVIA should be clear and colorless.
- Do not use either component of HYQVIA if either solution is cloudy or has particulates.
- Allow refrigerated product to come to room temperature before use. Do not apply heat or place in microwave.
- Do not shake HYQVIA.
- Do not mix the rHuPH20 and the Immune Globulin Infusion 10% (Human) of HYQVIA into the same container prior to administration.
- Do not mix or administer components of HYQVIA with other products. Administer components of HYQVIA sequentially. Do not use either component alone.
- Flush the infusion line with normal saline or Dextrose 5% in water (D5W) if required.
- HYQVIA contains no preservative. Discard any unused product according to local standards for biohazard products.
2.4 Instructions for Administration
Use aseptic technique when preparing and administering HYQVIA for infusion.
For more detail on steps, see accompanying Information for Patients.
1. Inspect the vials: Inspect for clarity, color, and expiration date(s).
2. Prepare for infusion:
- Gather supplies: HYQVIA dual vial unit(s), ancillary supplies, sharps container and infusion pump (program pump per physician recommendation following manufacturer's instructions).
- Prepare a clean work area.
- Wash hands.
- If the Immune Globulin Infusion 10% (Human) and rHuPH20 are pooled into separate containers, skip to Step 5.
3. Prepare the rHuPH20 of HYQVIA (Labeled as “HY”):
- Remove the purple protective cap and ensure the blue vial caps are removed.
- Wipe each stopper with a sterile alcohol wipe and allow to dry.
- Attach a syringe to a needle/needle-less transfer device. A sterile needle or needle-less transfer device [(18-22) gauge sterile needle] may be used for all vial sizes. Position the sharp tip of the needle/needle-less transfer device over the center of the vial stopper and insert it at a 90-degree angle. Inject air and then draw the full contents of each vial labeled "HY" into a single syringe, if possible.
- Attach the syringe containing the rHuPH20 to the subcutaneous needle set and prime up to the needle hub.
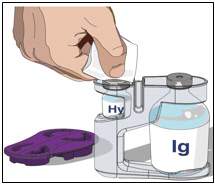
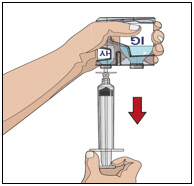
4. Prepare Immune Globulin Infusion 10% (Human) of HYQVIA (Labeled as “IG”):
- Wipe each stopper with a sterile alcohol wipe and allow to dry.
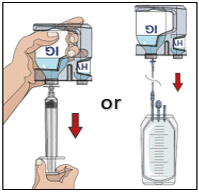
- Transfer the vial(s) labeled "IG" using one of the following methods:
- Pool into an infusion bag, using a transfer device per manufacturer's directions. Attach and prime the pump administrating tubing; or
- Directly spike the vial using a vented pump administration tubing and prime as directed.
If using a syringe driver, transfer into the syringe(s), preferably using a vented spike.
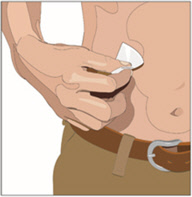
5. Prepare the infusion site(s):
- Potential sites for infusion include the middle to upper abdomen and thigh.
- Avoid: bony prominences, blood vessels, scars, or areas that are inflamed or infected.
- If two sites are desired, a bifurcated needle set may be used on opposite sides of the body.
- Rotate sites by choosing opposite sides of the body between successive infusions.
- Cleanse the infusion site(s) with a sterile alcohol wipe beginning at the center of each infusion site and moving outward in a circular motion. Allow the infusion site(s) to dry.
6. Insert and secure the 24-gauge subcutaneous needle:
- Pinch at least one inch of skin between two fingers. Insert the needle at a 90-degree angle into the subcutaneous tissue and secure the needle with sterile tape and check the placement.
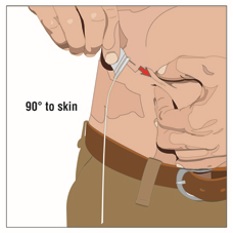
- If using the push method:
- Gently pull back on the plunger of the attached syringe and monitor for any blood return in the tubing.
- If blood is seen in the tubing, remove and discard the needle and repeat steps 3, 5, and 6 with a new subcutaneous needle and infusion site.
- If using the pump method:
- Close the clamp above the lower port of the pump tubing.
- Clean the lower port with an alcohol swab and allow to dry, for at least 30 seconds.
- Attach a 5 mL syringe to the lower port of the pump tubing.
- Pull back gently on the syringe plunger. If no blood, remove the syringe and open the lower port clamp.
- Secure the needle in place with a sterile protective dressing.
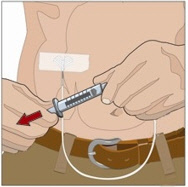
7. Administer the rHuPH20 of HYQVIA:
- If using push method:
- Infuse the rHuPH20manually at an initial rate of approximately 1 to 2 mL per minute per infusion site and increase as tolerated.
- If using a pump method:
- start at an initial rate at 60 mL to 120 mL/hr per infusion site and may increase as tolerated up to 300 mL/hr.
- If more than one site is used, divide the contents equally between sites. At the end of infusion, disconnect the empty syringe and attach the pump tubing/syringe containing the Immune Globulin Infusion 10% (Human) of HYQVIA to the same subcutaneous needle set.
8. Administer the Immune Globulin Infusion 10% (Human) of HYQVIA:
Within approximately 10 minutes of completing the infusion of the rHuPH20 of HYQVIA, start the variable rate program of the infusion pump to initiate the infusion of the full therapeutic dose of Immune Globulin Infusion 10% (Human) of HYQVIA. At the end of infusion, flush the infusion tubing up to the needle with normal saline or Dextrose 5% in water (D5W), if required.
9. Remove subcutaneous needle(s) from the infusion site(s):
After the infusion is complete, remove the needle set and cover with a protective dressing. Discard any partially used vial(s) and disposable supplies in accordance with local requirements.
10. Document the infusion:
Remove the peel-off label from each Immune Globulin Infusion 10% (Human) vial of HYQVIA used and affix to the patient’s treatment record or infusion log. In addition, record the time, date, dose, infusion site location and any reactions after each infusion.
For self-administration, provide the patient with instructions and training for infusion in the home or other appropriate setting.
-
3 DOSAGE FORMS AND STRENGTHS
HYQVIA is a dual vial unit consisting of one vial of a liquid solution containing Immune Globulin Infusion 10% (Human) and one vial of a liquid solution containing 160 U/mL rHuPH20.
HYQVIA is available in the following strengths:
Table 5: HYQVIA strengths Immune Globulin Infusion 10% (Human) rHuPH20 Volume Grams Protein Volume Units 25 mL 2.5 1.25 mL 200 50 mL 5.0 2.5 mL 400 100 mL 10.0 5.0 mL 800 200 mL 20.0 10.0 mL 1600 300 mL 30.0 15.0 mL 2400 -
4 CONTRAINDICATIONS
HYQVIA is contraindicated in:
- Patients who have had a history of anaphylactic or severe systemic reactions to the administration of IgG.
- IgA deficient patients with antibodies to IgA and a history of hypersensitivity.
- Patients with known systemic hypersensitivity to hyaluronidase including rHuPH20 of HYQVIA.
- Patients with known systemic hypersensitivity to human albumin (in the hyaluronidase solution)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Severe hypersensitivity reactions may occur, even in patients who have tolerated previous treatment with IgG. In case of hypersensitivity, discontinue the HYQVIA infusion immediately and institute appropriate treatment.
Immune Globulin Infusion 10% (Human) of HYQVIA contains trace amount of IgA (average concentration of 37µg/mL). Patients with antibodies to IgA potentially are at greater risk of developing potentially severe hypersensitivity and anaphylactic reactions.5.2 Thrombosis
Thrombosis has been reported to occur following treatment with immune globulin products, including HYQVIA. Risk factors may include advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors.
Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, such as those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. For patients at risk of thrombosis, administer HYQVIA at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity [see Boxed Warning, Dosage and Administration (2), Patient Counseling Information (17)].
5.3 Immunogenicity of rHuPH20
Eighteen percent (15 of 83) of subjects receiving HYQVIA in PID clinical studies developed non-neutralizing antibodies to the rHuPH20 component. The potential exists for such antibodies to cross-react with endogenous Hyaluronidase (PH20), which is known to be expressed in the adult male testes, epididymis, and sperm. It is unknown whether these antibodies can interfere with fertilization in humans. The clinical significance of these antibodies is not known.
Clinical studies in CIDP based on 3,188 rHuPH20 infusions in 132 patients revealed one incidence of anti-rHuPH20 neutralizing antibody positivity. Elevation in neutralizing antibody titers was transient and occurred at a single measurement over a 3-year follow-up period. No efficacy or safety issues were identified with the occurrence of neutralizing antibody positivity.
5.4 Aseptic Meningitis Syndrome (AMS)
AMS has been reported to occur with IGIV or IGSC, including HYQVIA. AMS may occur more frequently in female patients. Discontinuation of IgG treatment has resulted in remission of AMS within several days without sequelae. The syndrome usually begins within several hours to two days following immune globulin treatment.
AMS is characterized by the following signs and symptoms: severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea, and vomiting [see Patient Counseling Information (17)]. Cerebrospinal fluid (CSF) studies frequently reveal pleocytosis up to several thousand cells per mm3, predominantly from the granulocytic series, and elevated protein levels up to several hundred mg/dL, but negative culture results. Conduct a thorough neurological examination on patients exhibiting such symptoms and signs, including CSF studies, to rule out other causes of meningitis.
5.5 Hemolysis
IgG products, including HYQVIA, contain blood group antibodies which may act as hemolysins and induce in vivo coating of red blood cells (RBC) with IgG. These antibodies may cause a positive direct antiglobulin reaction and hemolysis.6 Acute intravascular hemolysis has been reported following intravenously administered IgG, including Immune Globulin Infusion 10% (Human) administered intravenously, and delayed hemolytic anemia can develop due to enhanced RBC sequestration [see Adverse Reactions (6)].
Monitor patients for clinical signs and symptoms of hemolysis. If signs and/or symptoms of hemolysis are present after HYQVIA infusion, perform appropriate confirmatory laboratory testing [see Patient Counseling Information (17)].
5.6 Renal Dysfunction/Failure
Acute renal dysfunction/failure, acute tubular necrosis, proximal tubular nephropathy, osmotic nephrosis, and death may occur upon use of IgG products administered intravenously, especially those containing sucrose4. HYQVIA does not contain sucrose. Acute renal dysfunction/failure has been reported in association with Immune Globulin Infusion 10% (Human) administered intravenously. Ensure that patients are not volume depleted prior to the initiation of infusion of HYQVIA. In patients who are at risk of developing renal dysfunction because of pre-existing renal insufficiency or predisposition to acute renal failure (such as diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs), monitor renal function and consider lower, more frequent dosing.
Periodic monitoring of renal function and urine output is particularly important in patients judged to be at increased risk for developing acute renal failure. Assess renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of HYQVIA and again at appropriate intervals thereafter. If renal function deteriorates, consider discontinuation of HYQVIA.
5.7 Spread of Localized Infection
Infusion into or around an infected area can spread a localized infection. Do not infuse HYQVIA into these areas due to potential risk of spreading a localized infection.
5.8 Transfusion-Related Acute Lung Injury (TRALI)
Non-cardiogenic pulmonary edema (TRALI) may occur with intravenously administered IgG and has been reported to occur with Immune Globulin Infusion 10% (Human) administered intravenously. TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Symptoms typically occur within 1 to 6 hours after treatment.
Monitor patients for pulmonary adverse reactions [see Patient Counseling Information (17)]. If TRALI is suspected, conduct an evaluation, including appropriate tests for the presence of anti-neutrophil and anti-HLA antibodies in both the product and patient serum. TRALI may be managed using oxygen therapy with adequate ventilatory support.
5.9 Transmissible Infectious Agents
Because Immune Globulin Infusion 10% (Human) of HYQVIA is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant CJD (vCJD) agent, and theoretically, the classic Creutzfeldt-Jakob disease agent. This also applies to unknown or emerging viruses and other pathogens. No cases of transmission of viral diseases or vCJD have been associated with HYQVIA.
Report all infections thought to be possibly transmitted by HYQVIA to Takeda Pharmaceuticals U.S.A, Inc. at 1-877-TAKEDA-7 (1-877-825-3327) (In the U.S.).
5.10 Interference with Laboratory Tests
- After infusion of IgG, the transitory rise of the various passively transferred antibodies in the patient's blood may yield false positive serological testing results, with the potential for misleading interpretation. Passive transmission of antibodies to erythrocyte antigens (e.g., A, B, and D) may cause a positive direct or indirect antiglobulin (Coombs') test.
- Infusions of immune globulin products can lead to false positive readings in assays that depend on detection of ß-D-glucans for diagnosis of fungal infections; this may persist during the weeks following infusion of the product.
-
6 ADVERSE REACTIONS
Primary Immunodeficiency (PI)
The most common adverse reactions observed in >5% of clinical study subjects receiving HYQVIA for PI were local reactions, headache, antibody formation against Recombinant Human Hyaluronidase (rHuPH20), fatigue, nausea, pyrexia, and vomiting.
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
The most common adverse reactions observed in >5% of study subjects in clinical studies (Study 1, Study 2) of HYQVIA for CIDP were local reactions, headache, pyrexia, nausea, fatigue, erythema, pruritus, increased lipase, abdominal pain, back pain, and pain in extremity.
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in clinical studies of a product cannot be directly compared to rates in the clinical studies of another product and may not reflect the rates observed in clinical practice.
Primary Immunodeficiency (PI)
Immune Globulin Infusion 10% (Human) administered intravenously: Prior to initiation of treatment with HYQVIA, 87 patients received 365 infusions of Immune Globulin Infusion 10% (Human) encompassing 22.2 patient-years. Among the 87 patients treated, 56 (64.4%) experienced 1 or more adverse reactions. Among the 365 intravenous infusions, 158 adverse reactions occurred for a rate per infusion of 0.43.
A total of 1,359 infusions of HYQVIA were administered during the trial; 230 of these infusions occurred during the ramp-up period and the other 1,129 occurred during the observation period. During the observation period, 81 patients received 1,129 infusions of HYQVIA; of those, 67 (82.7%) experienced one or more adverse reactions. Among the 1,129 HYQVIA infusions, 456 adverse reactions occurred for a rate per infusion of 0.40. Seven of these adverse reactions were severe, defined as marked impairment of function, can lead to temporary inability to resume normal life pattern, requires prolonged intervention and/or results in sequelae.
Adverse reactions occurring in greater than 5% of subjects associated with infusions of HYQVIA vs. Immune Globulin Infusion 10% (Human) given intravenously are shown in Table 6. The majority of these adverse reactions was mild to moderate in severity and did not necessitate discontinuing the infusions. Mild is defined as transient discomfort that resolves spontaneously or with minimal intervention; moderate is defined as limited impairment of function that resolves spontaneously or with minimal intervention with no sequelae. No serious adverse reactions occurred during the HYQVIA clinical trials.
Table 6: Adverse Reactions* in greater than 5% of Subjects Associated with Infusions of HYQVIA vs. Immune Globulin Infusion 10% (Human) (IGIV) Given Intravenously HYQVIA IGIV Given Intravenously Adverse Reactions† Number of Subjects (%)
N= 81Number of Adverse Reactions per Infusion (Rate‡)
N = 1129Number of Subjects (%)
N=87Number of Adverse Reactions per Infusion (Rate)
N = 365Local ARs 42 (51.9%) 234 (0.21) 4 (4.6%) 4 (0.01) Systemic ARs 55 (67.9%) 222 (0.20) 54 (62.1%) 154 (0.42) Headache 17 (21%) 40 (0.04) 22 (25.3%) 42 (0.12) Fatigue 9 (11.1%) 16 (0.01) 8 (9.2%) 10 (0.03) Nausea 6 (7.4%) 12 (0.01) 10 (11.5%) 10 (0.03) Pyrexia 6 (7.4%) 11 (0.01) 6 (6.9%) 7 (0.02) Vomiting 6 (7.4%) 11 (0.01) 5 (5.7%) 7 (0.02) Six subjects, 2 children and 4 adults, withdrew from the trial during the efficacy treatment period with HYQVIA due to mild to moderate adverse reactions. One child withdrew due to local pain and one due to fever, vomiting, and headaches. Of the four adults, two withdrew due to local pain and swelling, one had moderate swelling that transiently extended from the abdominal infusion site to the genitalia, and one had back injury.
Antibodies binding to rHuPH20: A total of 15 out of 83 subjects in the clinical trial who were treated with HYQVIA developed an antibody capable of binding to rHuPH20. These antibodies were not capable of neutralizing rHuPH20.
In the clinical trial, no temporal association between adverse reactions and the presence of antibodies capable of binding to the Recombinant Human Hyaluronidase of HYQVIA could be demonstrated. There was no increase in the incidence or severity of adverse reactions in subjects who developed antibodies to rHuPH20 of HYQVIA. In all subjects, antibody titers decreased despite continued treatment.
The effect of exposure to antibodies capable of binding to rHuPH20 of HYQVIA for periods longer than this clinical trial has not been evaluated.
The local adverse reactions are listed by frequency in Table 7. Mild swelling around the infusion site was present in most infusions due to the large volumes infused, but in general was not considered to be an adverse reaction unless it caused discomfort. Among the 234 local adverse reactions, three were severe (infusion site pain, infusion site swelling and infusion site edema that extended from the abdominal infusion site to the genitalia); all were transient and resolved without sequelae. More than 98% of local reactions were either mild (70.5%) or moderate (28.2%) in severity.
Table 7: Most Frequent Local Adverse Reactions Reported in greater than 1% of Infusion During Treatment With HYQVIA Infusion Site Reaction Number and Rate of Reactions per Infusion (N=1129) Rate per infusion = total number of events divided by total number of infusions. Discomfort/pain 122 (0.11) Erythema 32 (0.03) Swelling/Edema 35 (0.03) Pruritus 22 (0.02) During the combined efficacy and extension trials encompassing more than 3 years, the local adverse reaction rate was 2.6 per patient-year. During the first 12-month period (months 1- 12), the rate was 3.68 local adverse reactions per patient-year. During the subsequent 12-month period (months 13-24), the rate declined to 2.12 local adverse reactions per-patient year. Finally, during the third 12-month period (months 25-36), the rate further declined to 0.37 local adverse reactions per patient-year.
Sixty-six of the 68 subjects who completed the efficacy clinical trial enrolled in a prospective, open-label, multicenter extension trial to assess the long-term safety and tolerability of HYQVIA. Sixty-three of 66 subjects enrolled received HYQVIA and 3 received IGIV. Of the 63 subjects who received HYQVIA, 48 completed the extension trial. The cumulative exposure of HYQVIA across the two trials was 188 subject-years and 2,959 infusions, and a maximum exposure of 188 weeks or up to approximately 3.5 years. There were no clinically observable changes in the skin or subcutaneous tissue in either the efficacy or extension clinical trials.
Pediatric population in PI:
HYQVIA was evaluated in a pivotal efficacy and safety study in 44 pediatric subjects (aged 2 to 16 years of age). Results from the interim data analysis, where all subjects completed 12 months of participation (one year of observation period) in the study, indicated similar safety profiles to adults [see Clinical Pharmacology (12.3), and Clinical Studies (14.1)].
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
The safety of HYQVIA in adult subjects with CIDP was evaluated in 100 unique subjects in a randomized, placebo-controlled study (Study 1) and a single-arm, open-label, extension study (Study 2). A total of 3188 infusions of HYQVIA were administered during these 2 studies [see Clinical Studies (14.2)].
Development of antibodies to rHuPH20: A total of 15 subjects (15%) who were treated with HYQVIA in Studies 1 and 2 developed high-binding anti-rHuPH20 antibody titers (≥1:160). A total of 7 subjects (11.3%) who were treated with HYQVIA in Study 1 and a total of 14 subjects (17.7%) in Study 2 developed high-binding anti-rHuPH20 antibodies (≥1:160). Of the 7 subjects in Study 1, 6 continued to have high-binding antibodies in Study 2, and 1 subject did not enter Study 2. Eight of the 14 subjects developed high-binding antibodies during Study 2. The occurrence of high-binding antibodies was not associated with an increased rate of TEAEs. One subject had a transient positivity of neutralizing antibodies which were not associated with any TEAE consistent with decreased rHuPH20 effect (in Study 2). The binding antibody positivity rate was comparable to the rates reported in other development programs.
Study 1
A total of 62 subjects received HYQVIA and a total of 598 infusions of HYQVIA were administered. Adverse reactions (ARs) occurring in >5% of subjects with CIDP treated with HYQVIA in Study 1 are shown in Table 8.
Table 8: Adverse Reactions* in >5% of Subjects with CIDP Treated with HYQVIA (Study 1: All Safety Subjects) ARs† HYQVIA,
Number of
Subjects (%) N = 62HYQVIA
Number of ARs
per Infusion (Rate‡) N = 598Placebo
Number of Subjects (%)
N=70Placebo Number of ARs
per Infusion (Rate‡)
N=644AR=adverse reactions • Local ARs 17 (27.4) 103 (0.17) 5 (7.1) 13 (0.02) • Systemic ARs 18 (29.0) 44 (0.07) 7 (10.0) 14 (0.02) - Nausea 7 (11.3) 8 (0.01) 0 0 - Fatigue 5 (8.1) 7 (0.01) 1 (1.4) 1 (<0.01) - Pruritus 5 (8.1) 14 (0.02) 1 (1.4) 1 (<0.01) - Headache 4 (6.5) 9 (0.02) 5 (7.1) 12 (0.02) - Pyrexia 4 (6.5) 6 (0.01) 0 0 Blood pressure elevation was reported in 4 subjects (6.5%) treated with HYQVIA in this study, including 2 subjects with a history of hypertension on antihypertensive medications. Three of the 4 subjects had events which were causally related and/or temporally associated (occurring within 72 hours).
A total of 4 subjects (3.0%) experienced adverse events (AEs) that led to discontinuation from the study; Three (3) subjects in the HYQVIA group and 1 in the placebo group. The AEs leading to discontinuation in the 3 HYQVIA subjects included: a cerebrovascular accident in 1 subject (who had coexisting cardiovascular risk factors), infusion site edema and infusion site pain in another subject, and nausea and chills in the third subject.
The most frequent local ARs in Study 1 are listed by frequency in Table 9. All HYQVIA local reactions (100%) were either mild (88.41%) or moderate (11.59%) in severity.
Table 9: Most Frequent Local Adverse Reactions Reported in >1% of Infusion with CIDP Treated with HYQVIA (Study 1: All Safety Subjects) Infusion Site Reaction HYQVIA
Number of ARs per Infusion (%) N = 598Placebo
Number of ARs per Infusion (%)
N = 644AR=adverse reaction Discomfort/pain 39 (6.5%) 12 (1.9%) Swelling/edema 38 (6.4%) 6 (0.9%) Erythema 36 (6.0%) 0 Pruritus 15 (2.5%) 0 Study 2
At the time of the interim analysis, 79 subjects received HYQVIA and a total of 2,590 infusions of HYQVIA were administered. Adverse reactions occurring in >5% of subjects with CIDP treated with HYQVIA in Study 2 are shown in Table 10.
Table 10: Adverse Reactions* in >5% of Subjects with CIDP Treated with HYQVIA (Study 2: All Safety Subjects) ARs† Number of Subjects (%) N = 79 Number of ARs per Infusion (Rate‡) N = 2,590 AR=adverse reactions • Local ARs 19 (24.1) 251 (0.10) • Systemic ARs 32 (40.5) 249 (0.10) - Headache 16 (20.3) 71 (0.03) - Pyrexia 12 (15.2) 58 (0.02) - Nausea 7 (8.9) 9 (<0.01) - Erythema 6 (7.6) 75 (0.03) - Fatigue 6 (7.6) 9 (<0.01) - Abdominal pain 5 (6.3) 8 (<0.01) - Back pain 4 (5.1) 4 (<0.01) - Lipase increased 4 (5.1) 4 (<0.01) - Pain in extremity 4 (5.1) 11 (<0.01) Blood pressure elevation was reported in 5 subjects (6.3%) in this study, including 1 subject with a history of hypertension on antihypertensive medications. One (1) of the 5 subjects had an event which was causally related and/or temporally associated (occurring within 72 hours).
A total of 3 subjects (3.8%) experienced AEs that led to discontinuation from the study and 1 subject (1.3%) died prior to the time of the interim analysis. The AEs leading to discontinuation in the 3 subjects included mantle cell lymphoma in 1 subject, muscular weakness and worsening of CIDP in another subject, and abdominal pain in the third subject. The cause of death in the 1 subject was cholangiocarcinoma.
The most frequent local ARs in Study 161505 are listed by frequency in Table 11. Most of the local reactions (95%) were either mild (85.44%) or moderate (9.34%) in severity.
Table 11: Most Frequent Local Adverse Reactions Reported in >1% of Infusion with CIDP Treated with HYQVIA (Study 2: All Safety Subjects) Infusion Site Reaction Number of ARs per Infusion (%) N = 2,590 AR=adverse reaction Erythema 225 (8.7%) Swelling/edema 58 (2.2%) 6.2 Postmarketing Experience
Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
In addition to the adverse reactions listed above in clinical trials, the following adverse reactions have been reported in the postmarketing experience:
Immune System Disorder: Hypersensitivity
General Disorder and Administration Site Conditions: Influenza-like illness, Infusion site leaking
Postmarketing Experience of Immune Globulin Products
The following adverse reactions have been identified and reported during the postmarketing use of Immune Globulin products administered subcutaneously:
Anaphylactic reaction, Tremor, Tachycardia, Hypotension, Infusion related reaction, Dyspnea, Paresthesia oral, Dermatitis allergic, Injection site rash, and Alanine aminotransferase increased.
-
7 DRUG INTERACTIONS
Passive transfer of antibodies can transiently impair the immune responses to live attenuated virus vaccines, such as mumps, rubella, and varicella for up to 6 months and for a year or more to measles [see Patient Counseling Information (17)].
Admixtures of HYQVIA with other drugs solutions have not been evaluated. Do not mix or administer components of HYQVIA with other products.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited human data are available to assess the presence or absence of drug-associated risk in pregnancy. In a postmarketing pregnancy study, two out of 5 infants born to mothers taking HYQVIA during pregnancy had congenital abnormalities (1 cleft lip and 1 talipes calcaneovalgus). Animal reproduction studies have not been conducted with the Immune Globulin Infusion 10% (Human) component of HYQVIA. Immune globulins increasingly cross the placenta from maternal circulation after 30 weeks of gestation. There was no evidence of teratogenicity in animal studies for rHuPH20, (a component of HYQVIA). The effects of antibodies to the rHuPH20 on the human embryo or fetal development are unknown. It is not known whether HYQVIA can cause fetal harm when administered to a pregnant woman or if it can affect reproductive capacity. HYQVIA should be given to a pregnant woman only if clearly needed.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human data:
Nine women treated with HYQVIA were enrolled in a prospective, uncontrolled, open-label, multicenter post-authorization Pregnancy Registry. Seven mothers continued HYQVIA, and two mothers were treated with immune globulin other than HYQVIA during the pregnancy. One mother discontinued from the registry before the expected delivery. Of the eight pregnancies with known outcomes, there were eight live births. There were no specified labor or delivery complications. Two out of 5 infants whose mothers took HYQVIA during pregnancy had congenital abnormalities (cleft lip without cleft palate and talipes calcaneovalgus). Data from the HYQVIA Pregnancy Registry are insufficient to establish causality. The interpretation of the registry findings is limited by the small sample size, by the potential that selection bias may have increased enrollment of mothers of infants with congenital abnormalities, the absence of fetal outcomes in some exposed maternal-fetal pairs, and incomplete data on other potential etiologies.
The following adverse events were identified in post-approval reports: spontaneous abortions and fetal deaths. The following congenital anomalies were identified in post-approval reports in infants whose mothers took HYQVIA during pregnancy: cleft palate, atrial septal defect, ventricular septal defect, cleft lip, hypoplastic left heart syndrome (aortic atresia, mitral valve atresia), endocardial fibroelastosis, and tricuspid valve incompetence. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Animal data:
Animal reproduction studies have not been conducted with Immune Globulin Infusion 10% (Human) component of HYQVIA.
Development and reproductive toxicology studies have been conducted with rHuPH20 in mice and rabbits [see Nonclinical Toxicology (13.2)]. No adverse effects on pregnancy were associated with anti-rHuPH20 antibodies at a dose of 3 mg/kg rHuPH20 in mice, which is 4,800 times higher than the typical monthly human dose. No teratogenicity or signs of maternal toxicity were observed at doses up to 18 mg/kg, which is 28,800 times higher than the typical monthly human dose. Doses of 9 and 18 mg/kg (14,400 and 28,800 times higher than the typical monthly human dose) in mice were associated with reduced fetal weight and an increased number of fetal resorptions. In these studies, maternal antibodies to rHuPH20 were transferred to offspring in utero. The effects of antibodies to the rHuPH20 component of HYQVIA on the human embryo or on human fetal development are unknown.
8.2 Lactation
Risk Summary
It is not known whether HYQVIA can cause harm to the breastfed infant when administered to a lactating woman. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for HYQVIA and any potential adverse effects on the breastfed infant from HYQVIA or from the underlying maternal condition. Data from the HYQVIA Pregnancy Registry are insufficient to predict effects on the breastfed child from exposure to HYQVIA through human milk.
Data
Animal data:
In animal studies, maternal antibodies binding to rHuPH20 were transferred to offspring during lactation. No adverse effects on pregnancy or offspring development were associated with anti-rHuPH20 antibodies. The effects of antibodies that bind to rHuPH20 of HYQVIA transferred during human lactation are unknown.
8.3 Females and Males of Reproductive Potential
Risk Summary
Animal studies do not indicate direct or indirect harmful effects of (rHuPH20) with respect to reproductive potential at the doses used for facilitating administration of IG 10% [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Primary Immunodeficiency (PI)
The safety and effectiveness of HYQVIA for the treatment of Primary Immunodeficiency have been established in pediatric patients 2 years and older.
Use of HYQVIA for this indication is supported by evidence from the pivotal efficacy and safety study in 44 pediatric subjects (aged 2 to 16 years of age). Results from pre-specified interim data analysis, where all subjects completed 12 months of participation (one year of observation period) in the study, indicated similar safety profiles to adults. No pediatric-specific dose requirements were necessary to achieve the desired serum IgG levels. [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1)].
Safety and effectiveness of HYQVIA has not been evaluated in patients <2 years of age.
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
The safety and effectiveness of HYQVIA for the treatment of CIDP have not been established in pediatric patients under the age of 18 years.
8.5 Geriatric Use
Primary Immunodeficiency (PI)
HYQVIA was evaluated in 7 subjects over age 65 in the PI clinical trial. The available data are too limited to draw safety conclusions.
Chronic Inflammatory Demyelinating Polyneuropathy
HyQvia was evaluated in 13 subjects over age 65 in the pivotal clinical trial. No clinically significant differences in safety were observed between those 13 elderly subjects and the subjects 18 to 65 years of age.
Use caution when administering HYQVIA to patients age 65 and over who are judged to be at increased risk of developing thrombosis and acute renal insufficiency [see Boxed Warning, Warnings and Precautions (5.2, 5.6)]. Do not exceed recommended doses and administer HYQVIA at the minimum dose and infusion rate practicable.
-
11 DESCRIPTION
HYQVIA is a dual vial unit with one vial of Immune Globulin Infusion 10% (Human) and one vial of rHuPH20.
The Immune Globulin Infusion 10% (Human) of HYQVIA is a ready-for-use, sterile, liquid preparation of highly purified and concentrated IgG antibodies. The distribution of the IgG subclasses is similar to that of normal plasma. The Fc and Fab functions are maintained in the primary component. Pre-kallikrein activator activity is not detectable. The Immune Globulin Infusion 10% (Human) of HYQVIA contains 100 mg/mL protein. At least 98% of the protein is IgG, average immunoglobulin A (IgA) concentration is 37µg/mL, and immunoglobulin M (IgM) is present in trace amounts. The Immune Globulin Infusion 10% (Human) of HYQVIA contains a broad spectrum of IgG antibodies against bacterial and viral agents. Glycine (0.25M) serves as a stabilizing and buffering agent. There is no added sugar, sodium, or preservatives. The pH is 4.6 to 5.1. The osmolality is 240 to 300 mOsmol/kg.
The Immune Globulin Infusion 10% (Human) of HYQVIA is manufactured from large pools of human plasma. IgG preparations are purified from plasma pools using a modified Cohn Oncley cold ethanol fractionation process, as well as cation and anion exchange chromatography.
Screening against potentially infectious agents begins with the donor selection process and continues throughout plasma collection and plasma preparation. Each individual plasma donation used in the manufacture of the Immune Globulin Infusion 10% (Human) of HYQVIA is collected only at FDA approved blood establishments and is tested by FDA licensed serological tests for Hepatitis B Surface Antigen (HBsAg), and for antibodies to Human Immunodeficiency Virus (HIV-1/HIV-2) and Hepatitis C Virus (HCV) in accordance with U.S. regulatory requirements. As an additional safety measure, mini-pools of the plasma are tested for the presence of HIV-1 and HCV by FDA licensed Nucleic Acid Testing (NAT).
To further improve the margin of safety, three dedicated, independent and effective virus inactivation/removal steps have been integrated into the manufacturing and formulation processes, namely solvent/detergent (S/D) treatment, 35 nm nanofiltration, and a low pH incubation at elevated temperature. The S/D process includes treatment with an organic mixture of tri-n-butyl phosphate, octoxynol 9 and polysorbate 80 at 18°C to 25°C for a minimum of 60 minutes.7
In vitro virus spiking studies have been used to validate the capability of the manufacturing process to inactivate and remove viruses. To establish the minimum applicable virus clearance capacity of the manufacturing process, these virus clearance studies were performed under extreme conditions (e.g., at minimum S/D concentrations, incubation time and temperature for the S/D treatment). Virus clearance studies for the Immune Globulin Infusion 10% (Human) of HYQVIA performed in accordance with good laboratory practices (Table 12) have demonstrated that:
- S/D treatment inactivates the lipid-enveloped viruses investigated to below detection limits within minutes.
- 35 nm nanofiltration removes lipid-enveloped viruses to below detection limits and reduces the nonlipid enveloped viruses HAV and B19V. As determined by a polymerase chain reaction assay, nanofiltration reduced B19V by a mean log10 reduction factor of 4.8 genome equivalents.
- Treatment with low pH at elevated temperature of 30°C to 32°C inactivates lipid-enveloped viruses and encephalomyocarditis virus (EMCV, model for HAV) to below detection limits, and reduces mice minute virus (MMV, model for B19V).
Table 12: Three Dedicated Independent Virus Inactivation/Removal Steps Mean Log10 Reduction Factors* (RFs) For Each Virus and Manufacturing Step Virus type Enveloped
RNAEnveloped
DNANon-enveloped
RNANon-enveloped
DNAFamily Retroviridae Flaviviridae Herpesviridae Picornaviridae Parvoviridae Virus HIV-1 BVDV WNV PRV HAV EMCV MMV Abbreviations: HIV-1, Human Immunodeficiency Virus Type 1; BVDV, Bovine Viral Diarrhea Virus (model for Hepatitis C Virus and other lipid enveloped RNA viruses); WNV, West Nile Virus; PRV, Pseudorabies Virus (model for lipid enveloped DNA viruses, including Hepatitis B Virus); EMCV, Encephalomyocarditis Virus (model for non-lipid enveloped RNA viruses, including Hepatitis A virus [HAV]); MMV, Mice Minute Virus (model for non-lipid enveloped DNA viruses, including B19 virus [B19V]); n.d. (not done), n.a. (not applicable). - *
- For the calculation of these RF data from virus clearance study reports, applicable manufacturing conditions were used. Log10 RFs on the order of 4 or more are considered effective for virus clearance in accordance with the Committee for Medicinal Products for Human Use (CHMP, formerly CPMP) guidelines.
- †
- No RF obtained due to immediate neutralization of HAV by the anti-HAV antibodies present in the product.
SD treatment >4.5 >6.2 n.a. >4.8 n.d. n.d. n.d. 35 nm nanofiltration >4.5 >5.1 >6.2 >5.6 5.7 1.4 2.0 Low pH treatment >5.8 >5.5 >6.0 >6.5 n.d.† >6.3 3.1 Overall log reduction factor (ORF) >14.8 >16.8 >12.2 >16.9 5.7† >7.7 5.1 The rHuPH20 of HYQVIA is produced from genetically engineered Chinese Hamster Ovary (CHO) cells containing a DNA plasmid encoding for a soluble fragment of human hyaluronidase PH20. The purified hyaluronidase glycoprotein contains 447 amino acids with an approximate molecular weight of 61,000 Daltons [see Clinical Pharmacology (12.1)]. This component is supplied as a sterile, clear, colorless, ready-for-use solution and has an approximate pH of 7.4 and an osmolality of 290 to 350 mOsm. Each vial contains 160 U/mL of rHuPH20 with 8.5 mg/mL sodium chloride, 1.78 mg/mL sodium phosphate dibasic dihydrate, 1.0 mg/mL human albumin, 1.0 mg/mL edentate disodium dihydrate, 0.40 mg/mL calcium chloride dihydrate, and 0.17 mg/mL sodium hydroxide added for pH adjustment. It does not contain preservatives.
Due to comprehensive virus testing at the Master Cell Bank, Working Cell Bank and bulk harvest stage, effective virus reduction during the purification process and use of pharmaceutical grade human albumin as an excipient with no other materials of human or animal origin involved in the manufacturing process, rHuPH20 provides for high margins of safety with respect to viruses.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The Immune Globulin Infusion 10% (Human) provides the therapeutic effect of HYQVIA. The Recombinant Human Hyaluronidase of HYQVIA increases dispersion and absorption of the Immune Globulin Infusion 10% (Human). The Immune Globulin Infusion 10% (Human) of HYQVIA supplies a broad spectrum of opsonizing and neutralizing IgG antibodies against a wide variety of bacterial and viral agents. The Immune Globulin Infusion 10% (Human) also contains a spectrum of antibodies capable of interacting with and altering the activity of cells of the immune system as well as antibodies capable of reacting with cells such as erythrocytes. The role of these antibodies and the mechanisms of action of IgG in the Immune Globulin Infusion 10% (Human) of HYQVIA have not been fully elucidated.
The mechanism of action of immunoglobulins in the treatment of CIDP in adults has not been fully elucidated but may include immunomodulatory effects.
Hyaluronan is a polysaccharide found in the extracellular matrix of connective tissue8. It is depolymerized by the naturally occurring enzyme hyaluronidase. Unlike the stable structural components of the interstitial matrix, hyaluronan has a very fast turnover with a half-life of approximately 0.5 days. The rHuPH20 of HYQVIA increases permeability of the subcutaneous tissue by temporarily depolymerizing hyaluronan. In the doses administered, rHuPH20 of HYQVIA acts locally. The effects of the hyaluronidase are reversible, and permeability of the subcutaneous tissue is restored within 24 to 48 hours.
12.3 Pharmacokinetics
Primary Immunodeficiency (PI)
The pharmacokinetics (PK) of HYQVIA was evaluated during a clinical trial of adults with PI after they achieved steady state at their 3- or 4-week dosing interval and underwent individual dose adjustment [see Clinical Studies (14)]. For adults, dose adjustment was based on a comparison of the ratios of the area under the IgG concentration versus time curve (AUC) during intravenous treatment versus during HYQVIA treatment.
The AUC of HYQVIA compared to conventional IGSC administration was 20% higher. The bioavailability of HYQVIA based on weekly AUC was 93.3% relative to IGIV.
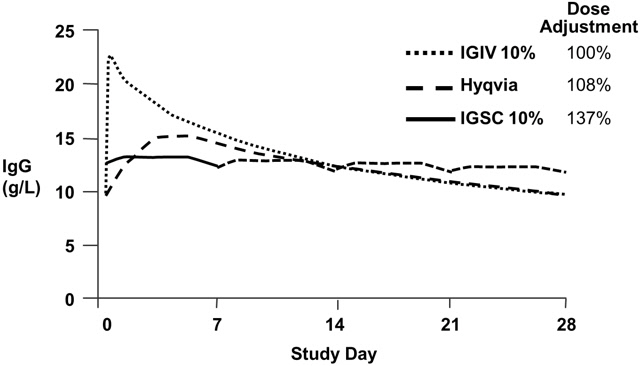
The pharmacokinetic parameters of HYQVIA compared with intravenously administered Immune Globulin Infusion 10% (Human) are shown in Table 13. The mean IgG dose in weekly equivalents was 147 mg/kg ± 50 (range 134 to 160 mg/kg). The serum IgG trough levels are comparable: mean serum IgG trough with HYQVIA was 1077 mg/dL compared with 1095 mg/dL with intravenously administered Immune Globulin Infusion 10% (Human). Cmax was lower with HYQVIA (1607 mg/dL) than with intravenously administered Immune Globulin Infusion 10% (Human) (2248 mg/dL). Time to reach maximum concentration of IgG following HYQVIA administration was 5 (3.3-5.1) days.
In the extension trial, reducing the dosing interval from 4 to 2 weeks resulted in a mean increase of 13% in serum IG trough levels.
Table 13: Pharmacokinetic Parameters of HYQVIA Compared to Intravenously Administered Immune Globulin Infusion 10% (Human) (IGIV) HYQVIA IGIV Number of Subjects 60 68 IgG Weekly Dose [mg/kg/week] Mean (SD) 147 (50) 139 (55) 95% CI 134 to 160 126 to 153 Cmax [mg/dL] Mean (SD) 1607 (382) 2248 (547) 95% CI 1508 to 1706 2116 to 2380 IgG Trough Levels [mg/dL]* Mean (SD) 1077 (275) 1095 (321) 95% CI 1004 to 1149 1017 to 1174 AUC/week [g*days/L]† Mean (SD) 91.4 (21) 98.7 (24.3) 95% CI 85.9 to 96.8 92.8 to 104.5 Bioavailability‡ Point estimate 93.3 100% (defined) 90% CI 91.4 to 95.2 N/A Clearance [mL/kg/day] Mean (SD) 1.6 (0.5)§ 1.4 (0.4) 95% CI 1.5 to 1.8 1.3 to 1.5 Terminal Half-life [days] Mean (SD) 59.3 (36.1) 41.6 (26.9) 95% CI 50 to 68.6 35.1 to 48.1 Tmax [days] Median 5.0 0.1 95% CI 3.3 to 5.1 0.1 to 0.1 Figure 1* shows a mean concentration-time plot of IgG in subjects 12 years and older. The concentration-time profile of HYQVIA is similar to that of intravenous administration but without the high peak. The peak to trough variation is more similar to subcutaneous administration.
Figure 1: Pharmacokinetic Comparison of Mean IgG Values for HYQVIA vs. Intravenously and Subcutaneously Administered Immune Globulin Infusion 10% (Human)* - *
- IGIV and HYQVIA data at 28-day dosing interval; IGSC data at 7-day dosing interval; IGSC dotted line shows weekly dose extrapolated over 21 additional days.
Pediatric Population
Results from the pivotal pediatric study suggest no clinically meaningful differences across age (2 to 16 years) groups with respect to total IgG PK (AUC/week and CL/F/BW) and serum trough levels.
Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
The pharmacokinetic profile of HYQVIA was not evaluated in clinical study 1 in patients with CIDP aged 18 years and older. Serum trough levels of total IgG were assessed throughout the study 1. Trough-level data at baseline, interim visit, and end of study were presented in Table 14.
Table 14: Summary of Serum Trough Total IgG Levels (mg/dL) in Epoch 1 of Study 1 Treatment Arm Baseline * Interim Visit † End of Epoch 1 Treatment / Early Termination SD = Standard deviation. N = Number of subjects who had a value at each time point. HYQVIA N 45 44 46 Mean (SD) 1465.3 (323.6) 1487.1 (348.0) 1569.5 (450.4) Min, Max 966, 2230 755, 2510 855, 2880 Placebo N 55 40 55 Mean (SD) 1477.6 (356.9) 1113.4 (275.9) 1240.8 (574.3) Min, Max 655, 2870 449, 1690 449, 4680 -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Immune Globulin Infusion 10% (Human)
Long-term animal studies have not been conducted to evaluate the carcinogenic potential of Immune Globulin Infusion 10% (Human) or its effect on fertility.
An in vitro mutagenicity test was performed for Immune Globulin Infusion 10% (Human). No evidence of mutagenicity was observed.
rHuPH20
Hyaluronidases are found in most tissues of the body. Long-term animal studies to evaluate the carcinogenic or mutagenic potential of rHuPH20 have not been conducted.
No adverse effects on fertility were observed in mice, rabbits and cynomolgus monkeys exposed to antibodies that bind to rHuPH20 and species-specific hyaluronidase. Reversible infertility has been observed in male and female guinea pigs immunized to produce antibodies to hyaluronidase. However, antibodies to hyaluronidase did not influence reproduction following immunization of mice, rabbits, sheep, or cynomolgus monkeys. The effects of antibodies that bind to rHuPH20 on human fertility are unknown.
13.2 Animal Toxicology and/or Pharmacology
Developmental studies in mice demonstrated that administration of rHuPH20 did not produce teratogenicity or signs of maternal toxicity at doses up to 18 mg/kg (2.2 × 106 U/kg), which is 28,800 times higher than the typical monthly human dose. Maternal doses of 9 and 18 mg/kg were associated with reduced fetal weight and an increased number of fetal resorptions. No adverse effects on fetal development were observed at a maternal dose of 3 mg/kg (360,000 U/kg) in mice and 0.76 mg/kg (91,000 U/kg) in rabbits, which is 4,800 and 1,200 times higher than the typical monthly human dose.
In a peri-and post-natal reproduction study, female mice were dosed daily with rHuPH20 from implantation through lactation and weaning. There were no adverse effects on gestation, parturition, lactation and maternal behavior or on the development of the male or female offspring of the treated female mice in terms of sexual maturation, learning and memory of offspring, or their ability to produce another generation of offspring at doses up to 9 mg/kg (1.1 ×106 Unit/kg), which is 14,400 times higher than the typical monthly human dose.
-
14 CLINICAL STUDIES
14.1 Primary Immunodeficiency (PI)
A prospective, open-label, non-controlled, multicenter trial was conducted in the US to determine the efficacy, tolerability, and pharmacokinetics (PK) of HYQVIA in subjects with PI. Two cohorts of subjects were enrolled. Thirty-one subjects had been treated intravenously for three months and then subcutaneously each week at 137% of the intravenous dose for approximately one year before transitioning to the HYQVIA trial. The remaining subjects also were treated intravenously for 3 months and then immediately began treatment with HYQVIA in the trial.
One week after the last intravenous or subcutaneous infusion, each subject began subcutaneous treatment with HYQVIA. After placing the subcutaneous needle set, the rHuPH20 of HYQVIA was infused through the needle set followed within 10 minutes by the immune globulin of HYQVIA at 108% of the intravenous dose. Dosing began with a 1-week equivalent dose. One week later, a 2-week dose was administered, followed 2 weeks later with a 3-week dose. For those subjects who were on a 4-week dose interval prior to entering the trial, 3 weeks later the 4-week dose was administered. This ramp-up period allowed subjects to become familiar with the large volumes required for a full 3- or 4-week treatment. Subsequently, subjects continued the 3- or 4-week dosing for the remainder of the trial. After 3 doses at the full volume, a serum IgG trough level was obtained for all subjects and used to individually adapt the subcutaneous dose of HYQVIA to compensate for individual variation from the mean value of 108% [see Clinical Pharmacology (12.3) and Administration (2.2)]. All subjects who completed the trial received a minimum of 12 infusions at this individually adapted dose. The period after the ramp-up was considered the efficacy period and used for safety and efficacy analyses.
Outcome measures included the rate of infections, adverse reactions, tolerability of the infusions of HYQVIA, number of infusion sites per month, and infusion rate. Eighty-nine subjects were enrolled, 87 treated intravenously and 83 treated with HYQVIA. The majority were Caucasian (79/87, 90.8%). Median age was 35.0 years (range 4 to 78 years). Forty-four of the subjects were naïve to subcutaneous treatment. Median serum IgG trough levels for the 6 months before enrollment were 1033.5 mg/dL (range: 405 to 3200 mg/dL) in subcutaneous-experienced subjects and 1000 mg/dL (range: 636 to 3200 mg/dL) in the subcutaneous-naïve subjects.
The 83 subjects received a total of 1,359 infusions of HYQVIA during the entire trial. Of these, 1,129 were administered after the ramp-up when the subjects were on a consistent interval of 3 or 4 weeks, which was predetermined to be the efficacy period for data analysis.
Median duration of treatment in the IGIV period was 91 days (range 84 to 122 days). Median duration of HYQVIA treatment during the dose ramp up period was 42 days (range 20 to 49), and during the efficacy period was 366 days (range 42 to 507 days). None of the subjects withdrew due to a severe or serious local or systemic adverse reaction [see Adverse Reactions (6.1)].
There were two acute serious bacterial infections (aSBI), both of which were episodes of pneumonia treated as outpatients with oral antibiotics during the 12-month efficacy period; an additional pneumonia requiring hospitalization occurred during the ramp-up. Based on this, the annualized rate of aSBI while treated with HYQVIA was 0.025, with an upper 99% confidence limit of 0.089, which is significantly less than (p<0.0001) the rate of one infection per year.10
The overall rates of infections throughout both the efficacy and extension trials are shown in Table 15. The secondary endpoints evaluated in the efficacy trial were the annual rate of all infections and other efficacy measures.
Table 15: Summary of Infections and Other Secondary Efficacy Endpoints Annual Rate Parameter Mean 95% CI Infections per patient per year (Efficacy Trial) 2.97 2.51 to 3.47 Infections per patient per year (Efficacy and Extension Trials) 2.99 2.60 to 3.42 Days off school/work 3.31 2.37 to 4.47 Days on antibiotics 20.26 15.45 to 25.98 Unscheduled physician visits for infections 4.78 3.83 to 5.88 Days in hospital due to infection 0.037 0.009 to 0.095 An objective of the trial was to achieve the same number or fewer infusions with HYQVIA per month as with intravenous administration and significantly fewer than with conventional subcutaneous administration. A summary of intravenous administration compared with HYQVIA administration is presented in Table 16.
Table 16: Summary of Infusions Parameter Intravenous HYQVIA Median monthly number of infusion sites 1.34
(1.2 to 1.7)1.09
(1.0 to 3.5)Mean volume per site (mL) 339
(75 to 800)292
(91 to 648)Dose per site (g) 33.9
(7.5 to 80.0)29.2
(9.1 to 64.8)Median duration of individual infusions (hr) 2.33
(0.92 to 6.33)2.08
(0.83 to 4.68)Monthly median infusion time (hr/month) 3.2 2.64 Median maximum infusion rate (mL/hr) 246
(60 to 668)300
(10 to 300)Percent (%) of infusion completed without change in rate, interruption, and discontinuation 95.9 97.7 Sixteen of 83 subjects (19.3%) were infused every 3 weeks and 67 (80.7%) were infused every 4 weeks. Seventy-eight of 83 (94%) subjects attained the same 3- or 4-week dosing as their previous IV treatment. One decreased from 4 to 3 weeks, one from 4 to 2 weeks and one from 3 to 2 weeks. The primary reason for decreasing the interval was discomfort due to swelling.
In a separate study evaluating subcutaneous treatment with Immune Globulin Infusion 10% (Human), a median of 21.43 sites were required each month with a median monthly infusion time of 5.35 hours.
Pediatric Study
A prospective, open-label, non-controlled, multicenter trial was conducted in the US to determine the efficacy, safety, immunogenicity and pharmacokinetics (PK) of HYQVIA in pediatric subjects with PI who had received IVIG or SCIG treatment before enrollment into the trial. A total of 44 subjects were enrolled, and the median age of pediatric subjects was 9.5 years (range: 3 to 15 years). Of the enrolled subjects, 26 subjects (59.1%) were male, 18 subjects (40.9%) were female, and 40 subjects (90.9%) were White. Most subjects were not Hispanic or Latino (39 subjects [88.6%]).
Pediatric subjects switched to HYQVIA SC immunoglobulin treatment schedule administered at doses (volumes and treatment intervals) typical for IVIG administration. Treatment intervals and doses gradually increased in a ramp-up phase to an interval of 3 or 4 weeks. Data were analyzed when all subjects completed 12 months of participation (one year of observation) in the trial. Overall, the median number of infusions per month was 1.1 (range: 1.0 to 1.5) and was comparable across the age groups. The median number of infusion sites per month was 2.2 (range: 1.1 to 2.9), with a similar median number for all age categories. There were no clinically meaningful differences in trough IgG levels across age groups.
The primary analysis for efficacy was based on the rate of aSBIs, defined as bacteremia/sepsis, bacterial meningitis, osteomyelitis/septic arthritis, bacterial pneumonia, and visceral abscess, per subject per year. Secondary analyses included the annual rate of other infections and health resource utilization outcome measures (e.g., days out of work/school/daycare).
During the 12-month trial period, the mean aSBI rate per year was 0.04 (with an upper 1-sided 99% confidence interval of 0.21, p<0.001), which met the predefined success rate of less than one aSBI per subject per year. One subject experienced two aSBIs of bacterial pneumonia, and there were no other episodes of serious bacterial infections reported in this trial. The mean rate of all infections per subject-year was 3.20, with an upper limit of the 95% CI of 4.05. The overall rate of infections per subject is consistent with results obtained in the pivotal clinical study. Pediatric subjects missed a mean (95% CI) of 5.0 (2.2 to 7.9) days of work/school/daycare.
14.2 Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
Safety and efficacy of HYQVIA in adult subjects with CIDP was evaluated in a randomized, placebo-controlled study (Study 1) and a single-arm, open-label, extension study (Study 2).
In a multicenter, randomized, placebo-controlled, phase 3 study, 132 adult subjects with CIDP underwent evaluation of safety and tolerability, 122 subjects underwent efficacy evaluation of HYQVIA as a maintenance therapy to prevent relapse that allowed self-infusion of a total therapeutic dose every 2 to 4 weeks. The study enrolled subjects ≥18 years of age (male or female) at the time of screening who had a documented diagnosis of definite or probable CIDP as per the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) 2010 criteria. All eligible subjects had responded to IgG treatment in the past (partial or complete resolution of neurological symptoms and deficits) and were on a stable dose of IGIV treatment within the dose range equivalent to a cumulative monthly dose of 0.4 to 2.4 g/kg body weight administered intravenously for at least 12 weeks before screening. The primary endpoint was the proportion of subjects who experienced a relapse, defined as an increase of ≥1 point relative to the pre-SC treatment baseline score in 2 consecutive adjusted inflammatory neuropathy cause and treatment (INCAT) disability scores obtained less than seven days apart.
Overall, a total of 184 subjects were screened, of which 46 (25%) were screen failures, 138 (75%) were randomized, and 132 (71.7%) received either HYQVIA (N=62) or placebo (N=70), 17 discontinued in Epoch 1, and 94 completed the study without developing a relapse in Epoch 1 and 21 rolled over to Epoch 2. All 132 treated subjects were analyzed for safety and 122 subjects (N= 57 for HYQVIA, N=65 for placebo) were analyzed for efficacy included in the modified intent-to-treat (mITT). The mean duration of exposure was 5.3 months in the HYQVIA group and 4.7 months in the placebo group. The mean monthly equivalent dose was 1.1g/kg. The average time to deliver the monthly HYQVIA dose was approximately 2 hrs. HYQVIA infusions were administered through 1 to 3 injection sites, and the majority of infusions (85.8%) were administered through 2 infusion sites using 12mm to 14mm needles.
The analysis of the primary endpoint demonstrated a statistically significant difference between the relapse rates in the HYQVIA group (N=57, 14.0%) compared to the placebo group (N=65, 32.3%) (p=0.0314). The treatment difference of –18.3% (two-sided 95% CI: -32.1%, -3.1%) indicated that HYQVIA demonstrated superiority over placebo in preventing relapse of CIDP.
Kaplan-Meier curves (Figure 2) demonstrated early separation between HYQVIA and placebo at approximately Week 4.
Figure 2: Kaplan-Meier Curves for Time to Relapse by Treatment Group 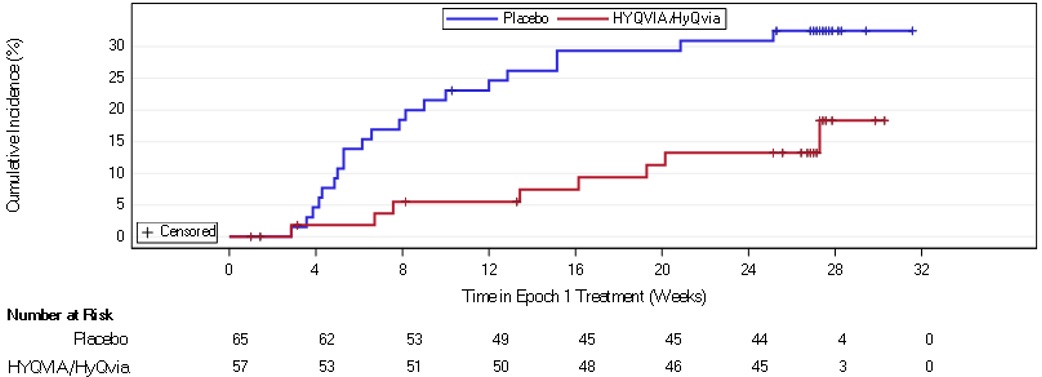
The effect of HYQVIA on activities of daily living was measured by the Rasch-built Overall Disability Scale (R ODS) centile score. The least squares (LS) mean changes were -1.2 in the HYQVIA group and -6.3 in the placebo group. The LS mean treatment difference between HYQVIA and placebo was 5.1.
A Phase 3b, single-arm, open-label, multicenter, extension study to assess the long-term safety, tolerability, and immunogenicity of HYQVIA for maintenance therapy for CIDP. The study was open to subjects who completed Study 1 without CIDP worsening or relapse. The dosing range was 0.4 g/kg to 2.4 g/kg, given in a frequency of 2-, 3-, or 4-week intervals. An interim analysis was performed, and the data included 79 subjects with a range of follow-ups from 0 to 5.1 years and a total follow-up of 169 patient-years. A total of 2,590 HYQVIA infusions were administered.
The proportion of subjects who developed CIDP relapse was 8.8% (5/57) during the interim study period. The 6-month relapse rate was 1.6%.
-
15 REFERENCES
- Orange JS, Hossny EM, Weiler CR, Ballow M, Berger M, Bonilla FA, Buckley R, Chinen J, El-Gamal Y, Mazer BD, Nelson Jr. RP, Patel DD, Secord E, Sorenson RU, Wasserman RL, Cunningham-Rundles C, Use of Intravenous Immunoglobulin in Human Disease: A Review of Evidence by Members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma, and Immunology. J Allergy Clin Immunol 2006; 117:S525-53.
- Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005; 94(suppl 1):S1-63.
- Eijkhout HW, Der Meer JW, Kallenberg CG, et al. The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. A randomized, double-blind, multicenter crossover trial. Ann Intern Med. 2001;135:165-174.
- Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Transfusion Med Rev. 2003;17:241-251.
- Katz U, Sheonfeld Y. Review: intravenous immunoglobulin therapy and thromboembolic complications. Lupus 2005;14:802-8
- Daw Z, Padmore R, Neurath D, Cober N, Tokessy M, Desjardins D, et al. Hemolytic transfusion reactions after administration of intravenous immune (gamma) globulin: a case series analysis. Transfusion 2008; 48:1598-601
- Kreil TR, Berting A, Kistner O, Kindermann J. West Nile virus and the safety of plasma derivatives: verification of high safety margins, and the validity of predictions based on model virus data. Transfusion 2003; 43:1023-1028.
- Bookbinder LH, Hofer A, Haller MF, Zepeda ML, Keller G-A, Lim JE, Edginton TS, Shepard HM, Patton JS, Frost GI. A recombinant human enzyme for enhanced interstitial transport of therapeutics. J of Controlled Release 2006; 114:230-241.
- Wasserman RL, Melamed I, Kobrynski L, Strausbaugh SD, Stein MR, Sharkhawy M, Engl W, Leibl H, Sobolevsky L, Gelmont D, Schiff RI, Grossman WJ. Efficacy, Safety, and Pharmacokinetics of a 10% Liquid Immune Globulin Preparation (GAMMAGARD LIQUID, 10%) Administered Subcutaneously in Subjects with Primary Immunodeficiency Disease. J Clin Immunol. 2011 Mar 22. [Epub ahead of print]
- Golding B. IGIV Clinical Endpoints. Presented at: Blood Products Advisory Committee, 65th Meeting. 17 March 2000. Silver Spring, MD.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
HYQVIA is supplied in a dual vial unit of two single dose vials containing the labeled amount of functionally active Immune Globulin Infusion 10% (Human) and rHuPH20. The packaging of this product is not made with natural rubber latex.
The following presentations of HYQVIA are available:
Immune Globulin Infusion 10% (Human) rHuPH20 NDC Number Volume Grams Protein Volume Units 0944-2510-02 25 mL 2.5 1.25 mL 200 0944-2511-02 50 mL 5.0 2.5 mL 400 0944-2512-02 100 mL 10.0 5.0 mL 800 0944-2513-02 200 mL 20.0 10.0 mL 1600 0944-2514-02 300 mL 30.0 15.0 mL 2400 Storage and Handling
- Do not freeze.
- Keep the vials in the carton in order to protect from light.
- Refrigeration: 2° to 8°C [36° to 46°F] for up to 36 months.
- Room Temperature: up to 25°C [77°F] for up to 3 months during the first 24 months from the date of manufacturing (Mfg date) printed on the carton.
- HYQVIA must be used within 3 months after removal to room temperature but within the expiration date on the carton and vial label.
- Do not return HYQVIA to the refrigerator after it has been stored at room temperature.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Inform patients to immediately report the following signs and symptoms to their healthcare professional:
- Acute respiratory distress, wheezing, swelling of the airway or severe hives or itching [see Warnings and Precautions (5.1)].
- Instruct patients to immediately report symptoms of thrombosis. These symptoms may include pain and/or swelling of an arm or leg with warmth over the affected area, discoloration of an arm or leg, unexplained shortness of breath, chest pain or discomfort that worsens on deep breathing, unexplained rapid pulse, numbness or weakness on one side of the body [see Warnings and Precautions (5.2)].
- Advise patients that antibodies against Recombinant Human Hyaluronidase (rHuPH20) can develop. The potential exists for such antibodies to cross-react with endogenous Hyaluronidase (PH20), which is known to be expressed in the adult male reproductive tract. The clinical significance of these antibodies is unknown [see Warnings and Precautions (5.3)].
- Severe headache, neck stiffness, drowsiness, fever, sensitivity to light, painful eye movements, nausea, and vomiting which suggest aseptic meningitis syndrome [see Warnings and Precautions (5.4)].
- Increased heart rate, fatigue, yellowing of the skin or eyes, and dark-colored urine may suggest hemolysis [see Warnings and Precautions (5.5)].
- Decreased urine output, sudden weight gain, fluid retention/edema, and/or shortness of breath may suggest kidney problems [see Warnings and Precautions (5.6)].
- Trouble breathing, chest pain, blue lips or extremities, or fever that can occur 1 to 6 hours after an infusion of HYQVIA, may suggest TRALI [see Warnings and Precautions (5.8)].
- Inform patients that HYQVIA is made from human plasma and may contain infectious agents that can cause disease (e.g., viruses and, theoretically, the vCJD agent). Patients should report any symptoms that concern them which might be caused by virus infections [see Warnings and Precautions (5.9)].
- Inform patients that HYQVIA can interfere with their immune response to live viral vaccines such as measles, mumps, rubella, and varicella, and instruct patients to notify their healthcare professional of this potential interaction when they are receiving vaccinations [see Drug Interactions (7)].
Self-administration – If self-administration is deemed appropriate by the physician, give clear instructions and training on how to administer HYQVIA. Document their ability to independently administer HYQVIA.
- Ensure the patient understands the importance of following regularly scheduled infusions to maintain appropriate steady IgG levels.
- Instruct the patient to keep a treatment infusion log. This infusion log should include information about each infusion such as, the lot number(s), infusion site location, the time, date, dose, and any reactions.
- Inform the patient that due to the volume that can be infused, swelling is common with HYQVIA. Mild to moderate local infusion-site reactions (e.g., swelling and redness) are common side effects of facilitated subcutaneous treatment with HYQVIA. Instruct the patient to contact their healthcare professional if a local reaction increases in severity or persists for more than a few days.
- Instruct the patient on the importance of following the directions for the pump for infusion of the Immune Globulin Infusion 10% (Human) of HYQVIA.
-
SPL UNCLASSIFIED SECTION
Takeda Pharmaceuticals U.S.A., Inc.
Cambridge, MA 02142U.S. License No. 1898
HYQVIA is a registered trademark of Baxalta Incorporated.
Takeda and
 are registered trademarks of Takeda Pharmaceutical Company Limited.
are registered trademarks of Takeda Pharmaceutical Company Limited.©2024 Takeda Pharmaceutical Company Limited. All rights reserved.
Patented: see www.takeda.com/en-us/patents
HYQ353
-
PATIENT PACKAGE INSERT
Patient Information
HYQVIA®
Immune Globulin Infusion 10% (Human)
With Recombinant Human Hyaluronidase (rHuPH20)
For Subcutaneous AdministrationThe following summarizes important information about HYQVIA (pronounced Hi-Q-via). Please read it carefully before using this medicine. This information does not take the place of talking with your healthcare professional, and it does not include all of the important information about HYQVIA. If you have any questions after reading this, ask your healthcare professional.
What is the most important information that I should know about HYQVIA?
- HYQVIA can cause blood clots.
- Call your healthcare professional if you have pain, swelling, warmth, redness, or a lump in your legs or arms, other than at the infusion site(s), unexplained shortness of breath, chest pain or discomfort that worsens on deep breathing, unexplained rapid pulse, numbness or weakness on one side of the body.
- Your healthcare professional may perform blood tests regularly to check your IgG level.
- Do not infuse HYQVIA into or around an infected or red swollen area because it can cause infection to spread.
What should I tell my healthcare professional before I start using HYQVIA?
Before starting HYQVIA, tell your healthcare professional if you:
- Have or had any kidney, liver, or heart problems or history of blood clots because HYQVIA can make these problems worse.
- Have IgA deficiency or a history of severe allergic reactions to IgG or other blood products.
- Are pregnant, trying to become pregnant or are breast feeding.
What is HYQVIA?
HYQVIA is a liquid medicine containing immune globulin and Recombinant Human Hyaluronidase. HYQVIA contains IgG antibodies, collected from human plasma donated by healthy people.
- The antibodies help your body to fight off bacterial and viral infections. People who have CIDP are believed to have an autoimmune disease in which the body's immune system targets the nerves, leading to muscle weakness and numbness, usually in the arms and legs. IgG is thought to reduce the damage to the nerve and assist in defending the nerve from harm.
- The hyaluronidase is found in your body naturally. It’s the first part of your two-part infusion. It temporarily opens the space under your skin (the subcutaneous space), allowing a larger amount of IgG to reach your subcutaneous tissue and be absorbed into your bloodstream.
Who should not take HYQVIA?
- Do not take HYQVIA if you: Are allergic to IgG, hyaluronidase, other blood products, or any ingredient in HYQVIA.
How should I take HYQVIA?
- HYQVIA is infused under the skin (subcutaneously) up to once every 4 weeks.
- You can get HYQVIA at your healthcare professional's office, clinic, or hospital.
- You can use HYQVIA at home. You and your healthcare professional will decide if home self-infusion is right for you.
What are the possible or reasonably likely side effects of HYQVIA?
After HYQVIA infusion a temporary, soft swelling may occur around the infusion site, which may last 1 to 3 days, due to the volume of fluid infused.
The following local reactions may occur at the site of infusion and generally go away in a few hours. Local reactions are less likely after the first few infusions.
- Mild or moderate pain
- Redness
- Swelling
- Itching
The most common side effects of HYQVIA are:
- Headache
- Vomiting
- Fatigue
- Nausea
- Fever
- Itching
- Redness
- Abdominal pain
- Back pain
- Pain in extremity
Antibodies to the hyaluronidase component of HYQVIA were formed in some patients taking HYQVIA. It is not known if there is any long-term effect. In theory, these antibodies could react with your body's own PH20. PH20 is present in the male reproductive tract. So far, these antibodies have not been associated with increased or new side-effects.
Call your healthcare professional or go to your emergency department right away if you get:
- Hives, swelling in the mouth or throat, itching, trouble breathing, wheezing, fainting or dizziness. These could be signs of a serious allergic reaction.
- Bad headache with nausea, vomiting, stiff neck, fever, and sensitivity to light. These could be signs of swelling in your brain.
- Reduced urination, sudden weight gain, or swelling in your legs. These could be signs of a kidney problem.
- Pain, swelling, warmth, redness, or a lump in your legs or arms, other than at the infusion site(s). These could be signs of a blood clot.
- Brown or red urine, fast heart rate, yellow skin or eyes. These could be signs of a liver or blood problem.
- Chest pain or trouble breathing, blue lips or extremities. These could be signs of a lung problem.
These are not all of the possible side effects for HYQVIA. You can ask your healthcare professional for information that is provided to healthcare professionals. Talk to your healthcare professional about any side effects that bother you or that don't go away.
How do I store HYQVIA?
Store HYQVIA refrigerated or at room temperature.
- You can store HYQVIA in the refrigerator (36° to 46°F [2° to 8°C]) for up to 36 months.
- You can store HYQVIA at room temperature (up to 77°F [25°C]) for up to 3 months during the first 24 months from the date of manufacturing (Mfg Date) printed on the carton.
- Do not return HYQVIA to the refrigerator if you take it out to room temperature.
Check the expiration date on the carton and vial label. Do not use HYQVIA after the expiration date.
Do not freeze.
Protect from light. You can use the original HYQVIA containers to protect it from light.
Resources at Takeda Available to the Patients
For more information on patient resources and education, please visit www.immunedisease.com.
-
Detailed Instructions for Administration
Do not use HYQVIA at home until you get instructions and training from your healthcare professional.
Your healthcare professional will decide which administration system that is right for you. You will take the hyaluronidase first. Then, within 10 minutes, you will take the immune globulin through an infusion pump.
Prepare HYQVIA vial(s):
- Remove HYQVIA from the box. Allow vials to reach room temperature. This may take up to 60 minutes.
- Do not apply heat or place in microwave.
- Do not shake the vials.
- 1.
- Check the vials:
- •
- Do not use after expiration date.
- •
- Do not use if the purple protective cap is missing or broken.
- •
- Look at the color:
- The Recombinant Human Hyaluronidase (HY) should be clear and colorless.
- The Immune Globulin Infusion 10% (Human) (IG) should be clear and colorless or pale yellow.
- If either liquid is cloudy or has particles, do not use.
- 2.
- Prepare for Infusion:
- •
- Gather all supplies:
Items include: dual vial unit(s) of HYQVIA, infusion supplies: subcutaneous needle set, solution container (bag or syringe), sterile clear bandage and tape, pump tubing, transfer devices, syringe(s), gauze, tape, sharps container, infusion pump, infusion log and other supplies as needed.
- •
- Clean work area
- •
- Program the infusion pump according to prescribed infusion rates and manufacturer's instructions.
- •
- Wash hands thoroughly.
- •
- Open supplies as shown by your healthcare professional.
- •
- If your HYQVIA vials are pooled together by your pharmacist, skip to step 5.
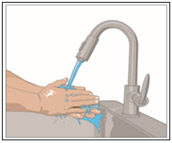
- 3.
- Prepare the Hyaluronidase (Labeled as “HY”):
- •
- Prior to use, take off the purple protective cap(s) and make sure the blue vial caps are removed. If not, manually remove the blue caps.
- •
- Wipe each of the HY vials (top of vial) with an alcohol wipe and allow to dry.
- •
- Remove sterile syringe from package and attach to a sterile needle/needle-less transfer device. A sterile needle or needle-less transfer device [(18-22) gauge sterile needle] may be used for all vial sizes.
- •
- Pull back on plunger of the syringe to fill the syringe with air equal to the amount in the HY vial.
- •
- Position the sharp tip of the needle/needle-less transfer device over the center of the vial stopper and insert it at a 90-degree angle.
- •
- Inject air into the vial and withdraw the full contents of the HY vial(s) into a syringe.
- •
- Repeat the above steps for each additional HY vial using the same syringe, if possible.
- •
- Remove the syringe from the needle/needle-less transfer device.
- •
- Point the syringe tip up and push the plunger of the syringe to remove the air
- •
-
IF using the push method to deliver “HY”:
- Attach the syringe filled with recombinant human hyaluronidase to the needle set
- Push the plunger of smaller syringe to remove the air and fill the needle set up to the needle wings with the recombinant human hyaluronidase.
- •
-
IF using the pump method to deliver “HY”:
- Clean the upper port of the pump administration tubing with alcohol swab and allow to dry for 30 seconds.
- Attach the syringe filled with recombinant human hyaluronidase “HY” to the upper port of pump administration tubing and attach the needle set to the opposite end.
- Close the clamp of lower port close to the needle set.
- Push the plunger of syringe (size may vary due to a larger volume) to remove the air and fill the pump administration tubing to the spike.
- Close the clamp above the upper port and open the clamps on the lower port and needle set. Fill up to the needle wings with the recombinant human hyaluronidase.
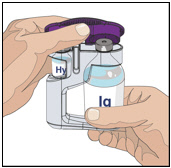

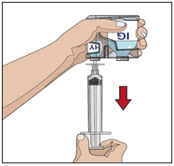
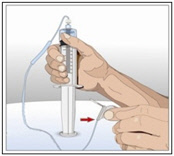
- 4.
- Prepare the immune globulin (Labeled as “IG”):
- •
- Wipe each stopper with a sterile alcohol wipe and allow to dry.
- •
- Transfer the vial(s) labeled IG into either syringe(s) or an infusion bag or as shown by your healthcare professional:
- o
- If using syringe(s)
- −
- Attach a sterile syringe to a vented spike.
- −
- Insert the vented spike into the center of the IG vials.
- −
- Turn the vial upside down and pull back on the plunger to pull the IG into the syringe(s).
- −
- Repeat these steps, if using multiple vials to achieve the desired dose.
- −
- If using a sterile needle, attach a sterile syringe to the sterile needle and pull back the plunger of syringe to fill with air which should equal amount of the liquid you will be taking from the vial. Insert the needle into the center of the stopper, and inject air in. Pull back on the plunger to withdraw the desired volume.
- o
- If using an infusion bag:
- −
- Insert the vented spike into the center of each IG vial. Open the vent.
- −
- Turn the vial upside down and fill the bag with the IG. Repeat this step, if using multiple vials to achieve the desired dose.
- −
- Remove the filling tube(s) of the bag and place a sterile cap over the open end of the bag and close the clamp on bag.
- −
- Insert the spike of the administration pump tubing into the bag and fill as directed by your healthcare professional.
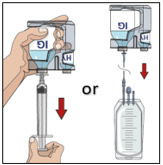
- 5.
- Prepare the infusion site:
- o
- Choose an infusion site(s) in either the middle or upper abdomen or thigh.
- o
- Avoid: bony areas, visible blood vessels, scars and any areas of inflammation (irritation) or infection.
- o
- Select sites on the opposite sides of the body if instructed to infuse in two sites.
- o
- If using three sites, the sites should be 10 cm apart.
- o
- Rotate sites by choosing opposite sides of the body between future infusions.
- o
- Wipe the infusion site(s) with a sterile alcohol wipe beginning at the center of each infusion site and moving outward in a circular motion. Allow the infusion site(s) to dry (at least 30 seconds).
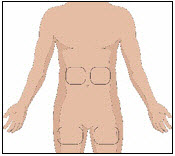
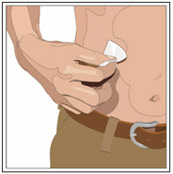
- 6.
- Insert and secure the 24-gauge subcutaneous needle:
- •
- Remove the needle cover. Firmly grasp and pinch at least 1 inch of skin between two fingers.
- •
- Insert needle with a rapid motion straight into the skin at a 90-degree angle. Tape needle in place with sterile tape.
- •
- Repeat these steps if you have a second or a third infusion site.
- •
- Open the clamp on the needle set.
- •
-
If using the push method:
- Check for proper needle placement by pulling back on the syringe plunger to check for blood return in the tubing of the needle set.
- •
-
If using the pump method:
- Close the clamp above the lower port of the pump tubing.
- Clean the lower port with an alcohol swab and allow to dry, for at least 30 seconds.
- Attach a 5 mL syringe to the lower port of the pump tubing.
- Pull back gently on the syringe plunger.
- •
- If blood is seen in the tubing, remove and discard the subcutaneous needle and repeat steps 3, 5, and 6 with a new subcutaneous needle and infusion site.
- •
- Secure the needle set in place by applying a sterile protective dressing over the site. If more than one site is used, repeat the steps.
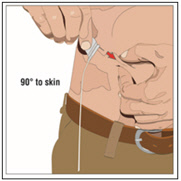
- 7.
- Administer the hyaluronidase (Labeled as “HY”):
- •
- Divide the contents equally between sites, if more than one site is used.
- •
-
If using the push method to deliver “HY”:
- Slowly push the plunger of the smaller syringe with the recombinant human hyaluronidase at an initial rate per infusion site to approximately 1 to 2 mL per minute and increase as tolerated.
- Attach the pump tubing or syringe filled with the IG to the same needle set.
- •
-
If using the pump method to deliver “HY”:
- If using a pump, prepare the pump to infuse the recombinant human hyaluronidase at an initial rate per infusion site of 60 to 120 mL/hour/site and increase as tolerated. Turn on the pump.
- Remove the syringe from pump administration tubing, after infusing all of the content of the syringe.
- Open the clamp on the upper port of the pump administration tubing to administer the IG to the same needle set.
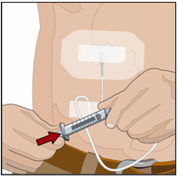
- 8.
- Administer the immune globulin (Labeled as “IG”):
Within 10 minutes of completing the infusion of the HY, start the pump to administer the IG.
- •
- Flush infusion tubing up to the needle wings with normal saline or Dextrose 5% in water (D5W) at the end of infusion, if directed by your healthcare professional.
- 9.
- Remove subcutaneous needle(s) from the infusion site(s):
- •
- Remove the needle set by loosening the tape on all edges.
- •
- Pull the needle wings straight up and out.
- •
- Gently press a small piece of gauze over the needle site and cover with a dressing.
- •
- Throw away the needle(s) into the sharps container.
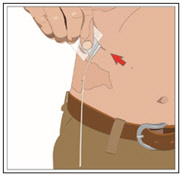
- 10.
- Record the infusion:
- •
- Remove the peel-off label from HYQVIA vial(s), which has the product lot number and expiration date, and place the label in your treatment record/ infusion log.
- •
- Write down the date, time, dose, site(s) of infusion (to assist in rotating sites) and any reactions after each infusion.
- •
- Throw away the disposable supplies, vials, and unused product as recommended by your healthcare professional.
Takeda Pharmaceuticals U.S.A., Inc.
Cambridge, MA 02142U.S. License No. 1898
HYQVIA is a registered trademark of Baxalta Incorporated.
Takeda and
are registered trademarks of Takeda Pharmaceutical Company Limited.©2024 Takeda Pharmaceutical Company Limited. All rights reserved.
Patented: see www.takeda.com/en-us/patents
Revised Date: 9/2024
HYQ353
- PRINCIPAL DISPLAY PANEL - 2.5 g/25 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 1.25 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 2.5 g/25 mL Kit Carton
- PRINCIPAL DISPLAY PANEL - 5 g/50 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 2.5 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 5 g/50 mL Kit Carton
- PRINCIPAL DISPLAY PANEL - 10 g/100 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 5 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 10 g/100 mL Kit Carton
- PRINCIPAL DISPLAY PANEL - 20 g/200 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 10 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 20 g/200 mL Kit Carton
- PRINCIPAL DISPLAY PANEL - 30 g/300 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 15 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 30 g/300 mL Kit Carton
-
INGREDIENTS AND APPEARANCE
HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0944-2510 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2510-02 1 in 1 CARTON; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, GLASS 25 mL Part 2 1 VIAL, GLASS 1.25 mL Part 1 of 2 IMMUNE GLOBULIN INFUSION (HUMAN), 10%
human immuneglobulin g solutionProduct Information Item Code (Source) NDC:0944-2715 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN IMMUNOGLOBULIN G (UNII: 66Y330CJHS) (HUMAN IMMUNOGLOBULIN G - UNII:66Y330CJHS) HUMAN IMMUNOGLOBULIN G 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2715-25 25 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 Part 2 of 2 RECOMBINANT HUMAN HYALURONIDASE
hyaluronidase (human recombinant) solutionProduct Information Item Code (Source) NDC:0944-2720 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HYALURONIDASE (HUMAN RECOMBINANT) (UNII: 743QUY4VD8) (HYALURONIDASE (HUMAN RECOMBINANT) - UNII:743QUY4VD8) HYALURONIDASE (HUMAN RECOMBINANT) 160 [USP'U] in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE (UNII: SE337SVY37) ALBUMIN HUMAN (UNII: ZIF514RVZR) EDETATE DISODIUM (UNII: 7FLD91C86K) CALCIUM CHLORIDE (UNII: M4I0D6VV5M) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2720-03 1.25 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0944-2511 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2511-02 1 in 1 CARTON; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, GLASS 50 mL Part 2 1 VIAL, GLASS 2.5 mL Part 1 of 2 IMMUNE GLOBULIN INFUSION (HUMAN), 10%
human immuneglobulin g solutionProduct Information Item Code (Source) NDC:0944-2716 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN IMMUNOGLOBULIN G (UNII: 66Y330CJHS) (HUMAN IMMUNOGLOBULIN G - UNII:66Y330CJHS) HUMAN IMMUNOGLOBULIN G 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2716-05 50 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 06/04/2012 Part 2 of 2 RECOMBINANT HUMAN HYALURONIDASE
hyaluronidase (human recombinant) solutionProduct Information Item Code (Source) NDC:0944-2721 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HYALURONIDASE (HUMAN RECOMBINANT) (UNII: 743QUY4VD8) (HYALURONIDASE (HUMAN RECOMBINANT) - UNII:743QUY4VD8) HYALURONIDASE (HUMAN RECOMBINANT) 160 [USP'U] in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE (UNII: SE337SVY37) ALBUMIN HUMAN (UNII: ZIF514RVZR) EDETATE DISODIUM (UNII: 7FLD91C86K) CALCIUM CHLORIDE (UNII: M4I0D6VV5M) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2721-03 2.5 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0944-2512 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2512-02 1 in 1 CARTON; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, GLASS 100 mL Part 2 1 VIAL, GLASS 5 mL Part 1 of 2 IMMUNE GLOBULIN INFUSION (HUMAN), 10%
human immuneglobulin g solutionProduct Information Item Code (Source) NDC:0944-2717 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN IMMUNOGLOBULIN G (UNII: 66Y330CJHS) (HUMAN IMMUNOGLOBULIN G - UNII:66Y330CJHS) HUMAN IMMUNOGLOBULIN G 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2717-10 100 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 06/04/2012 Part 2 of 2 RECOMBINANT HUMAN HYALURONIDASE
hyaluronidase (human recombinant) solutionProduct Information Item Code (Source) NDC:0944-2722 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HYALURONIDASE (HUMAN RECOMBINANT) (UNII: 743QUY4VD8) (HYALURONIDASE (HUMAN RECOMBINANT) - UNII:743QUY4VD8) HYALURONIDASE (HUMAN RECOMBINANT) 160 [USP'U] in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE (UNII: SE337SVY37) ALBUMIN HUMAN (UNII: ZIF514RVZR) EDETATE DISODIUM (UNII: 7FLD91C86K) CALCIUM CHLORIDE (UNII: M4I0D6VV5M) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2722-03 5 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0944-2513 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2513-02 1 in 1 CARTON; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, GLASS 200 mL Part 2 1 VIAL, GLASS 10 mL Part 1 of 2 IMMUNE GLOBULIN INFUSION (HUMAN), 10%
human immuneglobulin g solutionProduct Information Item Code (Source) NDC:0944-2718 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN IMMUNOGLOBULIN G (UNII: 66Y330CJHS) (HUMAN IMMUNOGLOBULIN G - UNII:66Y330CJHS) HUMAN IMMUNOGLOBULIN G 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2718-20 200 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 06/04/2012 Part 2 of 2 RECOMBINANT HUMAN HYALURONIDASE
hyaluronidase (human recombinant) solutionProduct Information Item Code (Source) NDC:0944-2723 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HYALURONIDASE (HUMAN RECOMBINANT) (UNII: 743QUY4VD8) (HYALURONIDASE (HUMAN RECOMBINANT) - UNII:743QUY4VD8) HYALURONIDASE (HUMAN RECOMBINANT) 160 [USP'U] in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE (UNII: SE337SVY37) ALBUMIN HUMAN (UNII: ZIF514RVZR) EDETATE DISODIUM (UNII: 7FLD91C86K) CALCIUM CHLORIDE (UNII: M4I0D6VV5M) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2723-03 10 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0944-2514 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2514-02 1 in 1 CARTON; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, GLASS 300 mL Part 2 1 VIAL, GLASS 15 mL Part 1 of 2 IMMUNE GLOBULIN INFUSION (HUMAN), 10%
human immuneglobulin g solutionProduct Information Item Code (Source) NDC:0944-2719 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN IMMUNOGLOBULIN G (UNII: 66Y330CJHS) (HUMAN IMMUNOGLOBULIN G - UNII:66Y330CJHS) HUMAN IMMUNOGLOBULIN G 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2719-30 300 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 06/04/2012 Part 2 of 2 RECOMBINANT HUMAN HYALURONIDASE
hyaluronidase (human recombinant) solutionProduct Information Item Code (Source) NDC:0944-2724 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HYALURONIDASE (HUMAN RECOMBINANT) (UNII: 743QUY4VD8) (HYALURONIDASE (HUMAN RECOMBINANT) - UNII:743QUY4VD8) HYALURONIDASE (HUMAN RECOMBINANT) 160 [USP'U] in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM PHOSPHATE (UNII: SE337SVY37) ALBUMIN HUMAN (UNII: ZIF514RVZR) EDETATE DISODIUM (UNII: 7FLD91C86K) CALCIUM CHLORIDE (UNII: M4I0D6VV5M) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2724-03 15 mL in 1 VIAL, GLASS; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125402 09/12/2014 Labeler - Takeda Pharmaceuticals America, Inc. (039997266) Establishment Name Address ID/FEI Business Operations Baxalta Belgium Manufacturing SA 370634700 MANUFACTURE(0944-2510, 0944-2511, 0944-2512, 0944-2513, 0944-2514) Establishment Name Address ID/FEI Business Operations Baxter Pharmaceutical Solutions, LLC 604719430 MANUFACTURE(0944-2510, 0944-2511, 0944-2512, 0944-2513, 0944-2514) Establishment Name Address ID/FEI Business Operations Takeda Manufacturing Austria AG 300434670 MANUFACTURE(0944-2510, 0944-2511, 0944-2512, 0944-2513, 0944-2514)