Label: POINT OF CARE L5- depo-medrol, lidocaine hci, isopropyl alcohol kit



-
Contains inactivated NDC Code(s)
NDC Code(s): 0009-3073-01, 70859-038-01 - Packager: NuCare Pharmaceuticals,Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated May 4, 2021
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
DEPO-MEDROL is an anti-inflammatory glucocorticoid for intramuscular, intra-articular, soft tissue or intralesional injection. It is available as single-dose vials in two strengths: 40 mg/mL, 80 mg/mL.
Each mL of these preparations contains:
Methylprednisolone acetate
40 mg
80 mg
Polyethylene glycol 3350
29 mg
28 mg
Myristyl-gamma-picolinium chloride
0.195 mg
0.189 mg
Sodium Chloride was added to adjust tonicity.
When necessary, pH was adjusted with sodium hydroxide and/or hydrochloric acid.
The pH of the finished product remains within the USP specified range (e.g., 3.0 to 7.0.)
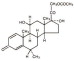
The chemical name for methylprednisolone acetate is pregna-1,4-diene-3,20-dione, 21-(acetyloxy)-11,17-dihydroxy-6-methyl-,(6α,11β)- and the molecular weight is 416.51. The structural formula is represented below:
DEPO-MEDROL Sterile Aqueous Suspension contains methylprednisolone acetate which is the 6-methyl derivative of prednisolone. Methylprednisolone acetate is a white or practically white, odorless, crystalline powder which melts at about 215° with some decomposition. It is soluble in dioxane, sparingly soluble in acetone, alcohol, chloroform, and methanol, and slightly soluble in ether. It is practically insoluble in water.
-
CLINICAL PHARMACOLOGY
Glucocorticoids, naturally occurring and synthetic, are adrenocortical steroids.
Naturally occurring glucocorticoids (hydrocortisone and cortisone), which also have salt retaining properties, are used in replacement therapy in adrenocortical deficiency states. Their synthetic analogs are used primarily for their anti-inflammatory effects in disorders of many organ systems.
-
INDICATIONS AND USAGE
A. For Intramuscular Administration
When oral therapy is not feasible and the strength, dosage form, and route of administration of the drug reasonably lend the preparation to the treatment of the condition, the intramuscular use of DEPO-MEDROL Sterile Aqueous Suspension is indicated as follows:
Allergic States: Control of severe or incapacitating allergic conditions intractable to adequate trials of conventional treatment in asthma, atopic dermatitis, contact dermatitis, drug hypersensitivity reactions, seasonal or perennial allergic rhinitis, serum sickness, transfusion reactions.
Dermatologic Diseases: Bullous dermatitis herpetiformis, exfoliative dermatitis, mycosis fungoides, pemphigus, severe erythema multiforme (Stevens-Johnson syndrome).
Endocrine Disorders: Primary or secondary adrenocortical insufficiency (hydrocortisone or cortisone is the drug of choice; synthetic analogs may be used in conjunction with mineralocorticoids where applicable; in infancy, mineralocorticoid supplementation is of particular importance), congenital adrenal hyperplasia, hypercalcemia associated with cancer, nonsupportive thyroiditis.
Gastrointestinal Diseases: To tide the patient over a critical period of the disease in regional enteritis (systemic therapy) and ulcerative colitis.
Hematologic Disorders: Acquired (autoimmune) hemolytic anemia, congenital (erythroid) hypoplastic anemia (Diamond Blackfan anemia), pure red cell aplasia, select cases of secondary thrombocytopenia.
Miscellaneous: Trichinosis with neurologic or myocardial involvement, tuberculous meningitis with subarachnoid block or impending block when used concurrently with appropriate antituberculous chemotherapy.
Neoplastic Diseases: For palliative management of: leukemias and lymphomas.
Nervous System: Acute exacerbations of multiple sclerosis; cerebral edema associated with primary or metastatic brain tumor or craniotomy.
Ophthalmic Diseases: Sympathetic opthalmia, temporal arteritis, uveitis, ocular inflammatory conditions unresponsive to topical corticosteroids.
Renal Diseases: To induce diuresis or remission of proteinuria in idiopathic nephrotic syndrome, or that due to lupus erythematosus.
Respiratory Diseases: Berylliosis, fulminating or disseminated pulmonary tuberculosis when used concurrently with appropriate antituberculous chemotherapy, idiopathic eosinophilic pneumonias, symptomatic sarcoidosis.
Rheumatic Disorders: As adjunctive therapy for short-term administration (to tide the patient over an acute episode or exacerbation) in acute gouty arthritis; acute rheumatic carditis; ankylosing spondylitis; psoriatic arthritis; rheumatoid arthritis, including juvenile rheumatoid arthritis (selected cases may require low-dose maintenance therapy). For the treatment of dermatomyositis, polymyositis, and systemic lupus erythematosus.
B. For Intra-articular Or Soft Tissue Administration
(See WARNINGS)
DEPO-MEDROL is indicated as adjunctive therapy for short-term administration (to tide the patient over an acute episode or exacerbation) in acute gouty arthritis, acute and subacute bursitis, acute nonspecific tenosynovitis, epicondylitis, rheumatoid arthritis, synovitis of osteoarthritis.
C. For Intralesional Administration
DEPO-MEDROL is indicated for intralesional use in alopecia areata, discoid lupus erythematosus; keloids, localized hypertrophic, infiltrated inflammatory lesions of granuloma annulare, lichen planus, lichen simplex chronicus (neurodermatitis) and psoriatic plaques; necrobiosis lipoidica diabeticorum.
DEPO-MEDROL also may be useful in cystic tumors of an aponeurosis or tendon (ganglia).
-
CONTRAINDICATIONS
DEPO-MEDROL is contraindicated in patients with known hypersensitivity to the product and its constituents.
Intramuscular corticosteroid preparations are contraindicated for idiopathic thrombocytopenic purpura.
DEPO-MEDROL is contraindicated for intrathecal administration. This formulation of methylprednisolone acetate has been associated with reports of severe medical events when administered by this route.
DEPO-MEDROL is contraindicated in systemic fungal infections, except when administered as an intra-articular injection for localized joint conditions (see WARNINGS: Infections, Fungal Infections).
-
WARNINGS
General
This product is not suitable for multi-dose use. Following administration of the desired dose, any remaining suspension should be discarded.
Injection of DEPO-MEDROL may result in dermal and/or subdermal changes forming depressions in the skin at the injection site.
In order to minimize the incidence of dermal and subdermal atrophy, care must be exercised not to exceed recommended doses in injections. Multiple small injections into the area of the lesion should be made whenever possible. The technique of intra-articular and intramuscular injection should include precautions against injection or leakage into the dermis. Injection into the deltoid muscle should be avoided because of a high incidence of subcutaneous atrophy.
It is critical that, during administration of DEPO-MEDROL, appropriate technique be used and care taken to ensure proper placement of drug.
Rare instances of anaphylactoid reactions have occurred in patients receiving corticosteroid therapy (see ADVERSE REACTIONS).
Increased dosage of rapidly acting corticosteroids is indicated in patients on corticosteroid therapy subjected to any unusual stress before, during, and after the stressful situation.
Results from one multicenter, randomized, placebo-controlled study with methylprednisolone hemisuccinate, an IV corticosteroid, showed an increase in early (at 2 weeks) and late (at 6 months) mortality in patients with cranial trauma who were determined not to have other clear indications for corticosteroid treatment. High doses of systemic corticosteroids, including DEPO-MEDROL, should not be used for the treatment of traumatic brain injury.
Cardio-renal
Average and large doses of corticosteroids can cause elevation of blood pressure, salt and water retention, and increased excretion of potassium. These effects are less likely to occur with synthetic derivatives when used in large doses. Dietary salt restriction and potassium supplementation may be necessary. All corticosteroids increase calcium excretion.
Literature reports suggest an apparent association between use of corticosteroids and left ventricular free wall rupture after a recent myocardial infarction; therefore, therapy with corticosteroids should be used with great caution in these patients.
Endocrine
Hypothalamic-pituitary adrenal (HPA) axis suppression. Cushing's syndrome, and hyperglycemia: Monitor patients for these conditions with chronic use.
Corticosteroids can produce reversible HPA axis suppression with the potential for glucocorticosteroid insufficiency after withdrawal of treatment. Drug induced secondary adrenocortical insufficiency may be minimized by gradual reduction of dosage. This type of relative insufficiency may persist for months after discontinuation of therapy; therefore, in any situation of stress occurring during that period, hormone therapy should be reinstituted.
Infections
General
Persons who are on corticosteroids are more susceptible to infections than are healthy individuals. There may be decreased resistance and inability to localize infection when corticosteroids are used. Infections with any pathogen (viral, bacterial, fungal, protozoan, or helminthic) in any location of the body, may be associated with the use of corticosteroids alone or in combination with other immunosuppressive agents.
These infections may be mild, but can be severe and at times fatal. With increasing doses of corticosteroids, the rate of occurrence of infectious complications increases. Do not use intra-articularly, intrabursally, or for intratendinous administration for local effect in the presence of an acute infection. Corticosteroids may mask some signs of infection and new infections may appear during their use.
Fungal Infections
Corticosteroids may exacerbate systemic fungal infections and therefore should not be used in the presence of such infections unless they are needed to control drug interactions. There have been cases reported in which concomitant use of amphotericin B and hydrocortisone was followed by cardiac enlargement and congestive heart failure (see CONTRAINDICATIONS and PRECAUTIONS: Drug Interactions, Amphotericin B injection and potassium-depleting agents).
Special Pathogens
Latent disease may be activated or there may be an exacerbation of intercurrent infections due to pathogens, including those caused by Amoeba, Candida, Cryptococcus, Mycobacterium, Nocardia, Pneumocystis, and Toxoplasma.
It is recommended that latent amebiasis or active amebiasis be ruled out before initiating corticosteroid therapy in any patient who has spent time in the tropics or in any patient with unexplained diarrhea.
Similarly, corticosteroids should be used with great care in patients with known or suspected Strongyloides (threadworm) infestation. In such patients, corticosteroid-induced immunosuppression may lead to Strongyloides hyperinfection and dissemination with widespread larval migration, often accompanied by severe enterocolitis and potentially fatal gram-negative septicemia.
Corticosteroids should not be used in cerebral malaria. There is currently no evidence of benefit from steroids in this condition.
Tuberculosis
The use of corticosteroids in active tuberculosis should be restricted to those cases of fulminating or disseminated tuberculosis in which the corticosteroid is used for the management of the disease in conjunction with an appropriate antituberculous regimen.
If corticosteroids are indicated in patients with latent tuberculosis or tuberculin reactivity, close observation is necessary, as reactivation of the disease may occur. During prolonged corticosteroid therapy, these patients should receive chemoprophylaxis.
Vaccinations
Administration of live or live, attenuated vaccines is contraindicated in patients receiving immunosuppressive doses of corticosteroids. Killed or inactivated vaccines may be administered. However, the response to such vaccines cannot be predicted.
Immunization procedures may be undertaken in patients who are receiving corticosteroids as replacement therapy (e.g., for Addison's disease).
Viral Infections
Chicken pox and measles can have a more serious or even fatal course in pediatric and adult patients on corticosteroids. In pediatric and adult patients who have not had these diseases, particular care should be taken to avoid exposure. The contribution of the underlying disease and/or prior corticosteroid treatment to the risk is also not known. If exposed to chicken pox, prophylaxis with varicella zoster immune globulin (VZIG) may be indicated. If exposed to measles, prophylaxis with immunoglobulin (IG) may be indicated (see the respective package inserts for complete VZIG and IG prescribing information). If chicken pox develops, treatment with antiviral agents should be considered.
Ophthalmic
Use of corticosteroids may produce posterior subcapsular cataracts, glaucoma with possible damage to the optic nerves, and may enhance the establishment of secondary ocular infections due to bacteria, fungi, or viruses. The use of systemic corticosteroids is not recommended in the treatment of optic neuritis and may lead to an increase in the risk of new episodes. Corticosteroids should be used cautiously in patients with ocular herpes simplex because of corneal perforation. Corticosteroids should not be used in active ocular herpes simplex.
-
PRECAUTIONS
General
This product, like many other corticosteroids, is sensitive to heat. Therefore, it should not be autoclaved when it is desirable to sterilize the exterior of the vial.
The lowest possible dose of corticosteroid should be used to control the condition under treatment. When reduction in dosage is possible, the reduction should be gradual.
Since complications of treatment with glucocorticosteroids are dependent on the size of the dose and the duration of treatment, a risk/benefit decision must be made in each individual case as to dose and duration of treatment and as to whether daily or intermittent therapy should be used.
Karposi's sarcoma has been reported to occur in patients receiving corticosteroid therapy, most often for chronic conditions. Discontinuation of corticosteroids may result in clinical improvement.
Cardio-renal
As sodium retention with resultant edema and potassium loss may occur in patients receiving corticosteroids, these agents should be used with caution in patients with congestive heart failure or renal insufficiency.
Endocrine
Drug-induced secondary adrenocortical insufficiency may be minimized by gradual reduction of dosage. This type of relative insufficiency may persist for months after discontinuation of therapy; therefore, in any situation of stress occurring during that period, hormone therapy should be reinstituted. Since mineralocorticoid secretion may be impaired, salt and/or a mineralocorticoid should be administered concurrently.
Metabolic clearance of corticosteroids is decreased in hypothyroid patients and increased in hyperthyroid patients. Changes in thyroid status of the patient may necessitate adjustment in dosage.
Gastrointestinal
Steroids should be used with caution in active or latent peptic ulcers, diverticulitis, fresh intestinal anastomoses, and nonspecific ulcerative colitis, since they may increase the risk of perforation.
Signs of peritoneal irritation following gastrointestinal perforation in patients receiving corticosteroids may be minimal or absent.
Parenteral Administration
Intra-articularly injected corticosteroids may be systemically absorbed.
Appropriate examination of any joint fluid present is necessary to exclude a septic process.
A marked increase in pain accompanied by local swelling, further restriction of joint motion, fever, and malaise are suggestive of septic arthritis. If this complication occurs and diagnosis of sepsis is confirmed, appropriate antimicrobial therapy should be instituted.
Injection of a steroid into an infected site is to be avoided. Local injection of a steroid into a previously infected joint is not usually recommended.
Musculoskeletal
Corticosteroids decrease bone formation and increase bone resorption both through their effect on calcium regulation (e.g., decreasing absorption and increasing excretion) and inhibition of osteoblast function. This, together with a decrease in the protein matrix of the bone secondary to an increase in protein catabolism, and reduced sex hormone production, may lead to an inhibition of bone growth in pediatric patients and the development of osteoporosis at any age. Special consideration should be given to patients at increased risk of osteoporosis (i.e., postmenopausal women) before initiating corticosteroid therapy.
Neuro-psychiatric
Although controlled clinical trials have shown corticosteroids to be effective in speeding the resolution of acute exacerbations of multiple sclerosis, they do not show that corticosteroids affect the ultimate outcome or natural history of the disease. The studies do show that relatively high doses of corticosteroids are necessary to demonstrate a significant effect (see DOSAGE AND ADMINISTRATION).
An acute myopathy has been observed with the use of high doses of corticosteroids, most often occurring in patients with disorders of neuromuscular transmission (e.g., myasthenia gravis), or in patients receiving concomitant therapy with neuromuscular blocking drugs (e.g., pancuronium). This acute myopathy is generalized, may involve ocular and respiratory muscles, and may result in quadriparesis. Elevation of creatine kinase may occur. Clinical improvement or recovery after stopping corticosteroids may require weeks to years.
Psychic derangements may appear when corticosteroids are used, ranging from euphoria, insomnia, mood swings, personality changes, and severe depression to frank psychotic manifestations. Also, existing emotional instability or psychotic tendencies may be aggravated by corticosteroids.
Ophthalmic
Intraocular pressure may become elevated in some individuals. If steroid therapy is continued long-term, intraocular pressure should be monitored.
Corticosteroids should be used cautiously in patients with ocular herpes simplex for fear of corneal perforation.
Information for the Patient
Patients should be warned not to discontinue the use of corticosteroids abruptly or without medical supervision, to advise any medical attendants that they are taking corticosteroids and to seek medical advice at once should they develop a fever or other signs of infection.
Persons who are on corticosteroids should be warned to avoid exposure to chicken pox or measles. Patients should also be advised that if they are exposed, medical advice should be sought without delay.
Drug Interactions
Aminoglutethimide: Aminoglutethimide may lead to a loss of corticosteroid-induced adrenal suppression.
Amphotericin B injection and potassium-depleting agents: When corticosteroids are administered concomitantly with potassium depleting agents (e.g., amphotericin B, diuretics), patients should be observed closely for development of hypokalemia. There have been cases reported in which concomitant use of amphotericin B and hydrocortisone was followed by cardiac enlargement and congestive heart failure.
Antibiotics: Macrolide antibiotics have been reported to cause a significant decrease in corticosteroid clearance (see PRECAUTIONS: Drug Interactions, Hepatic Enzyme Inhibitors).
Anticholinesterases: Concomitant use of anticholinesterase agents and corticosteroids may produce severe weakness in patients with myasthenia gravis. If possible, anticholinesterase agents should be withdrawn at least 24 hours before initiating corticosteroid therapy.
Anticoagulants, oral: Coadministration of corticosteroids and warfarin usually results in inhibition of response to warfarin, although there have been some conflicting reports. Therefore, coagulation indices should be monitored frequently to maintain the desired anticoagulant effect.
Antidiabetics: Because corticosteroids may increase blood glucose concentration, dosage adjustments of antidiabetic agents may be required.
Cyclosporine: Increased activity of both cyclosporine and corticosteroids may occur when the two are used concurrently. Convulsions have been reported with this concurrent use.
Digitalis glycosides: Patients on digitalis glycosides may be at risk of arrhythmias due to hypokalemia.
Estrogens, including oral contraceptives: Estrogens may decrease the hepatic metabolism of certain corticosteroids, thereby increasing their effect.
Hepatic Enzyme Inducers (e.g., barbiturates, phenytoin, carbamazepine, rifampin): Drugs which induce cytochrome P450 3A4 enzyme activity may enhance the metabolism of corticosteroids and require that the dosage of the corticosteroid be increased.
Hepatic Enzyme Inhibitors (e.g., ketoconazole, macrolide antibiotics such as erythromycin and troleandomycin): Drugs which inhibit cytochrome P450 3A4 have the potential to result in increased plasma concentrations of corticosteroids.
Ketoconazole: Ketoconazole has been reported to significantly decrease the metabolism of certain corticosteroids by up to 60%, leading to an increased risk of corticosteroid side effects.
Nonsteroidal anti-inflammatory drugs (NSAIDs): Concomitant use of aspirin (or other nonsteroidal anti-inflammatory agents) and corticosteroids increases the risk of gastrointestinal side effects. Aspirin should be used cautiously in conjunction with corticosteroids in hypoprothrombinemia. The clearance of salicylates may be increased with concurrent use of corticosteroids.
Vaccines: Patients on prolonged corticosteroid therapy may exhibit a diminished response to toxoids and live or inactivated vaccines due to inhibition of antibody response. Corticosteroids may also potentiate the replication of some organisms contained in live attenuated vaccines. Routine administration of vaccines or toxoids should be deferred until corticosteroid therapy is discontinued if possible (see WARNINGS: Infections, Vaccinations).
Carcinogenesis, Mutagenesis, Impairment of Fertility
No adequate studies have been conducted in animals to determine whether corticosteroids have a potential for carcinogenesis or mutagenesis.
Steroids may increase or decrease motility and number of spermatozoa in some patients.
Pregnancy: Teratogenic Effects: Pregnancy Category C
Corticosteroids have been shown to be teratogenic in many species when given in doses equivalent to the human dose. Animal studies in which corticosteroids have been given to pregnant mice, rats, and rabbits have yielded an increased incidence of cleft palate in the offspring. There are no adequate and well-controlled studies in pregnant women. Corticosteroids should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Infants born to mothers who have received corticosteroids during pregnancy should be carefully observed for signs of hypoadrenalism.
Nursing Mothers
Systemically administered corticosteroids appear in human milk and could suppress growth, interfere with endogenous corticosteroid production, or cause other untoward effects. Because of the potential for serious adverse reactions in nursing infants from corticosteroids, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
The efficacy and safety of corticosteroids in the pediatric population are based on the well-established course of effect of corticosteroids which is similar in pediatric and adult populations. Published studies provide evidence of efficacy and safety in pediatric patients for the treatment of nephritic syndrome (patients >2 years of age) and aggressive lymphomas and leukemias (patients >1 month of age). Other indications for pediatric use of corticosteroids (e.g., severe asthma and wheezing) are based on adequate and well-controlled clinical trials conducted in adults, on the premises that the course of the diseases and their pathophysiology are considered to be substantially similar in both populations.
The adverse effects of corticosteroids in pediatric patients are similar to those in adults (see ADVERSE REACTIONS). Like adults, pediatric patients should be carefully observed with frequent measurements of blood pressure, weight, height, intraocular pressure, and clinical evaluation for the presence of infection, psychosocial disturbances, thromboembolism, peptic ulcers, cataracts, and osteoporosis. Pediatric patients who are treated with corticosteroids by any route, including systemically administered corticosteroids, may experience a decrease in their growth velocity. This negative impact of corticosteroids on growth has been observed at low systemic doses and in the absence of laboratory evidence of HPA axis suppression (i.e., cosyntropin stimulation and basal cortisol plasma levels). Growth velocity may therefore be a more sensitive indicator of systemic corticosteroid exposure in pediatric patients than some commonly used tests of HPA axis function. The linear growth of pediatric patients treated with corticosteroids should be monitored, and the potential growth effects of prolonged treatment should be weighed against clinical benefits obtained and the availability of treatment alternatives. In order to minimize the potential growth effects of corticosteroids, pediatric patients should be titrated to the lowest effective dose.
Geriatric Use
Clinical studies did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
The following adverse reactions have been reported with DEPO-MEDROL or other corticosteroids:
Allergic reactions: Allergic or hypersensitivity reactions, anaphylactoid reaction, anaphylaxis, angioedema.
Cardiovascular: Bradycardia, cardiac arrest, cardiac arrhythmias, cardiac enlargement, circulatory collapse, congestive heart failure, fat embolism, hypertension, hypertrophic cardiomyopathy in premature infants, myocardial rupture following recent myocardial infarction (see WARNINGS), pulmonary edema, syncope, tachycardia, thromboembolism, thrombophlebitis, vasculitis.
Dermatologic: Acne, allergic dermatitis, cutaneous and subcutaneous atrophy, dry scaly skin, ecchymoses and petechiae, edema, erythema, hyperpigmentation, hypopigmentation, impaired wound healing, increased sweating, rash, sterile abscess, striae, suppressed reactions to skin tests, thin fragile skin, thinning scalp hair, urticaria.
Endocrine: Decreased carbohydrate and glucose tolerance, development of cushingoid state, glycosuria, hirsutism, hypertrichosis, increased requirements for insulin or oral hypoglycemic agents in diabetes, manifestations of latent diabetes mellitus, menstrual irregularities, secondary adrenocortical and pituitary unresponsiveness (particularly in times of stress, as in trauma, surgery, or illness), suppression of growth in pediatric patients.
Fluid and electrolyte disturbances: Congestive heart failure in susceptible patients, fluid retention, hypokalemic alkalosis, potassium loss, sodium retention.
Gastrointestinal: Abdominal distention, bowel/bladder dysfunction (after intrathecal administration), elevation in serum liver enzyme levels (usually reversible upon discontinuation), hepatomegaly, increased appetite, nausea, pancreatitis, peptic ulcer with possible subsequent perforation and hemorrhage, perforation of the small and large intestine (particularly in patients with inflammatory bowel disease), ulcerative esophagitis.
Metabolic: Negative nitrogen balance due to protein catabolism.
Musculoskeletal: Aseptic necrosis of femoral and humeral heads, calcinosis (following intra-articular or intra-lesional use), Charcot-like arthropathy, loss of muscle mass, muscle weakness, osteoporosis, pathologic fracture of long bones, postinjection flare (following intra-articular use), steroid myopathy, tendon rupture, vertebral compression fractures.
Neurologic/Psychiatric: Convulsions, depression, emotional instability, euphoria, headache, increased intracranial pressure with papilledema (pseudotumor cerebri) usually following discontinuation of treatment, insomnia, mood swings, neuritis, neuropathy, paresthesia, personality changes, psychic disorders, vertigo.
Ophthalmic: Exophthalmoses, glaucoma, increased intraocular pressure, posterior subcapsular cataracts.
Other: Abnormal fat deposits, decreased resistance to infection, hiccups, increased or decreased motility and number of spermatozoa, injection site infections following non-sterile administration (see WARNINGS), malaise, moon face, weight gain.
The following adverse reactions have been reported with the following routes of administration:
Intrathecal/Epidural: Arachnoiditis, bowel/bladder dysfunction, headache, meningitis, parapareisis/paraplegia, seizures, sensory disturbances.
Intranasal: Allergic reactions, rhinitis, temporary/permanent visual impairment including blindness.
Ophthalmic: Increased intraocular pressure, infection, ocular and periocular inflammation including allergic reactions, residue or slough at injection site, temporary/permanent visual impairment including blindness.
Miscellaneous injection sites (scalp, tonsillar fauces, sphenopalatine ganglion): blindness.
- OVERDOSAGE
-
DOSAGE AND ADMINISTRATION
Because of possible physical incompatibilities, DEPO-MEDROL Sterile Aqueous Suspension should not be diluted or mixed with other solutions.
The initial dosage of parenterally administered DEPO-MEDROL will vary from 4 to 120 mg, depending on the specific disease entity being treated. However, in certain overwhelming, acute, life-threatening situations, administration in dosages exceeding the usual dosages may be justified and may be in multiples of the oral dosages.
It Should Be Emphasized that Dosage Requirements Are Variable and Must Be Individualized on the Basis of the Disease Under Treatment and the Response of the Patient. After a favorable response is noted, the proper maintenance dosage should be determined by decreasing the initial drug dosage in small decrements at appropriate time intervals until the lowest dosage which will maintain an adequate clinical response is reached. Situations which may make dosage adjustments necessary are changes in clinical status secondary to remissions or exacerbations in the disease process, the patient's individual drug responsiveness, and the effect of patient exposure to stressful situations not directly related to the disease entity under treatment. In this latter situation, it may be necessary to increase the dosage of the corticosteroid for a period of time consistent with the patient's condition. If after long-term therapy the drug is to be stopped, it is recommended that it be withdrawn gradually rather than abruptly.
A. Administration for Local Effect
Therapy with DEPO-MEDROL does not obviate the need for the conventional measures usually employed. Although this method of treatment will ameliorate symptoms, it is in no sense a cure and the hormone has no effect on the cause of the inflammation.
1. Rheumatoid Arthritis and Osteoarthritis. The dose for intra-articular administration depends upon the size of the joint and varies with the severity of the condition in the individual patient. In chronic cases, injections may be repeated at intervals ranging from one to five or more weeks, depending upon the degree of relief obtained from the initial injection. The doses in the following table are given as a general guide:
Size of Joint Examples Range of Dosage Large
Knees
Ankles
Shoulders20 to 80 mg
Medium
Elbows
Wrists10 to 40 mg
Small
Metacarpophalangeal
Interphalangeal
Sternoclavicular
Acromioclavicular4 to 10 mg
Procedure: It is recommended that the anatomy of the joint involved be reviewed before attempting intra-articular injection. In order to obtain the full anti-inflammatory effect, it is important that the injection be made into the synovial space. Employing the same sterile technique as for a lumbar puncture, a sterile 20 to 24 gauge needle (on a dry syringe) is quickly inserted into the synovial cavity. Procaine infiltration is elective. The aspiration of only a few drops of joint fluid proves the joint space has been entered by the needle. The injection site for each joint is determined by that location where the synovial cavity is most superficial and most free of large vessels and nerves. With the needle in place, the aspirating syringe is removed and replaced by a second syringe containing the desired amount of DEPO-MEDROL. The plunger is then pulled outward slightly to aspirate synovial fluid and to make sure the needle is still in the synovial space. After injection, the joint is moved gently a few times to aid mixing of the synovial fluid and the suspension. The site is covered with a small sterile dressing.
Suitable sites for intra-articular injection are the knee, ankle, wrist, elbow, shoulder, phalangeal, and hip joints. Since difficulty is not infrequently encountered in entering the hip joint, precautions should be taken to avoid any large blood vessels in the area. Joints not suitable for injection are those that are anatomically inaccessible such as the spinal joints and those like the sacroiliac joints that are devoid of synovial space. Treatment failures are most frequently the result of failure to enter the joint space. Little or no benefit follows injection into surrounding tissue. If failures occur when injections into the synovial spaces are certain, as determined by aspiration of fluid, repeated injections are usually futile.
If a local anesthetic is used prior to injection of DEPO-MEDROL, the anesthetic package insert should be read carefully and all the precautions observed.
2. Bursitis. The area around the injection site is prepared in a sterile way and a wheal at the site made with 1 percent procaine hydrochloride solution. A 20 to 24 gauge needle attached to a dry syringe is inserted into the bursa and the fluid aspirated. The needle is left in place and the aspirating syringe changed for a small syringe containing the desired dose. After injection, the needle is withdrawn and a small dressing applied.
3. Miscellaneous: Ganglion, Tendinitis, Epicondylitis. In the treatment of conditions such as tendinitis or tenosynovitis, care should be taken following application of a suitable antiseptic to the overlying skin to inject the suspension into the tendon sheath rather than into the substance of the tendon. The tendon may be readily palpated when placed on a stretch. When treating conditions such as epicondylitis, the area of greatest tenderness should be outlined carefully and the suspension infiltrated into the area. For ganglia of the tendon sheaths, the suspension is injected directly into the cyst. In many cases, a single injection causes a marked decrease in the size of the cystic tumor and may effect disappearance. The usual sterile precautions should be observed, of course, with each injection.
The dose in the treatment of the various conditions of the tendinous or bursal structures listed above varies with the condition being treated and ranges from 4 to 30 mg. In recurrent or chronic conditions, repeated injections may be necessary.
4. Injections for Local Effect in Dermatologic Conditions. Following cleansing with an appropriate antiseptic such as 70% alcohol, 20 to 60 mg of the suspension is injected into the lesion. It may be necessary to distribute doses ranging from 20 to 40 mg by repeated local injections in the case of large lesions. Care should be taken to avoid injection of sufficient material to cause blanching since this may be followed by a small slough. One to four injections are usually employed, the intervals between injections varying with the type of lesion being treated and the duration of improvement produced by the initial injection.
B. Administration for Systemic Effect
The intramuscular dosage will vary with the condition being treated. When employed as a temporary substitute for oral therapy, a single injection during each 24-hour period of a dose of the suspension equal to the total daily oral dose of MEDROL ® Tablets (methylprednisolone tablets, USP) is usually sufficient. When a prolonged effect is desired, the weekly dose may be calculated by multiplying the daily oral dose by 7 and given as a single intramuscular injection.
In pediatric patients, the initial dose of methylprednisolone may vary depending on the specific disease entity being treated. Dosage must be individualized according to the severity of the disease and response of the patient. The recommended dosage may be reduced for pediatric patients, but dosage should be governed by the severity of the condition rather than by strict adherence to the ratio indicated by age or body weight.
In patients with the adrenogenital syndrome, a single intramuscular injection of 40 mg every two weeks may be adequate. For maintenance of patients with rheumatoid arthritis, the weekly intramuscular dose will vary from 40 to 120 mg. The usual dosage for patients with dermatologic lesions benefited by systemic corticoid therapy is 40 to 120 mg of methylprednisolone acetate administered intramuscularly at weekly intervals for one to four weeks. In acute severe dermatitis due to poison ivy, relief may result within 8 to 12 hours following intramuscular administration of a single dose of 80 to 120 mg. In chronic contact dermatitis, repeated injections at 5 to 10 day intervals may be necessary. In seborrheic dermatitis, a weekly dose of 80 mg may be adequate to control the condition.
Following intramuscular administration of 80 to 120 mg to asthmatic patients, relief may result within 6 to 48 hours and persist for several days to two weeks. Similarly, in patients with allergic rhinitis (hay fever), an intramuscular dose of 80 to 120 mg may be followed by relief of coryzal symptoms within six hours persisting for several days to three weeks.
If signs of stress are associated with the condition being treated, the dosage of the suspension should be increased. If a rapid hormonal effect of maximum intensity is required, the intravenous administration of highly soluble methylprednisolone sodium succinate is indicated.
In treatment of acute exacerbations of multiple sclerosis, daily doses of 160 mg of methylprednisolone for a week followed by 64 mg every other day for 1 month have been shown to be effective.
For the purpose of comparison, the following is the equivalent milligram dose of the various glucocorticoids:
Cortisone, 25
Triamcinolone, 4
Hydrocortisone, 20
Paramethasone, 2
Prednisolone, 5
Betamethasone, 0.75
Prednisone, 5
Dexamethasone, 0.75
Methylprednisolone, 4
These dose relationships apply only to oral or intravenous administration of these compounds. When these substances or their derivatives are injected intramuscularly or into joint spaces, their relative properties may be greatly altered.
- HOW SUPPLIED
- SPL UNCLASSIFIED SECTION
- SPL UNCLASSIFIED SECTION
-
LIDOCAINE DESCRIPTION
Lidocaine Hydrochloride Injection, USP is a sterile, nonpyrogenic solution of lidocaine hydrochloride in water for injection for parenteral administration in various concentrations with characteristics as follows:
Multiple-dose vials contain 0.1% of methylparaben added as preservative. May contain sodium hydroxide and/or hydrochloric acid for pH adjustment. The pH is 6.5 (5.0 to 7.0). See HOW SUPPLIED section for various sizes and strengths.
Lidocaine is a local anesthetic of the amide type.
Lidocaine Hydrochloride, USP is chemically designated 2-(diethylamino)-N-(2,6-dimethylphenyl)-acetamide monohydrochloride monohydrate, a white powder freely soluble in water. The molecular weight is 288.82. It has the following structural formula:
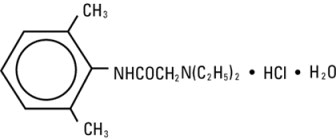
The semi-rigid vial used for the plastic vials is fabricated from a specially formulated polyolefin. It is a copolymer of ethylene and propylene. The safety of the plastic has been confirmed by tests in animals according to USP biological standards for plastic containers. The container requires no vapor barrier to maintain the proper drug concentration.
-
CLINICAL PHARMACOLOGY
Mechanism of action: Lidocaine stabilizes the neuronal membrane by inhibiting the ionic fluxes required for the initiation and conduction of impulses, thereby effecting local anesthetic action.
Hemodynamics: Excessive blood levels may cause changes in cardiac output, total peripheral resistance, and mean arterial pressure. With central neural blockade these changes may be attributable to block of autonomic fibers, a direct depressant effect of the local anesthetic agent on various components of the cardiovascular system and/or the beta-adrenergic receptor stimulating action of epinephrine when present. The net effect is normally a modest hypotension when the recommended dosages are not exceeded.
Pharmacokinetics and metabolism: Information derived from diverse formulations, concentrations and usages reveals that lidocaine is completely absorbed following parenteral administration, its rate of absorption depending, for example, upon various factors such as the site of administration and the presence or absence of a vasoconstrictor agent. Except for intravascular administration, the highest blood levels are obtained following intercostal nerve block and the lowest after subcutaneous administration.
The plasma binding of lidocaine is dependent on drug concentration, and the fraction bound decreases with increasing concentration. At concentrations of 1 to 4 mcg of free base per mL, 60 to 80 percent of lidocaine is protein bound. Binding is also dependent on the plasma concentration of the alpha-1-acid glycoprotein.
Lidocaine crosses the blood-brain and placental barriers, presumably by passive diffusion.
Lidocaine is metabolized rapidly by the liver, and metabolites and unchanged drug are excreted by the kidneys. Biotransformation includes oxidative N-dealkylation, ring hydroxylation, cleavage of the amide linkage, and conjugation. N-dealkylation, a major pathway of biotransformation, yields the metabolites monoethylglycinexylidide and glycinexylidide. The pharmacological/toxicological actions of these metabolites are similar to, but less potent than, those of lidocaine. Approximately 90% of lidocaine administered is excreted in the form of various metabolites, and less than 10% is excreted unchanged. The primary metabolite in urine is a conjugate of 4-hydroxy-2, 6-dimethylaniline.
The elimination half-life of lidocaine following an intravenous bolus injection is typically 1.5 to 2.0 hours. Because of the rapid rate at which lidocaine is metabolized, any condition that affects liver function may alter lidocaine kinetics. The half-life may be prolonged two-fold or more in patients with liver dysfunction. Renal dysfunction does not affect lidocaine kinetics but may increase the accumulation of metabolites.
Factors such as acidosis and the use of CNS stimulants and depressants affect the CNS levels of lidocaine required to produce overt systemic effects. Objective adverse manifestations become increasingly apparent with increasing venous plasma levels above 6.0 mcg free base per mL. In the rhesus monkey arterial blood levels of 18-21 mcg/mL have been shown to be threshold for convulsive activity.
-
INDICATIONS AND USAGE
Lidocaine Hydrochloride Injection, USP is indicated for production of local or regional anesthesia by infiltration techniques such as percutaneous injection and intravenous regional anesthesia by peripheral nerve block techniques such as brachial plexus and intercostal and by central neural techniques such as lumbar and caudal epidural blocks, when the accepted procedures for these techniques as described in standard textbooks are observed.
- CONTRAINDICATIONS
-
PRECAUTIONS
General:
The safety and effectiveness of lidocaine depend on proper dosage, correct technique, adequate precautions, and readiness for emergencies. Standard textbooks should be consulted for specific techniques and precautions for various regional anesthetic procedures.
Resuscitative equipment, oxygen, and other resuscitative drugs should be available for immediate use. (See WARNINGS and ADVERSE REACTIONS). The lowest dosage that results in effective anesthesia should be used to avoid high plasma levels and serious adverse effects. Syringe aspirations should also be performed before and during each supplemental injection when using indwelling catheter techniques. During the administration of epidural anesthesia, it is recommended that a test dose be administered initially and that the patient be monitored for central nervous system toxicity and cardiovascular toxicity, as well as for signs of unintended intrathecal administration before proceeding. When clinical conditions permit, consideration should be given to employing local anesthetic solutions that contain epinephrine for the test dose because circulatory changes compatible with epinephrine may also serve as a warning sign of unintended intravascular injection. An intravascular injection is still possible even if aspirations for blood are negative. Repeated doses of lidocaine may cause significant increases in blood levels with each repeated dose because of slow accumulation of the drug or its metabolites. Tolerance to elevated blood levels varies with the status of the patient. Debilitated, elderly patients, acutely ill patients and children should be given reduced doses commensurate with their age and physical condition. Lidocaine should also be used with caution in patients with severe shock or heart block. Lumbar and caudal epidural anesthesia should be used with extreme caution in persons with the following conditions: existing neurological disease, spinal deformities, septicemia and severe hypertension.
Local anesthetic solutions containing a vasoconstrictor should be used cautiously and in carefully circumscribed quantities in areas of the body supplied by end arteries or having otherwise compromised blood supply. Patients with peripheral vascular disease and those with hypertensive vascular disease may exhibit exaggerated vasoconstrictor response. Ischemic injury or necrosis may result. Preparations containing a vasoconstrictor should be used with caution in patients during or following the administration of potent general anesthetic agents, since cardiac arrhythmias may occur under such conditions.
Careful and constant monitoring of cardiovascular and respiratory (adequacy of ventilation) vital signs and the patient’s state of consciousness should be accomplished after each local anesthetic injection. It should be kept in mind at such times that restlessness, anxiety, tinnitus, dizziness, blurred vision, tremors, depression or drowsiness may be early warning signs of central nervous system toxicity.
Since amide-type local anesthetics are metabolized by the liver, lidocaine should be used with caution in patients with hepatic disease. Patients with severe hepatic disease, because of their inability to metabolize local anesthetics normally, are at greater risk of developing toxic plasma concentrations. Lidocaine should also be used with caution in patients with impaired cardiovascular function since they may be less able to compensate for functional changes associated with the prolongation of A-V conduction produced by these drugs. Many drugs used during the conduct of anesthesia are considered potential triggering agents for familial malignant hyperthermia. Since it is not known whether amide-type local anesthetics may trigger this reaction and since the need for supplemental general anesthesia cannot be predicted in advance, it is suggested that a standard protocol for the management of malignant hyperthermia should be available. Early unexplained signs of tachycardia, tachypnea, labile blood pressure and metabolic acidosis may precede temperature elevation. Successful outcome is dependent on early diagnosis, prompt discontinuance of the suspect triggering agent(s) and institution of treatment, including oxygen therapy, indicated supportive measures and dantrolene (consult dantrolene sodium intravenous package insert before using).
Proper tourniquet technique, as described in publications and standard textbooks, is essential in the performance of intravenous regional anesthesia. Solutions containing epinephrine or other vasoconstrictors should not be used for this technique.
Lidocaine should be used with caution in persons with known drug sensitivities. Patients allergic to para-aminobenzoic acid derivatives (procaine, tetracaine, benzocaine, etc.) have not shown cross sensitivity to lidocaine.
Use in the Head and Neck Area: Small doses of local anesthetics injected into the head and neck area, including retrobulbar, dental and stellate ganglion blocks, may produce adverse reactions similar to systemic toxicity seen with unintentional intravascular injections of larger doses. Confusion, convulsions, respiratory depression and/or respiratory arrest and cardiovascular stimulation or depression have been reported. These reactions may be due to intra-arterial injections of the local anesthetic with retrograde flow to the cerebral circulation. Patients receiving these blocks should have their circulation and respiration monitored and be constantly observed. Resuscitative equipment and personnel for treating adverse reactions should be immediately available. Dosage recommendations should not be exceeded. (See DOSAGE AND ADMINISTRATION).
Information for Patients:
When appropriate, patients should be informed in advance that they may experience temporary loss of sensation and motor activity, usually in the lower half of the body following proper administration of epidural anesthesia.
Clinically Significant Drug Interactions:
The administration of local anesthetic solutions containing epinephrine or norepinephrine to patients receiving monoamine oxidase inhibitors or tricyclic antidepressants may produce severe prolonged hypertension.
Phenothiazines and butyrophenones may reduce or reverse the pressor effect of epinephrine.
Concurrent use of these agents should generally be avoided. In situations when concurrent therapy is necessary, careful patient monitoring is essential.
Concurrent administration of vasopressor drugs (for the treatment of hypotension related to obstetric blocks) and ergot-type oxytoxic drugs may cause severe persistent hypertension or cerebrovascular accidents.
Drug Laboratory Test Interactions:
The intramuscular injection of lidocaine may result in an increase in creatine phosphokinase levels. Thus, the use of this enzyme determination without isoenzyme separation as a diagnostic test for the presence of acute myocardial infarction may be compromised by the intramuscular injection of lidocaine.
Carcinogenesis, Mutagenesis, Impairment of Fertility:
Studies of lidocaine in animals to evaluate the carcinogenic and mutagenic potential or the effect on fertility have not been conducted.
Pregnancy:
Teratogenic Effects. Pregnancy Category B. Reproduction studies have been performed in rats at doses up to 6.6 times the human dose and have revealed no evidence of harm to the fetus caused by lidocaine. There are, however, no adequate and well-controlled studies in pregnant women. Animal reproduction studies are not always predictive of human response. General consideration should be given to this fact before administering lidocaine to women of childbearing potential, especially during early pregnancy when maximum organogenesis takes place.
Labor and Delivery:
Local anesthetics rapidly cross the placenta and when used for epidural, paracervical, pudendal or caudal block anesthesia, can cause varying degrees of maternal, fetal and neonatal toxicity (See CLINICAL PHARMACOLOGY— Pharmacokinetics). The potential for toxicity depends upon the procedure performed, the type and amount of drug used, and the technique of drug administration. Adverse reactions in the parturient, fetus and neonate involve alterations of the central nervous system peripheral vascular tone and cardiac function.
Maternal hypotension has resulted from regional anesthesia. Local anesthetics produce vasodilation by blocking sympathetic nerves. Elevating the patient’s legs and positioning her on her left side will help prevent decreases in blood pressure. The fetal heart rate also should be monitored continuously, and electronic fetal monitoring is highly advisable.
Epidural, spinal, paracervical, or pudendal anesthesia may alter the forces of parturition through changes in uterine contractility or maternal expulsive efforts. In one study, paracervical block anesthesia was associated with a decrease in the mean duration of first stage labor and facilitation of cervical dilation. However, spinal and epidural anesthesia have also been reported to prolong the second stage of labor by removing the parturient’s reflex urge to bear down or by interfering with motor function. The use of obstetrical anesthesia may increase the need for forceps assistance.
The use of some local anesthetic drug products during labor and delivery may be followed by diminished muscle strength and tone for the first day or two of life. The long-term significance of these observations is unknown. Fetal bradycardia may occur in 20 to 30 percent of patients receiving paracervical nerve block anesthesia with the amide-type local anesthetics and may be associated with fetal acidosis. Fetal heart rate should always be monitored during paracervical anesthesia. The physician should weigh the possible advantages against risks when considering paracervical block in prematurity, toxemia of pregnancy and fetal distress. Careful adherence to recommended dosage is of the utmost importance in obstetrical paracervical block. Failure to achieve adequate analgesia with recommended doses should arouse suspicion of intravascular or fetal intracranial injection. Cases compatible with unintended fetal intracranial injection of local anesthetic solution have been reported following intended paracervical or pudendal block or both. Babies so affected present with unexplained neonatal depression at birth, which correlates with high local anesthetic serum levels, and often manifest seizures within six hours. Prompt use of supportive measures combined with forced urinary excretion of the local anesthetic has been used successfully to manage this complication.
Case reports of maternal convulsions and cardiovascular collapse following use of some local anesthetics for paracervical block in early pregnancy (as anesthesia for elective abortion) suggest that systemic absorption under these circumstances may be rapid. The recommended maximum dose of each drug should not be exceeded. Injection should be made slowly and with frequent aspiration. Allow a 5-minute interval between sides.
-
WARNINGS
LIDOCAINE HYDROCHLORIDE INJECTION, FOR INFILTRATION AND NERVE BLOCK, SHOULD BE EMPLOYED ONLY BY CLINICIANS WHO ARE WELL VERSED IN DIAGNOSIS AND MANAGEMENT OF DOSE-RELATED TOXICITY AND OTHER ACUTE EMERGENCIES THAT MIGHT ARISE FROM THE BLOCK TO BE EMPLOYED AND THEN ONLY AFTER ENSURING THE IMMEDIATE AVAILABILITY OF OXYGEN, OTHER RESUSCITATIVE DRUGS, CARDIOPULMONARY EQUIPMENT, AND THE PERSONNEL NEEDED FOR PROPER MANAGEMENT OF TOXIC REACTIONS AND RELATED EMERGENCIES (See also ADVERSE REACTIONS and PRECAUTIONS). DELAY IN PROPER MANAGEMENT OF DOSE-RELATED TOXICITY, UNDERVENTILATION FROM ANY CAUSE AND/OR ALTERED SENSITIVITY MAY LEAD TO THE DEVELOPMENT OF ACIDOSIS, CARDIAC ARREST AND, POSSIBLY, DEATH.
Intra-articular infusions of local anesthetics following arthroscopic and other surgical procedures is an unapproved use, and there have been post-marketing reports of chondrolysis in patients receiving such infusions. The majority of reported cases of chondrolysis have involved the shoulder joint; cases of gleno-humeral chondrolysis have been described in pediatric and adult patients following intra-articular infusions of local anesthetics with and without epinephrine for periods of 48 to 72 hours. There is insufficient information to determine whether shorter infusion periods are not associated with these findings. The time of onset of symptoms, such as joint pain, stiffness and loss of motion can be variable, but may begin as early as the 2nd month after surgery. Currently, there is no effective treatment for chondrolysis; patients who experienced chondrolysis have required additional diagnostic and therapeutic procedures and some required arthroplasty or shoulder replacement.
To avoid intravascular injection, aspiration should be performed before the local anesthetic solution is injected. The needle must be repositioned until no return of blood can be elicited by aspiration. Note, however, that the absence of blood in the syringe does not guarantee that intravascular injection has been avoided.
Local anesthetic solutions containing antimicrobial preservatives (e.g., methylparaben) should not be used for epidural or spinal anesthesia because the safety of these agents has not been established with regard to intrathecal injection, either intentional or accidental.
-
ADVERSE REACTIONS
Systemic: Adverse experiences following the administration of lidocaine are similar in nature to those observed with other amide local anesthetic agents. These adverse experiences are, in general, dose-related and may result from high plasma levels caused by excessive dosage, rapid absorption or inadvertent intravascular injection, or may result from a hypersensitivity, idiosyncrasy or diminished tolerance on the part of the patient. Serious adverse experiences are generally systemic in nature. The following types are those most commonly reported:
Central Nervous System: CNS manifestations are excitatory and/or depressant and may be characterized by lightheadedness, nervousness, apprehension, euphoria, confusion, dizziness, drowsiness, tinnitus, blurred or double vision, vomiting, sensations of heat, cold or numbness, twitching, tremors, convulsions, unconsciousness, respiratory depression and arrest. The excitatory manifestations may be very brief or may not occur at all, in which case the first manifestation of toxicity may be drowsiness merging into unconsciousness and respiratory arrest.
Drowsiness following the administration of lidocaine is usually an early sign of a high blood level of the drug and may occur as a consequence of rapid absorption.
Cardiovascular System: Cardiovascular manifestations are usually depressant and are characterized by bradycardia, hypotension, and cardiovascular collapse, which may lead to cardiac arrest.
Allergic: Allergic reactions are characterized by cutaneous lesions, urticaria, edema or anaphylactoid reactions. Allergic reactions may occur as a result of sensitivity either to local anesthetic agents or to the methylparaben used as a preservative in multiple dose vials. Allergic reactions as a result of sensitivity to lidocaine are extremely rare and, if they occur, should be managed by conventional means. The detection of sensitivity by skin testing is of doubtful value.
Neurologic: The incidences of adverse reactions associated with the use of local anesthetics may be related to the total dose of local anesthetic administered and are also dependent upon the particular drug used, the route of administration and the physical status of the patient. In a prospective review of 10,440 patients who received lidocaine for spinal anesthesia, the incidences of adverse reactions were reported to be about 3 percent each for positional headaches, hypotension and backache; 2 percent for shivering; and less than 1 percent each for peripheral nerve symptoms, nausea, respiratory inadequacy and double vision. Many of these observations may be related to local anesthetic techniques, with or without a contribution from the local anesthetic.
In the practice of caudal or lumbar epidural block, occasional unintentional penetration of the subarachnoid space by the catheter may occur. Subsequent adverse effects may depend partially on the amount of drug administered subdurally.
These may include spinal block of varying magnitude (including total spinal block), hypotension secondary to spinal block, loss of bladder and bowel control, and loss of perineal sensation and sexual function. Persistent motor, sensory and/or autonomic (sphincter control) deficit of some lower spinal segments with slow recovery (several months) or incomplete recovery have been reported in rare instances when caudal or lumbar epidural block has been attempted. Backache and headache have also been noted following use of these anesthetic procedures.
There have been reported cases of permanent injury to extraocular muscles requiring surgical repair following retrobulbar administration.
-
OVERDOSAGE
Acute emergencies from local anesthetics are generally related to high plasma levels encountered during therapeutic use of local anesthetics or to unintended subarachnoid injection of local anesthetic solution (see ADVERSE REACTIONS, WARNINGS and PRECAUTIONS).
Management of Local Anesthetic Emergencies: The first consideration is prevention, best accomplished by careful monitoring of cardiovascular and respiratory vital signs and the patient’s state of consciousness after each local anesthetic injection. At the first sign of change, oxygen should be administered.
The first step in the management of convulsions, as well as underventilation or apnea due to unintended subarachnoid injection of drug solution, consists of immediate attention to the maintenance of a patent airway and assisted or controlled ventilation with oxygen and a delivery system capable of permitting immediate positive airway pressure by mask. Immediately after the institution of these ventilatory measures, the adequacy of the circulation should be evaluated, keeping in mind that drugs used to treat convulsions sometimes depress the circulation when administered intravenously. Should convulsions persist despite adequate respiratory support, and if the status of the circulation permits, small increments of an ultra-short acting barbiturate (such as thiopental or thiamylal) or a benzodiazepine (such as diazepam) may be administered intravenously. The clinician should be familiar, prior to use of local anesthetics, with these anticonvulsant drugs. Supportive treatment of circulatory depression may require administration of intravenous fluids and, when appropriate, a vasopressor as directed by the clinical situation (e.g., ephedrine).
If not treated immediately, both convulsions and cardiovascular depression can result in hypoxia, acidosis, bradycardia, arrhythmias and cardiac arrest. Underventilation or apnea due to unintentional subarachnoid injection of local anesthetic solution may produce these same signs and also lead to cardiac arrest if ventilatory support is not instituted. If cardiac arrest should occur standard cardiopulmonary resuscitative measures should be instituted.
Endotracheal intubation, employing drugs and techniques familiar to the clinician, may be indicated, after initial administration of oxygen by mask, if difficulty is encountered in the maintenance of a patent airway or if prolonged ventilatory support (assisted or controlled) is indicated.
Dialysis is of negligible value in the treatment of acute overdosage with lidocaine.
The oral LD 50 of lidocaine HCl in non-fasted female rats is 459 (346−773) mg/kg (as the salt) and 214 (159−324) mg/kg (as the salt) in fasted female rats.
-
DOSAGE AND ADMINISTRATION
Table 1 (Recommended Dosages) summarizes the recommended volumes and concentrations of Lidocaine Hydrochloride Injection, USP for various types of anesthetic procedures. The dosages suggested in this table are for normal healthy adults and refer to the use of epinephrine-free solutions. When larger volumes are required only solutions containing epinephrine should be used, except in those cases where vasopressor drugs may be contraindicated.
There have been adverse event reports of chondrolysis in patients receiving intra-articular infusions of local anesthetics following arthroscopic and other surgical procedures. Lidocaine is not approved for this use (see WARNINGS and DOSAGE AND ADMINISTRATION).
These recommended doses serve only as a guide to the amount of anesthetic required for most routine procedures. The actual volumes and concentrations to be used depend on a number of factors such as type and extent of surgical procedure, depth of anesthesia and degree of muscular relaxation required, duration of anesthesia required, and the physical condition of the patient. In all cases the lowest concentration and smallest dose that will produce the desired result should be given. Dosages should be reduced for children and for elderly and debilitated patients and patients with cardiac and/or liver disease.
The onset of anesthesia, the duration of anesthesia and the degree of muscular relaxation are proportional to the volume and concentration (i.e., total dose) of local anesthetic used. Thus, an increase in volume and concentration of Lidocaine Hydrochloride Injection will decrease the onset of anesthesia, prolong the duration of anesthesia, provide a greater degree of muscular relaxation and increase the segmental spread of anesthesia. However, increasing the volume and concentration of Lidocaine Hydrochloride Injection may result in a more profound fall in blood pressure when used in epidural anesthesia. Although the incidence of side effects with lidocaine is quite low, caution should be exercised when employing large volumes and concentrations, since the incidence of side effects is directly proportional to the total dose of local anesthetic agent injected.
For intravenous regional anesthesia, only the 50 mL single-dose vial containing 0.5% Lidocaine Hydrochloride Injection, USP should be used.
Epidural Anesthesia
For epidural anesthesia, only the following available specific products of Lidocaine Hydrochloride Injection by Hospira are recommended:
1%. . . . . . . . . . . . . . . . . . . . 30 mL single-dose teartop vials
1.5%. . . . . . . . . . . . . . . . . . . . . . . 20 mL single-dose ampuls
2%. . . . . . . . . . . . . . . . . . . . . . . . . 10 mL single-dose ampuls
Although these solutions are intended specifically for epidural anesthesia, they may also be used for infiltration and peripheral nerve block provided they are employed as single dose units. These solutions contain no bacteriostatic agent. In epidural anesthesia, the dosage varies with the number of dermatomes to be anesthetized (generally 2−3 mL of the indicated concentration per dermatome).
Caudal and Lumbar Epidural Block: As a precaution against the adverse experiences sometimes observed following unintentional penetration of the subarachnoid space, a test dose such as 2−3 mL of 1.5% lidocaine hydrochloride should be administered at least 5 minutes prior to injecting the total volume required for a lumbar or caudal epidural block. The test dose should be repeated if the patient is moved in a manner that may have displaced the catheter. Epinephrine, if contained in the test dose (10−15 mcg have been suggested), may serve as a warning of unintentional intravascular injection. If injected into a blood vessel, this amount of epinephrine is likely to produce a transient "epinephrine response" within 45 seconds, consisting of an increase in heart rate and systolic blood pressure, circumoral pallor, palpitations and nervousness in the unsedated patient. The sedated patient may exhibit only a pulse rate increase of 20 or more beats per minute for 15 or more seconds. Patients on beta-blockers may not manifest changes in heart rate, but blood pressure monitoring can detect an evanescent rise in systolic blood pressure. Adequate time should be allowed for onset of anesthesia after administration of each test dose. The rapid injection of a large volume of Lidocaine Hydrochloride Injection through the catheter should be avoided, and, when feasible, fractional doses should be administered.
In the event of the known injection of a large volume of local anesthetic solutions into the subarachnoid space, after suitable resuscitation and if the catheter is in place, consider attempting the recovery of drug by draining a moderate amount of cerebrospinal fluid (such as 10 mL) through the epidural catheter.
Maximum Recommended Dosages
NOTE: The products accompanying this insert do not contain epinephrine.
Adults: For normal healthy adults, the individual maximum recommended dose of lidocaine HCl with epinephrine should not exceed 7 mg/kg (3.5 mg/lb) of body weight and in general it is recommended that the maximum total dose not exceed 500 mg. When used without epinephrine, the maximum individual dose should not exceed 4.5 mg/kg (2 mg/lb) of body weight and in general it is recommended that the maximum total dose does not exceed 300 mg. For continuous epidural or caudal anesthesia, the maximum recommended dosage should not be administered at intervals of less than 90 minutes. When continuous lumbar or caudal epidural anesthesia is used for non-obstetrical procedures, more drug may be administered if required to produce adequate anesthesia.
The maximum recommended dose per 90 minute period of lidocaine hydrochloride for paracervical block in obstetrical patients and non-obstetrical patients is 200 mg total. One-half of the total dose is usually administered to each side. Inject slowly five minutes between sides. (See also discussion of paracervical block in PRECAUTIONS).
For intravenous regional anesthesia, the dose administered should not exceed 4 mg/kg in adults.
Children: It is difficult to recommend a maximum dose of any drug for children, since this varies as a function of age and weight. For children over 3 years of age who have a normal lean body mass and normal body development, the maximum dose is determined by the child’s age and weight. For example, in a child of 5 years weighing 50 lbs., the dose of lidocaine HCl should not exceed 75 — 100 mg (1.5 — 2 mg/lb). The use of even more dilute solutions (i.e., 0.25 — 0.5%) and total dosages not to exceed 3 mg/kg (1.4 mg/lb) are recommended for induction of intravenous regional anesthesia in children.
In order to guard against systemic toxicity, the lowest effective concentration and lowest effective dose should be used at all times. In some cases it will be necessary to dilute available concentrations with 0.9% sodium chloride injection in order to obtain the required final concentration.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever the solution and container permit. Solutions that are discolored and/or contain particulate matter should not be used.
Table 1
Recommended Dosages of Lidocaine Hydrochloride Injection, USP for Various Anesthetic
Procedures in Normal Healthy Adults
Lidocaine Hydrochloride Injection, USP (without Epinephrine)
Procedure
Conc. (%)
Vol. (mL)
Total Dose (mg)
Infiltration
Percutaneous
0.5 or 1.0
1−60
5−300
Intravenous Regional
0.5
10−60
50−300
Peripheral Nerve Blocks, e.g.
Brachial
1.5
15−20
225−300
Dental
2.0
1−5
20−100
Intercostal
1.0
3
30
Paravertebral
1.0
3−5
30−50
Pudendal (each side)
1.0
10
100
Paracervical
Obstetrical Analgesia
(each side)
1.0
10
100
Sympathetic Nerve Blocks, e.g.
Cervical (stellate ganglion)
1.0
5
50
Lumbar
1.0
5−10
50−100
Central Neural Blocks
Epidural*
Thoracic
1.0
20−30
200−300
Lumbar
Analgesia
1.0
25−30
250−300
Anesthesia
1.5
15−20
225−300
2.0
10−15
200−300
Caudal
Obstetrical Analgesia
1.0
20−30
200−300
Surgical Anesthesia
1.5
15−20
225−300
*Dose determined by number of dermatomes to be anesthetized (2 to 3 mL/dermatome).
THE ABOVE SUGGESTED CONCENTRATIONS AND VOLUMES SERVE ONLY AS A GUIDE. OTHER VOLUMES AND CONCENTRATIONS MAY BE USED PROVIDED THE TOTAL MAXIMUM RECOMMENDED DOSE IS NOT EXCEEDED.
Sterilization, Storage and Technical Procedures: Disinfecting agents containing heavy metals, which cause release of respective ions (mercury, zinc, copper, etc.) should not be used for skin or mucous membrane disinfection as they have been related to incidence of swelling and edema. When chemical disinfection of multi-dose vials is desired, either isopropyl alcohol (91%) or 70% ethyl alcohol is recommended. Many commercially available brands of rubbing alcohol, as well as solutions of ethyl alcohol not of USP grade, contain denaturants which are injurious to rubber and, therefore, are not to be used. It is recommended that chemical disinfection be accomplished by wiping the vial stopper thoroughly with cotton or gauze that has been moistened with the recommended alcohol just prior to use.
-
HOW SUPPLIED
Lidocaine Hydrochloride Injection, USP is supplied as follows:
Single-dose products are preservative-free.
NDC 55150-0159-74 Injection 5mL
Store at 20 to 25°C (68 to 77°F). [See USP Controlled Room Temperature.]
Lidocaine Hydrochloride Injection, USP solutions packaged in ampuls and glass teartop vials may be autoclaved one time only. Autoclave at 15 pounds pressure, 121°C (250°F) for 15 minutes. DO NOT AUTOCLAVE PRODUCT IN PLASTIC VIALS.
Revised: February, 2010
Printed in USA EN-2421
Hospira, Inc., Lake Forest, IL 60045 USA
- DEPO-MEDROL Principal Display Panel – Vial Label
- LIDOCAINE INJECTION
- NDC 70859-038-01
-
INGREDIENTS AND APPEARANCE
POINT OF CARE L5
depo-medrol, lidocaine hci, isopropyl alcohol kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70859-038 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70859-038-01 1 in 1 CARTON; Type 1: Convenience Kit of Co-Package 08/07/2018 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 2 VIAL, SINGLE-DOSE 2 mL Part 2 2 AMPULE 10 mL Part 3 3 POUCH 15 mL Part 1 of 3 DEPO-MEDROL
methylprednisolone acetate injection, suspensionProduct Information Item Code (Source) NDC:0009-3073 Route of Administration INTRAMUSCULAR, INTRA-ARTICULAR, INTRALESIONAL, SOFT TISSUE Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METHYLPREDNISOLONE ACETATE (UNII: 43502P7F0P) (METHYLPREDNISOLONE - UNII:X4W7ZR7023) METHYLPREDNISOLONE ACETATE 40 mg in 1 mL Inactive Ingredients Ingredient Name Strength POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) 29 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0009-3073-01 1 in 1 PACKAGE 1 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA011757 05/28/1959 Part 2 of 3 LIDOCAINE HYDROCHLORIDE
lidocaine hydrochloride injection, solutionProduct Information Item Code (Source) NDC:55150-159 Route of Administration INFILTRATION Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LIDOCAINE HYDROCHLORIDE (UNII: V13007Z41A) (LIDOCAINE - UNII:98PI200987) LIDOCAINE HYDROCHLORIDE ANHYDROUS 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 7 mg in 1 mL WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 5 mL in 1 AMPULE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA203040 03/14/2010 Part 3 of 3 ISOPROPYL ALCOHOL
isopropyl alcohol dressingProduct Information Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ISOPROPYL ALCOHOL (UNII: ND2M416302) (ISOPROPYL ALCOHOL - UNII:ND2M416302) ISOPROPYL ALCOHOL 70 mL in 100 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 5 mL in 1 POUCH; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date OTC monograph not final part333A 01/01/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA011757 05/28/1959 Labeler - NuCare Pharmaceuticals,Inc. (010632300) Establishment Name Address ID/FEI Business Operations NuCare Pharmaceuticals,Inc. 010632300 manufacture(70859-038)