Label: MELPHALAN HYDROCHLORIDE kit

- NDC Code(s): 45963-686-02, 45963-687-49, 45963-688-57
- Packager: Actavis Pharma, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated July 27, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
WARNING
Melphalan Hydrochloride for Injection should be administered under the supervision of a qualified physician experienced in the use of cancer chemotherapeutic agents. Severe bone marrow suppression with resulting infection or bleeding may occur. Controlled trials comparing intravenous to oral melphalan have shown more myelosuppression with the intravenous formulation. Hypersensitivity reactions, including anaphylaxis, have occurred in approximately 2% of patients who received the intravenous formulation. Melphalan is leukemogenic in humans. Melphalan produces chromosomal aberrations in vitro and in vivo and, therefore, should be considered potentially mutagenic in humans.
-
DESCRIPTION
Melphalan, also known as L-phenylalanine mustard, phenylalanine mustard, L-PAM, or L-sarcolysin, is a phenylalanine derivative of nitrogen mustard. Melphalan is a bifunctional alkylating agent that is active against selected human neoplastic diseases. It is known chemically as 4-[bis(2-chloroethyl)amino]-L-phenylalanine. The molecular formula is C13H18Cl2N2O2 and the molecular weight is 305.20. The structural formula is:
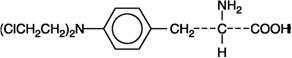
Melphalan is the active L-isomer of the compound and was first synthesized in 1953 by Bergel and Stock; the D-isomer, known as medphalan, is less active against certain animal tumors, and the dose needed to produce effects on chromosomes is larger than that required with the L-isomer. The racemic (DL-) form is known as merphalan or sarcolysin.
Melphalan is practically insoluble in water and has a pKa1 of ~2.5.
Melphalan Hydrochloride for Injection is supplied as a sterile, nonpyrogenic, freeze-dried powder. Each single-dose vial contains melphalan hydrochloride equivalent to 50 mg melphalan and 20 mg povidone K12. Melphalan Hydrochloride for Injection is reconstituted using the sterile diluent provided. Each vial of sterile diluent contains sodium citrate 0.2 g, propylene glycol 6.0 mL, dehydrated alcohol 0.51 mL, and Water for Injection to a total of 10 mL. Melphalan Hydrochloride for Injection is administered intravenously.
-
CLINICAL PHARMACOLOGY
Melphalan is an alkylating agent of the bischloroethylamine type. As a result, its cytotoxicity appears to be related to the extent of its interstrand cross-linking with DNA, probably by binding at the N7 position of guanine. Like other bifunctional alkylating agents, it is active against both resting and rapidly dividing tumor cells.
Pharmacokinetics
Following injection, drug plasma concentrations declined rapidly in a biexponential manner with distribution phase and terminal elimination phase half-lives of approximately 10 and 75 minutes, respectively. Estimates of average total body clearance varied among studies, but typical values of approximately 7 to 9 mL/min/kg (250 to 325 mL/min/m2) were observed. One study has reported that on repeat dosing of 0.5 mg/kg every 6 weeks, the clearance of melphalan decreased from 8.1 mL/min/kg after the first course, to 5.5 mL/min/kg after the third course, but did not decrease appreciably after the third course. Mean (±SD) peak melphalan plasma concentrations in myeloma patients given intravenous melphalan at doses of 10 or 20 mg/m2 were 1.2 ± 0.4 and 2.8 ± 1.9 mcg/mL, respectively.
The steady-state volume of distribution of melphalan is 0.5 L/kg. Penetration into cerebrospinal fluid (CSF) is low. The average melphalan binding to plasma proteins is highly variable (range: 53% to 92%). Serum albumin is the major binding protein, accounting for approximately 40% to 60% of the plasma protein binding, while α1-acid glycoprotein accounts for about 20% of the plasma protein binding. Approximately 30% of melphalan is (covalently) irreversibly bound to plasma proteins. Interactions with immunoglobulins have been found to be negligible.
Melphalan is eliminated from plasma primarily by chemical hydrolysis to monohydroxymelphalan and dihydroxymelphalan. Aside from these hydrolysis products, no other melphalan metabolites have been observed in humans. Although the contribution of renal elimination to melphalan clearance appears to be low, one study noted an increase in the occurrence of severe leukopenia in patients with elevated BUN after 10 weeks of therapy.
-
Clinical Trial
A randomized trial compared prednisone plus intravenous melphalan to prednisone plus oral melphalan in the treatment of myeloma. As discussed below, overall response rates at week 22 were comparable; however, because of changes in trial design, conclusions as to the relative activity of the 2 formulations after week 22 are impossible to make.
Both arms received oral prednisone starting at 0.8 mg/kg/day with doses tapered over 6 weeks. Melphalan doses in each arm were:
Arm 1: Oral melphalan 0.15 mg/kg/day x 7 followed by 0.05 mg/kg/day when WBC began to rise.
Arm 2: Intravenous melphalan 16 mg/m2 q 2 weeks x 4 (over 6 weeks) followed by the same dose every 4 weeks.
Doses of melphalan were adjusted according to the following criteria:
Table 1. Criteria for Dosage Adjustment in a Randomized Clinical Trial WBC/mm3 Platelets Percent of Full Dose ≥4,000 ≥100,000 100 ≥3,000 ≥75,000 75 ≥2,000 ≥50,000 50 <2,000 <50,000 0 One hundred seven patients were randomized to the oral melphalan arm and 203 patients to the intravenous melphalan arm. More patients had a poor-risk classification (58% versus 44%) and high tumor load (51% versus 34%) on the oral compared to the intravenous arm (P<0.04). Response rates at week 22 are shown in the following table:
Table 2. Response Rates at Week 22 Evaluable Responders Initial Arm Patients n (%) P Oral melphalan 100 44 (44%) P>0.2 Intravenous melphalan 195 74 (38%) Because of changes in protocol design after week 22, other efficacy parameters such as response duration and survival cannot be compared.
Severe myelotoxicity (WBC ≤1,000 and/or platelets ≤25,000) was more common in the intravenous melphalan arm (28%) than in the oral melphalan arm (11%).
An association was noted between poor renal function and myelosuppression; consequently, an amendment to the protocol required a 50% reduction in intravenous melphalan dose if the BUN was ≥30 mg/dL. The rate of severe leukopenia in the intravenous arm in the patients with BUN over 30 mg/dL decreased from 50% (8/16) before protocol amendment to 11% (3/28) (P = 0.01) after the amendment.
Before the dosing amendment, there was a 10% (8/77) incidence of drug-related death in the intravenous arm. After the dosing amendment, this incidence was 3% (3/108). This compares to an overall 1% (1/100) incidence of drug-related death in the oral arm.
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
Melphalan Hydrochloride for Injection may cause local tissue damage should extravasation occur, and consequently it should not be administered by direct injection into a peripheral vein. It is recommended that Melphalan Hydrochloride for Injection be administered by injecting slowly into a fast-running intravenous infusion via an injection port, or via a central venous line (see DOSAGE AND ADMINISTRATION: Administration Precautions).
Melphalan Hydrochloride for Injection should be administered in carefully adjusted dosage by or under the supervision of experienced physicians who are familiar with the drug's actions and the possible complications of its use.
As with other nitrogen mustard drugs, excessive dosage will produce marked bone marrow suppression. Bone marrow suppression is the most significant toxicity associated with Melphalan Hydrochloride for Injection in most patients. Therefore, the following tests should be performed at the start of therapy and prior to each subsequent dose of Melphalan Hydrochloride for Injection: platelet count, hemoglobin, white blood cell count, and differential. Thrombocytopenia and/or leukopenia are indications to withhold further therapy until the blood counts have sufficiently recovered. Frequent blood counts are essential to determine optimal dosage and to avoid toxicity. Dose adjustment on the basis of blood counts at the nadir and day of treatment should be considered.
Hypersensitivity reactions including anaphylaxis have occurred in approximately 2% of patients who received the intravenous formulation (see ADVERSE REACTIONS). These reactions usually occur after multiple courses of treatment. Treatment is symptomatic. The infusion should be terminated immediately, followed by the administration of volume expanders, pressor agents, corticosteroids, or antihistamines at the discretion of the physician. If a hypersensitivity reaction occurs, intravenous or oral melphalan should not be readministered since hypersensitivity reactions have also been reported with oral melphalan.
Carcinogenesis
Secondary malignancies, including acute nonlymphocytic leukemia, myeloproliferative syndrome, and carcinoma, have been reported in patients with cancer treated with alkylating agents (including melphalan). Some patients also received other chemotherapeutic agents or radiation therapy. Precise quantitation of the risk of acute leukemia, myeloproliferative syndrome, or carcinoma is not possible. Published reports of leukemia in patients who have received melphalan (and other alkylating agents) suggest that the risk of leukemogenesis increases with chronicity of treatment and with cumulative dose. In one study, the 10-year cumulative risk of developing acute leukemia or myeloproliferative syndrome after oral melphalan therapy was 19.5% for cumulative doses ranging from 730 to 9,652 mg. In this same study, as well as in an additional study, the 10-year cumulative risk of developing acute leukemia or myeloproliferative syndrome after oral melphalan therapy was less than 2% for cumulative doses under 600 mg. This does not mean that there is a cumulative dose below which there is no risk of the induction of secondary malignancy. The potential benefits from melphalan therapy must be weighed on an individual basis against the possible risk of the induction of a second malignancy.
Adequate and well-controlled carcinogenicity studies have not been conducted in animals. However, intraperitoneal (IP) administration of melphalan in rats (5.4 to 10.8 mg/m2) and in mice (2.25 to 4.5 mg/m2) 3 times per week for 6 months followed by 12 months post-dose observation produced peritoneal sarcoma and lung tumors, respectively.
Mutagenesis
Melphalan has been shown to cause chromatid or chromosome damage in humans. Intramuscular administration of melphalan at 6 and 60 mg/m2 produced structural aberrations of the chromatid and chromosomes in bone marrow cells of Wistar rats.
Impairment of Fertility
Melphalan causes suppression of ovarian function in premenopausal women, resulting in amenorrhea in a significant number of patients. Reversible and irreversible testicular suppression have also been reported.
Pregnancy
Pregnancy Category D. Melphalan Hydrochloride for Injection may cause fetal harm when administered to a pregnant woman. While adequate animal studies have not been conducted with intravenous melphalan, oral (6 to 18 mg/m2/day for 10 days) and IP (18 mg/m2) administration in rats was embryolethal and teratogenic. Malformations resulting from melphalan included alterations of the brain (underdevelopment, deformation, meningocele, and encephalocele) and eye (anophthalmia and microphthalmos), reduction of the mandible and tail, as well as hepatocele (exomphaly). There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
-
PRECAUTIONS
General
In all instances where the use of Melphalan Hydrochloride for Injection is considered for chemotherapy, the physician must evaluate the need and usefulness of the drug against the risk of adverse events. Melphalan Hydrochloride for Injection should be used with extreme caution in patients whose bone marrow reserve may have been compromised by prior irradiation or chemotherapy or whose marrow function is recovering from previous cytotoxic therapy.
Dose reduction should be considered in patients with renal insufficiency receiving intravenous melphalan. In one trial, increased bone marrow suppression was observed in patients with BUN levels ≥30 mg/dL. A 50% reduction in the intravenous melphalan dose decreased the incidence of severe bone marrow suppression in the latter portion of this study.
Administration of live vaccines to immunocompromised patients should be avoided.
Information for Patients
Patients should be informed that the major acute toxicities of melphalan are related to bone marrow suppression, hypersensitivity reactions, gastrointestinal toxicity, and pulmonary toxicity. The major long-term toxicities are related to infertility and secondary malignancies. Patients should never be allowed to take the drug without close medical supervision and should be advised to consult their physicians if they experience skin rash, signs or symptoms of vasculitis, bleeding, fever, persistent cough, nausea, vomiting, amenorrhea, weight loss, or unusual lumps/masses. Women of childbearing potential should be advised to avoid becoming pregnant.
Laboratory Tests
Periodic complete blood counts with differentials should be performed during the course of treatment with Melphalan Hydrochloride for Injection. At least 1 determination should be obtained prior to each dose. Patients should be observed closely for consequences of bone marrow suppression, which include severe infections, bleeding, and symptomatic anemia (see WARNINGS).
Drug Interactions
The development of severe renal failure has been reported in patients treated with a single dose of intravenous melphalan followed by standard oral doses of cyclosporine. Cisplatin may affect melphalan kinetics by inducing renal dysfunction and subsequently altering melphalan clearance. Intravenous melphalan may also reduce the threshold for BCNU lung toxicity. When nalidixic acid and intravenous melphalan are given simultaneously, the incidence of severe hemorrhagic necrotic enterocolitis has been reported to increase in pediatric patients.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Intravenous melphalan should not be given to nursing mothers.
Geriatric Use
Clinical studies of Melphalan Hydrochloride for Injection did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
(SEE OVERDOSAGE)
The following information on adverse reactions is based on data from both oral and intravenous administration of melphalan as a single agent, using several different dose schedules for treatment of a wide variety of malignancies.
Hematologic
The most common side effect is bone marrow suppression leading to leukopenia, thrombocytopenia, and anemia. White blood cell count and platelet count nadirs usually occur 2 to 3 weeks after treatment, with recovery in 4 to 5 weeks after treatment. Irreversible bone marrow failure has been reported.
Gastrointestinal
Gastrointestinal disturbances such as nausea and vomiting, diarrhea, and oral ulceration occur infrequently. Hepatic disorders ranging from abnormal liver function tests to clinical manifestations such as hepatitis and jaundice have been reported. Hepatic veno-occlusive disease has been reported.
Hypersensitivity
Acute hypersensitivity reactions including anaphylaxis were reported in 2.4% of 425 patients receiving Melphalan Hydrochloride for Injection for myeloma (see WARNINGS). These reactions were characterized by urticaria, pruritus, edema, skin rashes, and in some patients, tachycardia, bronchospasm, dyspnea, and hypotension. These patients appeared to respond to antihistamine and corticosteroid therapy. If a hypersensitivity reaction occurs, intravenous or oral melphalan should not be readministered since hypersensitivity reactions have also been reported with oral melphalan. Cardiac arrest has also been reported rarely in association with such reports.
Miscellaneous
Other reported adverse reactions include skin hypersensitivity, skin ulceration at injection site, skin necrosis rarely requiring skin grafting, maculopapular rashes, vasculitis, alopecia, hemolytic anemia, allergic reaction, pulmonary fibrosis (including fatal outcomes), and interstitial pneumonitis. Temporary significant elevation of the blood urea has been seen in the early stages of therapy in patients with renal damage. Subjective and transient sensation of warmth and/or tingling.
-
OVERDOSAGE
Overdoses resulting in death have been reported. Overdoses, including doses up to 290 mg/m2, have produced the following symptoms: severe nausea and vomiting, decreased consciousness, convulsions, muscular paralysis, and cholinomimetic effects. Severe mucositis, stomatitis, colitis, diarrhea, and hemorrhage of the gastrointestinal tract occur at high doses (>100 mg/m2). Elevations in liver enzymes and veno-occlusive disease occur infrequently. Significant hyponatremia caused by an associated inappropriate secretion of ADH syndrome has been observed. Nephrotoxicity and adult respiratory distress syndrome have been reported rarely. The principal toxic effect is bone marrow suppression. Hematologic parameters should be closely followed for 3 to 6 weeks. An uncontrolled study suggests that administration of autologous bone marrow or hematopoietic growth factors (i.e., sargramostim, filgrastim) may shorten the period of pancytopenia. General supportive measures together with appropriate blood transfusions and antibiotics should be instituted as deemed necessary by the physician. This drug is not removed from plasma to any significant degree by hemodialysis or hemoperfusion. A pediatric patient survived a 254-mg/m2 overdose treated with standard supportive care.
-
DOSAGE AND ADMINISTRATION
The usual intravenous dose is 16 mg/m2. Dosage reduction of up to 50% should be considered in patients with renal insufficiency (BUN ≥ 30 mg/dL) (see PRECAUTIONS: General). The drug is administered as a single infusion over 15 to 20 minutes. Melphalan Hydrochloride for Injection is administered at 2-week intervals for 4 doses, then, after adequate recovery from toxicity, at 4-week intervals. Available evidence suggests about one third to one half of the patients with multiple myeloma show a favorable response to the drug. Experience with oral melphalan suggests that repeated courses should be given since improvement may continue slowly over many months, and the maximum benefit may be missed if treatment is abandoned prematurely. Dose adjustment on the basis of blood cell counts at the nadir and day of treatment should be considered.
Administration Precautions
As with other toxic compounds, caution should be exercised in handling and preparing the solution of Melphalan Hydrochloride for Injection. Skin reactions associated with accidental exposure may occur. The use of gloves is recommended. If the solution of Melphalan Hydrochloride for Injection contacts the skin or mucosa, immediately wash the skin or mucosa thoroughly with soap and water.
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published.1-4 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
Parenteral drug products should be visually inspected for particulate matter and discoloration prior to administration whenever solution and container permit. If either occurs, do not use this product.
Care should be taken to avoid possible extravasation of melphalan and in cases of poor peripheral venous access, consideration should be given to use of a central venous line (see WARNINGS).
Preparation for Administration/Stability
- Melphalan Hydrochloride for Injection must be reconstituted by rapidly injecting 10 mL of the supplied diluent directly into the vial of lyophilized powder using a sterile needle (20-gauge or larger needle diameter) and syringe. Immediately shake vial vigorously until a clear solution is obtained. This provides a 5-mg/mL solution of melphalan. Rapid addition of the diluent followed by immediate vigorous shaking is important for proper dissolution.
- Immediately dilute the dose to be administered in 0.9% Sodium Chloride Injection, USP, to a concentration not greater than 0.45 mg/mL.
- Administer the diluted product over a minimum of 15 minutes.
- Complete administration within 60 minutes of reconstitution.
The time between reconstitution/dilution and administration of Melphalan Hydrochloride for Injection should be kept to a minimum because reconstituted and diluted solutions of Melphalan Hydrochloride for Injection are unstable. Over as short a time as 30 minutes, a citrate derivative of melphalan has been detected in reconstituted material from the reaction of melphalan hydrochloride with Sterile Diluent for Melphalan Hydrochloride for Injection. Upon further dilution with saline, nearly 1% label strength of melphalan hydrolyzes every 10 minutes.
A precipitate forms if the reconstituted solution is stored at 5°C. DO NOT REFRIGERATE THE RECONSTITUTED PRODUCT.
-
HOW SUPPLIED
Melphalan Hydrochloride for Injection is supplied in a carton containing one single-dose clear glass vial of freeze-dried melphalan hydrochloride equivalent to 50 mg melphalan and one 10-mL clear glass vial of sterile diluent (NDC 45963-686-02).
Store at controlled room temperature 15° to 30°C (59° to 86°F) and protect from light.
Retain in carton.
Discard unused portion.
Sterile, Nonpyrogenic, Preservative-free
The vial stopper is not made with natural rubber latex.
-
REFERENCES
- NIOSH Alert: Preventing Occupational Exposures to Antineoplastic and Other Hazardous Drugs in Healthcare Settings. U.S. Department of Health and Human Services, Public Health Service. Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
- American Society of Health-System Pharmacists. (2006) ASHP Guidelines on Handling Hazardous Drugs. Am J Health-Syst Pharm. 2006;63:1172-1193.
- Polovich M, White JM, Kelleher LO (eds.) 2005. Chemotherapy and Biotherapy Guidelines and Recommendations for Practice. (2nd ed.) Pittsburgh, PA: Oncology Nursing Society.
Manufactured In Romania By:
Sindan Pharma SRL
11 Ion Mihalache Blvd.
Bucharest 1, Romania 011171Manufactured For:
Teva Pharmaceuticals
Parsippany, NJ 07054Rev. B 7/2022
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
NDC 45963-686-02
SINGLE-DOSE
Melphalan Hydrochloride for Injection
50 mg*
For Intravenous Infusion
*One vial of Melphalan Hydrochloride for Injection containing sterile, nonpyrogenic, freeze-dried melphalan hydrochloride equivalent to 50 mg melphalan and 20 mg povidone K12.
One vial of sterile, nonpyrogenic diluent containing 0.2 g sodium citrate, 6.0 mL propylene glycol, 0.51 mL dehydrated alcohol, and Water for Injection to a total of 10 mL.CAUTION: Cytotoxic Agent
Rx only

-
INGREDIENTS AND APPEARANCE
MELPHALAN HYDROCHLORIDE
melphalan hydrochloride kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:45963-686 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:45963-686-02 1 in 1 PACKAGE, COMBINATION; Type 0: Not a Combination Product 01/13/2017 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-DOSE 10 mL Part 2 1 VIAL, SINGLE-USE 10 mL Part 1 of 2 MELPHALAN HYDROCHLORIDE
melphalan hydrochloride injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC:45963-687 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MELPHALAN HYDROCHLORIDE (UNII: 1VXP4V453T) (MELPHALAN - UNII:Q41OR9510P) MELPHALAN 50 mg in 10 mL Inactive Ingredients Ingredient Name Strength POVIDONE K12 (UNII: 333AG72FWJ) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:45963-687-49 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA206018 01/13/2017 Part 2 of 2 DILUENT
diluent injectionProduct Information Item Code (Source) NDC:45963-688 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) SODIUM CITRATE (UNII: 1Q73Q2JULR) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) ALCOHOL (UNII: 3K9958V90M) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:45963-688-57 10 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA206018 01/13/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA206018 01/13/2017 Labeler - Actavis Pharma, Inc. (119723554)