Label: SOLU-MEDROL- methylprednisolone sodium succinate injection, powder, for solution

- NDC Code(s): 55154-3944-5
- Packager: Cardinal Health 107, LLC
- This is a repackaged label.
- Source NDC Code(s): 0009-0047
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated March 19, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
SOLU-MEDROL Sterile Powder is an anti-inflammatory glucocorticoid, which contains methylprednisolone sodium succinate as the active ingredient. Methylprednisolone sodium succinate, USP, is the sodium succinate ester of methylprednisolone, and it occurs as a white, or nearly white, odorless hygroscopic, amorphous solid. It is very soluble in water and in alcohol; it is insoluble in chloroform and is very slightly soluble in acetone.
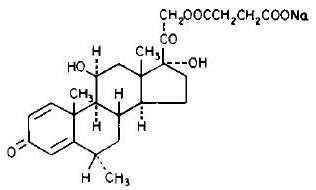
The chemical name for methylprednisolone sodium succinate is pregna-1,4-diene-3,20-dione,21-(3-carboxy-1-oxopropoxy)-11,17-dihydroxy-6-methyl-monosodium salt, (6α, 11β), and the molecular weight is 496.53. The structural formula is represented below:
Methylprednisolone sodium succinate is soluble in water; it may be administered in a small volume of diluent and is well suited for intravenous use in situations where high blood levels of methylprednisolone are required rapidly.
SOLU-MEDROL is available in preservative and preservative-free formulations:
Preservative-free Formulations 40 mg Act-O-Vial System (Single-Dose Vial)—Each mL (when mixed) contains methylprednisolone sodium succinate equivalent to 40 mg methylprednisolone; also
1.6 mg monobasic sodium phosphate anhydrous; 17.46 mg dibasic sodium phosphate dried; and 25 mg lactose hydrous.
125 mg Act-O-Vial System (Single-Dose Vial)—Each 2 mL (when mixed) contains methylprednisolone sodium succinate equivalent to 125 mg methylprednisolone; also
1.6 mg monobasic sodium phosphate anhydrous; and 17.4 mg dibasic sodium phosphate dried.500 mg Act-O-Vial System (Single-Dose Vial)—Each 4 mL (when mixed) contains methylprednisolone sodium succinate equivalent to 500 mg methylprednisolone; also
6.4 mg monobasic sodium phosphate anhydrous; and 69.6 mg dibasic sodium phosphate dried.
1 gram Act-O-Vial System (Single-Dose Vial)—Each 8 mL (when mixed) contains methylprednisolone sodium succinate equivalent to 1 gram methylprednisolone; also
12.8 mg monobasic sodium phosphate anhydrous; and 139.2 mg dibasic sodium phosphate dried.
Formulations preserved with Benzyl Alcohol 500 mg Vial—Each 8 mL (when mixed as directed) contains methylprednisolone sodium succinate equivalent to 500 mg methylprednisolone; also 6.4 mg monobasic sodium phosphate anhydrous; 69.6 mg dibasic sodium phosphate dried.
This package does not contain diluent. Recommended diluent (Bacteriostatic water) contains benzyl alcohol as a preservative.1 gram Vial—Each 16 mL (when mixed as directed) contains methylprednisolone sodium succinate equivalent to 1 gram methylprednisolone; also 12.8 mg monobasic sodium phosphate anhydrous; 139.2 mg dibasic sodium phosphate dried.
This package does not contain diluent. Recommended diluent (Bacteriostatic water) contains benzyl alcohol as a preservative.2 gram Vial—Each 30.6 mL (when mixed as directed) contains methylprednisolone sodium succinate equivalent to 2 grams methylprednisolone; also 25.6 mg monobasic sodium phosphate anhydrous; 278 mg dibasic sodium phosphate dried.
This package does not contain diluent. Recommended diluent (Bacteriostatic water) contains benzyl alcohol as a preservative.2 gram Vial with Diluent—Each 30.6 mL (when mixed as directed) contains methylprednisolone sodium succinate equivalent to 2 grams methylprednisolone; also 25.6 mg monobasic sodium phosphate anhydrous; 278 mg dibasic sodium phosphate dried.
The packaged diluent (Bacteriostatic Water for Injection) contains benzyl alcohol as a preservative.IMPORTANT — Use only the accompanying diluent
or Bacteriostatic Water For Injection with
Benzyl Alcohol when reconstituting SOLU-MEDROL.
Use within 48 hours after mixing.When necessary, the pH of each formula was adjusted with sodium hydroxide so that the pH of the reconstituted solution is within the USP specified range of 7 to 8.
-
CLINICAL PHARMACOLOGY
Glucocorticoids, naturally occurring and synthetic, are adrenocortical steroids that are readily absorbed from the gastrointestinal tract.
Naturally occurring glucocorticoids (hydrocortisone and cortisone), which also have salt-retaining properties, are used as replacement therapy in adrenocortical deficiency states. Their synthetic analogs are primarily used for their potent anti-inflammatory effects in disorders of many organ systems.
Glucocorticoids cause profound and varied metabolic effects. In addition, they modify the body's immune responses to diverse stimuli.
Methylprednisolone is a potent anti-inflammatory steroid with greater anti-inflammatory potency than prednisolone and even less tendency than prednisolone to induce sodium and water retention.
Methylprednisolone sodium succinate has the same metabolic and anti-inflammatory actions as methylprednisolone. When given parenterally and in equimolar quantities, the two compounds are equivalent in biologic activity. Following the intravenous injection of methylprednisolone sodium succinate, demonstrable effects are evident within one hour and persist for a variable period. Excretion of the administered dose is nearly complete within 12 hours. Thus, if constantly high blood levels are required, injections should be made every 4 to 6 hours. This preparation is also rapidly absorbed when administered intramuscularly and is excreted in a pattern similar to that observed after intravenous injection.
-
INDICATIONS AND USAGE
When oral therapy is not feasible, and the strength, dosage form, and route of administration of the drug reasonably lend the preparation to the treatment of the condition, the intravenous or intramuscular use of SOLU-MEDROL Sterile Powder is indicated as follows:
Allergic states
Control of severe or incapacitating allergic conditions intractable to adequate trials of conventional treatment in asthma, atopic dermatitis, contact dermatitis, drug hypersensitivity reactions, serum sickness, transfusion reactions.
Dermatologic diseases
Bullous dermatitis herpetiformis, exfoliative erythroderma, mycosis fungoides, pemphigus, severe erythema multiforme (Stevens-Johnson syndrome).
Endocrine disorders
Primary or secondary adrenocortical insufficiency (hydrocortisone or cortisone is the drug of choice; synthetic analogs may be used in conjunction with mineralocorticoids where applicable; in infancy, mineralocorticoid supplementation is of particular importance), congenital adrenal hyperplasia, hypercalcemia associated with cancer, nonsuppurative thyroiditis.
Gastrointestinal diseases
To tide the patient over a critical period of the disease in regional enteritis (systemic therapy) and ulcerative colitis.
Hematologic disorders
Acquired (autoimmune) hemolytic anemia, congenital (erythroid) hypoplastic anemia (Diamond-Blackfan anemia), idiopathic thrombocytopenic purpura in adults (intravenous administration only; intramuscular administration is contraindicated), pure red cell aplasia, selected cases of secondary thrombocytopenia.
Miscellaneous
Trichinosis with neurologic or myocardial involvement, tuberculous meningitis with subarachnoid block or impending block when used concurrently with appropriate antituberculous chemotherapy.
Nervous System
Acute exacerbations of multiple sclerosis; cerebral edema associated with primary or metastatic brain tumor, or craniotomy.
Ophthalmic diseases
Sympathetic ophthalmia, uveitis and ocular inflammatory conditions unresponsive to topical corticosteroids.
Renal diseases
To induce diuresis or remission of proteinuria in idiopathic nephrotic syndrome or that due to lupus erythematosus.
Respiratory diseases
Berylliosis, fulminating or disseminated pulmonary tuberculosis when used concurrently with appropriate antituberculous chemotherapy, idiopathic eosinophilic pneumonias, symptomatic sarcoidosis.
Rheumatic disorders
As adjunctive therapy for short-term administration (to tide the patient over an acute episode or exacerbation) in acute gouty arthritis; acute rheumatic carditis; ankylosing spondylitis; psoriatic arthritis; rheumatoid arthritis, including juvenile rheumatoid arthritis (selected cases may require low-dose maintenance therapy). For the treatment of dermatomyositis, temporal arteritis, polymyositis, and systemic lupus erythematosus.
-
CONTRAINDICATIONS
SOLU-MEDROL Sterile Powder is contraindicated:
- •
- in systemic fungal infections and patients with known hypersensitivity to the product and its constituents. The SOLU-MEDROL 40 mg presentation includes lactose monohydrate produced from cow's milk. This presentation is therefore contraindicated in patients with a known or suspected hypersensitivity to cow's milk or its components or other dairy products because it may contain trace amounts of milk ingredients.
- •
- for intrathecal administration. Reports of severe medical events have been associated with this route of administration.
Intramuscular corticosteroid preparations are contraindicated for idiopathic thrombocytopenic purpura.
Additional contraindication for the use of SOLU-MEDROL Sterile Powder preserved with benzyl alcohol:
Formulations preserved with benzyl alcohol are contraindicated for use in premature infants. (See WARNINGS and PRECAUTIONS, Pediatric Use.)
-
WARNINGS
Serious Neurologic Adverse Reactions with Epidural Administration
Serious neurologic events, some resulting in death, have been reported with epidural injection of corticosteroids. Specific events reported include, but are not limited to, spinal cord infarction, paraplegia, quadriplegia, cortical blindness, and stroke. These serious neurologic events have been reported with and without use of fluoroscopy. The safety and effectiveness of epidural administration of corticosteroids have not been established, and corticosteroids are not approved for this use.
General
Formulations with preservative (see DESCRIPTION) contain benzyl alcohol, which is potentially toxic when administered locally to neural tissue. Exposure to excessive amounts of benzyl alcohol has been associated with toxicity (hypotension, metabolic acidosis), particularly in neonates, and an increased incidence of kernicterus, particularly in small preterm infants. There have been rare reports of deaths, primarily in preterm infants, associated with exposure to excessive amounts of benzyl alcohol. The amount of benzyl alcohol from medications is usually considered negligible compared to that received in flush solutions containing benzyl alcohol. Administration of high dosages of medications containing this preservative must take into account the total amount of benzyl alcohol administered. The amount of benzyl alcohol at which toxicity may occur is not known. If the patient requires more than the recommended dosages or other medications containing this preservative, the practitioner must consider the daily metabolic load of benzyl alcohol from these combined sources (see PRECAUTIONS, Pediatric Use).
Injection of SOLU-MEDROL may result in dermal and/or subdermal changes forming depressions in the skin at the injection site. In order to minimize the incidence of dermal and subdermal atrophy, care must be exercised not to exceed recommended doses in injections. Injection into the deltoid muscle should be avoided because of a high incidence of subcutaneous atrophy.
Rare instances of anaphylactoid reactions have occurred in patients receiving corticosteroid therapy (see ADVERSE REACTIONS).
In patients receiving the 40 mg presentation of SOLU-MEDROL during the treatment for acute allergic conditions and where these symptoms worsen or any new allergic symptoms occur, consideration should be given to the potential for hypersensitivity reactions to cow's milk ingredients (see CONTRAINDICATIONS). If appropriate, administration of SOLU-MEDROL should be stopped, and the patient's condition should be treated accordingly. Alternative treatments, including the use of corticosteroid formulations that do not contain ingredients produced from cow's milk, should be considered for acute allergy management, where appropriate.
Increased dosage of rapidly acting corticosteroids is indicated in patients on corticosteroid therapy who are subjected to any unusual stress before, during, and after the stressful situation.
Results from one multicenter, randomized, placebo-controlled study with methylprednisolone hemisuccinate, an intravenous corticosteroid, showed an increase in early (at 2 weeks) and late (at 6 months) mortality in patients with cranial trauma who were determined not to have other clear indications for corticosteroid treatment. High doses of systemic corticosteroids, including SOLU-MEDROL, should not be used for the treatment of traumatic brain injury.
Cardio-renal
Average and large doses of corticosteroids can cause elevation of blood pressure, salt and water retention, and increased excretion of potassium. These effects are less likely to occur with the synthetic derivatives except when used in large doses. Dietary salt restriction and potassium supplementation may be necessary. All corticosteroids increase calcium excretion.
Literature reports suggest an apparent association between the use of corticosteroids and left ventricular free wall rupture after a recent myocardial infarction; therefore, therapy with corticosteroids should be used with great caution in these patients.
Endocrine
Hypothalamic-pituitary adrenal (HPA) axis suppression, Cushing's syndrome, and hyperglycemia. Monitor patients for these conditions with chronic use.
Corticosteroids can produce reversible HPA axis suppression with the potential for glucocorticosteroid insufficiency after withdrawal of treatment. Drug induced secondary adrenocortical insufficiency may be minimized by gradual reduction of dosage. This type of relative insufficiency may persist for months after discontinuation of therapy; therefore, in any situation of stress occurring during that period, hormone therapy should be reinstituted.
Drug-Induced Liver Injury
Rarely, high doses of cyclically pulsed intravenous methylprednisolone (usually for the treatment of exacerbations of multiple sclerosis at doses of 1 gram/day) can induce a toxic form of acute hepatitis. The time to onset of this form of steroid-induced liver injury can be several weeks or longer. Resolution has been observed after discontinuation of treatment. However, serious liver injury can occur, sometimes resulting in acute liver failure and death. Discontinue intravenous methylprednisolone if toxic hepatitis occurs. Since recurrence has occurred after re-challenge, avoid use of high dose intravenous methylprednisolone in patients with a history of toxic hepatitis caused by methylprednisolone.
Infections
General
Patients who are on corticosteroids are more susceptible to infections than are healthy individuals. There may be decreased resistance and inability to localize infection when corticosteroids are used. Infections with any pathogen (viral, bacterial, fungal, protozoan, or helminthic) in any location of the body may be associated with the use of corticosteroids alone or in combination with other immunosuppressive agents.
These infections may be mild, but can be severe and at times fatal. With increasing doses of corticosteroids, the rate of occurrence of infectious complications increases. Corticosteroids may also mask some signs of current infection. Do not use intra-articularly, intrabursally or for intratendinous administration for local effect in the presence of acute local infection.
A study has failed to establish the efficacy of methylprednisolone sodium succinate in the treatment of sepsis syndrome and septic shock. The study also suggests that treatment of these conditions with methylprednisolone sodium succinate may increase the risk of mortality in certain patients (i.e., patients with elevated serum creatinine levels or patients who develop secondary infections after methylprednisolone sodium succinate).
Fungal infections
Corticosteroids may exacerbate systemic fungal infections and therefore should not be used in the presence of such infections unless they are needed to control drug reactions. There have been cases reported in which concomitant use of amphotericin B and hydrocortisone was followed by cardiac enlargement and congestive heart failure (see CONTRAINDICATIONS and PRECAUTIONS, Drug Interactions, Amphotericin B injection and potassium-depleting agents).
Special pathogens
Latent disease may be activated or there may be an exacerbation of intercurrent infections due to pathogens, including those caused by Amoeba, Candida, Cryptococcus, Mycobacterium, Nocardia, Pneumocystis, Toxoplasma.
It is recommended that latent amebiasis or active amebiasis be ruled out before initiating corticosteroid therapy in any patient who has spent time in the tropics or in any patient with unexplained diarrhea.
Similarly, corticosteroids should be used with great care in patients with known or suspected Strongyloides (threadworm) infestation. In such patients, corticosteroid-induced immunosuppression may lead to Strongyloides hyperinfection and dissemination with widespread larval migration, often accompanied by severe enterocolitis and potentially fatal gram-negative septicemia.
Corticosteroids should not be used in cerebral malaria. There is currently no evidence of benefit from steroids in this condition.
Tuberculosis
The use of corticosteroids in active tuberculosis should be restricted to those cases of fulminating or disseminated tuberculosis in which the corticosteroid is used for the management of the disease in conjunction with appropriate antituberculous regimen.
If corticosteroids are indicated in patients with latent tuberculosis or tuberculin reactivity, close observation is necessary as reactivation of the disease may occur. During prolonged corticosteroid therapy, these patients should receive chemoprophylaxis.
Vaccination
Administration of live or live, attenuated vaccines is contraindicated in patients receiving immunosuppressive doses of corticosteroids. Killed or inactivated vaccines may be administered. However, the response to such vaccines can not be predicted. Immunization procedures may be undertaken in patients receiving corticosteroids as replacement therapy, e.g., for Addison's disease.
Viral infections
Chicken pox and measles can have a more serious or even fatal course in pediatric and adult patients on corticosteroids. In pediatric and adult patients who have not had these diseases, particular care should be taken to avoid exposure. The contribution of the underlying disease and/or prior corticosteroid treatment to the risk is also not known. If exposed to chicken pox, prophylaxis with varicella zoster immune globulin (VZIG) may be indicated. If exposed to measles, prophylaxis with immunoglobulin (IG) may be indicated. (See the respective package inserts for complete VZIG and IG prescribing information.) If chicken pox develops, treatment with antiviral agents should be considered.
Neurologic
Reports of severe medical events have been associated with the intrathecal route of administration (see ADVERSE REACTIONS, Gastrointestinal and Neurologic/Psychiatric).
Ophthalmic
Use of corticosteroids may produce posterior subcapsular cataracts, glaucoma with possible damage to the optic nerves, and may enhance the establishment of secondary ocular infections due to bacteria, fungi, or viruses. The use of oral corticosteroids is not recommended in the treatment of optic neuritis and may lead to an increase in the risk of new episodes. Corticosteroids should be used cautiously in patients with ocular herpes simplex because of corneal perforation. Corticosteroids should not be used in active ocular herpes simplex.
-
PRECAUTIONS
General
This product, like many other steroid formulations, is sensitive to heat. Therefore, it should not be autoclaved when it is desirable to sterilize the exterior of the vial.
The lowest possible dose of corticosteroid should be used to control the condition under treatment. When reduction in dosage is possible, the reduction should be gradual.
Since complications of treatment with glucocorticoids are dependent on the size of the dose and the duration of treatment, a risk/benefit decision must be made in each individual case as to dose and duration of treatment and as to whether daily or intermittent therapy should be used.
Kaposi's sarcoma has been reported to occur in patients receiving corticosteroid therapy, most often for chronic conditions. Discontinuation of corticosteroids may result in clinical improvement.
Cardio-renal
Caution is required in patients with systemic sclerosis because an increased incidence of scleroderma renal crisis has been observed with corticosteroids, including methylprednisolone.
As sodium retention with resultant edema and potassium loss may occur in patients receiving corticosteroids, these agents should be used with caution in patients with congestive heart failure, hypertension, or renal insufficiency.
Endocrine
Drug-induced secondary adrenocortical insufficiency may be minimized by gradual reduction of dosage. This type of relative insufficiency may persist for months after discontinuation of therapy; therefore, in any situation of stress occurring during that period, hormone therapy should be reinstituted.
Metabolic clearance of corticosteroids is decreased in hypothyroid patients and increased in hyperthyroid patients. Changes in thyroid status of the patient may necessitate adjustment in dosage.
Gastrointestinal
Steroids should be used with caution in active or latent peptic ulcers, diverticulitis, fresh intestinal anastomoses, and nonspecific ulcerative colitis, since they may increase the risk of a perforation. Signs of peritoneal irritation following gastrointestinal perforation in patients receiving corticosteroids may be minimal or absent.
There is an enhanced effect due to decreased metabolism of corticosteroids in patients with cirrhosis.
Musculoskeletal
Corticosteroids decrease bone formation and increase bone resorption both through their effect on calcium regulation (i.e., decreasing absorption and increasing excretion) and inhibition of osteoblast function. This, together with a decrease in the protein matrix of the bone secondary to an increase in protein catabolism, and reduced sex hormone production, may lead to inhibition of bone growth in pediatric patients and the development of osteoporosis at any age. Special consideration should be given to patients at increased risk of osteoporosis (i.e., postmenopausal women) before initiating corticosteroid therapy.
Local injection of a steroid into a previously infected site is not usually recommended.
Neurologic-psychiatric
Although controlled clinical trials have shown corticosteroids to be effective in speeding the resolution of acute exacerbations of multiple sclerosis, they do not show that corticosteroids affect the ultimate outcome or natural history of the disease. The studies do show that relatively high doses of corticosteroids are necessary to demonstrate a significant effect. (See DOSAGE AND ADMINISTRATION.)
An acute myopathy has been observed with the use of high doses of corticosteroids, most often occurring in patients with disorders of neuromuscular transmission (e.g., myasthenia gravis), or in patients receiving concomitant therapy with neuromuscular blocking drugs (e.g., pancuronium). This acute myopathy is generalized, may involve ocular and respiratory muscles, and may result in quadriparesis. Elevations of creatine kinase may occur. Clinical improvement or recovery after stopping corticosteroids may require weeks to years.
Psychic derangements may appear when corticosteroids are used, ranging from euphoria, insomnia, mood swings, personality changes, and severe depression, to frank psychotic manifestations. Also, existing emotional instability or psychotic tendencies may be aggravated by corticosteroids.
Ophthalmic
Intraocular pressure may become elevated in some individuals. If steroid therapy is continued for more than 6 weeks, intraocular pressure should be monitored.
Information for Patients
Patients should be warned not to discontinue the use of corticosteroids abruptly or without medical supervision, to advise any medical attendants that they are taking corticosteroids, and to seek medical advice at once should they develop fever or other signs of infection.
Persons who are on corticosteroids should be warned to avoid exposure to chicken pox or measles. Patients should also be advised that if they are exposed, medical advice should be sought without delay.
Drug Interactions
Aminoglutethimide
Aminoglutethimide may lead to a loss of corticosteroid-induced adrenal suppression.
Amphotericin B injection and potassium-depleting agents
When corticosteroids are administered concomitantly with potassium-depleting agents (i.e., amphotericin B, diuretics), patients should be observed closely for development of hypokalemia. There have been cases reported in which concomitant use of amphotericin B and hydrocortisone was followed by cardiac enlargement and congestive heart failure.
Antibiotics
Macrolide antibiotics have been reported to cause a significant decrease in corticosteroid clearance (see Drug Interactions, Hepatic Enzyme Inhibitors).
Anticholinesterases
Concomitant use of anticholinesterase agents and corticosteroids may produce severe weakness in patients with myasthenia gravis. If possible, anticholinesterase agents should be withdrawn at least 24 hours before initiating corticosteroid therapy.
Anticoagulants, oral
Coadministration of corticosteroids and warfarin usually results in inhibition of response to warfarin, although there have been some conflicting reports. Therefore, coagulation indices should be monitored frequently to maintain the desired anticoagulant effect.
Antidiabetics
Because corticosteroids may increase blood glucose concentrations, dosage adjustments of antidiabetic agents may be required.
Cyclosporine
Increased activity of both cyclosporine and corticosteroids may occur when the two are used concurrently. Convulsions have been reported with this concurrent use.
Digitalis glycosides
Patients on digitalis glycosides may be at increased risk of arrhythmias due to hypokalemia.
Estrogens, including oral contraceptives
Estrogens may decrease the hepatic metabolism of certain corticosteroids, thereby increasing their effect.
Hepatic Enzyme Inducers (e.g., barbiturates, phenytoin, carbamazepine, rifampin)
Drugs which induce cytochrome P450 3A4 enzyme activity may enhance the metabolism of corticosteroids and require that the dosage of the corticosteroid be increased.
Hepatic Enzyme Inhibitors (e.g., ketoconazole, macrolide antibiotics such as erythromycin and troleandomycin)
Drugs which inhibit cytochrome P450 3A4 have the potential to result in increased plasma concentrations of corticosteroids.
Ketoconazole
Ketoconazole has been reported to significantly decrease the metabolism of certain corticosteroids by up to 60%, leading to an increased risk of corticosteroid side effects.
Nonsteroidal anti-inflammatory agents (NSAIDs)
Concomitant use of aspirin (or other nonsteroidal anti-inflammatory agents) and corticosteroids increases the risk of gastrointestinal side effects. Aspirin should be used cautiously in conjunction with corticosteroids in hypoprothrombinemia. The clearance of salicylates may be increased with concurrent use of corticosteroids.
Vaccines
Patients on prolonged corticosteroid therapy may exhibit a diminished response to toxoids and live or inactivated vaccines due to inhibition of antibody response. Corticosteroids may also potentiate the replication of some organisms contained in live attenuated vaccines. Routine administration of vaccines or toxoids should be deferred until corticosteroid therapy is discontinued if possible (see WARNINGS, Infections, Vaccination).
Carcinogenesis, Mutagenesis, Impairment of Fertility
No adequate studies have been conducted in animals to determine whether corticosteroids have a potential for carcinogenesis or mutagenesis.
Steroids may increase or decrease motility and number of spermatozoa in some patients.
Corticosteroids have been shown to impair fertility in male rats.
Pregnancy
Teratogenic effects
Corticosteroids have been shown to be teratogenic in many species when given in doses equivalent to the human dose. Animal studies in which corticosteroids have been given to pregnant mice, rats, and rabbits have yielded an increased incidence of cleft palate in the offspring. There are no adequate and well-controlled studies in pregnant women. Corticosteroids should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Infants born to mothers who have received corticosteroids during pregnancy should be carefully observed for signs of hypoadrenalism.
This product contains benzyl alcohol as a preservative.
Benzyl alcohol can cross the placenta. See PRECAUTIONS: Pediatric use.
Nursing Mothers
Systemically administered corticosteroids appear in human milk and could suppress growth, interfere with endogenous corticosteroid production, or cause other untoward effects. Because of the potential for serious adverse reactions in nursing infants from corticosteroids, a decision should be made whether to continue nursing, or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Some formulations of this product contain benzyl alcohol as a preservative (see DESCRIPTION). Carefully examine vials to determine formulation that is being used.
Benzyl alcohol, a component of this product, has been associated with serious adverse events and death, particularly in pediatric patients. The "gasping syndrome" (characterized by central nervous system depression, metabolic acidosis, gasping respirations, and high levels of benzyl alcohol and its metabolites found in the blood and urine) has been associated with benzyl alcohol dosages >99 mg/kg/day in neonates and low-birth-weight neonates. Additional symptoms may include gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Although normal therapeutic doses of this product ordinarily delivers amounts of benzyl alcohol that are substantially lower than those reported in association with the "gasping syndrome", the minimum amount of benzyl alcohol at which toxicity may occur is not known. The risk of benzyl alcohol toxicity depends on the quantity administered and the hepatic capacity to detoxify the chemical. Premature and low-birth-weight infants, as well as patients receiving high dosages, may be more likely to develop toxicity. Practitioners administering this and other medications containing benzyl alcohol should consider the combined daily metabolic load of benzyl alcohol from all sources.
The efficacy and safety of corticosteroids in the pediatric population are based on the well-established course of effect of corticosteroids which is similar in pediatric and adult populations. Published studies provide evidence of efficacy and safety in pediatric patients for the treatment of nephrotic syndrome (>2 years of age), and aggressive lymphomas and leukemias (>1 month of age). Other indications for pediatric use of corticosteroids, e.g., severe asthma and wheezing, are based on adequate and well-controlled trials conducted in adults, on the premises that the course of the diseases and their pathophysiology are considered to be substantially similar in both populations.
The adverse effects of corticosteroids in pediatric patients are similar to those in adults (see ADVERSE REACTIONS). Like adults, pediatric patients should be carefully observed with frequent measurements of blood pressure, weight, height, intraocular pressure, and clinical evaluation for the presence of infection, psychosocial disturbances, thromboembolism, peptic ulcers, cataracts, and osteoporosis. Pediatric patients who are treated with corticosteroids by any route, including systemically administered corticosteroids, may experience a decrease in their growth velocity. This negative impact of corticosteroids on growth has been observed at low systemic doses and in the absence of laboratory evidence of HPA axis suppression (i.e., cosyntropin stimulation and basal cortisol plasma levels). Growth velocity may therefore be a more sensitive indicator of systemic corticosteroid exposure in pediatric patients than some commonly used tests of HPA axis function. The linear growth of pediatric patients treated with corticosteroids should be monitored, and the potential growth effects of prolonged treatment should be weighed against clinical benefits obtained and the availability of treatment alternatives. In order to minimize the potential growth effects of corticosteroids, pediatric patients should be titrated to the lowest effective dose.
Hypertrophic cardiomyopathy may develop after administration of methylprednisolone to prematurely born infants, therefore appropriate diagnostic evaluation and monitoring of cardiac function and structure should be performed.
Geriatric Use
Clinical studies did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
The following adverse reactions have been reported with SOLU-MEDROL or other corticosteroids:
Allergic reactions: Allergic or hypersensitivity reactions, anaphylactoid reaction, anaphylaxis, angioedema.
Blood and lymphatic system disorders: Leukocytosis.
Cardiovascular: Bradycardia, cardiac arrest, cardiac arrhythmias, cardiac enlargement, circulatory collapse, congestive heart failure, fat embolism, hypertension, hypertrophic cardiomyopathy in premature infants, myocardial rupture following recent myocardial infarction (see WARNINGS), pulmonary edema, syncope, tachycardia, thromboembolism, thrombophlebitis, vasculitis.
Dermatologic: Acne, allergic dermatitis, burning or tingling (especially in the perineal area after intravenous injection), cutaneous and subcutaneous atrophy, dry scaly skin, ecchymoses and petechiae, edema, erythema, hyperpigmentation, hypopigmentation, impaired wound healing, increased sweating, rash, sterile abscess, striae, suppressed reactions to skin tests, thin fragile skin, thinning scalp hair, urticaria.
Endocrine: Decreased carbohydrate and glucose tolerance, development of cushingoid state, glycosuria, hirsutism, hypertrichosis, increased requirements for insulin or oral hypoglycemic agents in diabetes, manifestations of latent diabetes mellitus, menstrual irregularities, secondary adrenocortical and pituitary unresponsiveness (particularly in times of stress, as in trauma, surgery, or illness), suppression of growth in pediatric patients.
Fluid and electrolyte disturbances: Congestive heart failure in susceptible patients, fluid retention, hypokalemic alkalosis, potassium loss, sodium retention.
Gastrointestinal: Abdominal distention, bowel/bladder dysfunction (after intrathecal administration), elevation in serum liver enzyme levels (usually reversible upon discontinuation), hepatomegaly, increased appetite, nausea, pancreatitis, peptic ulcer with possible perforation and hemorrhage, perforation of the small and large intestine (particularly in patients with inflammatory bowel disease), ulcerative esophagitis.
Hepatobiliary: Hepatitis (see WARNINGS, Drug-Induced Liver Injury).
Metabolic: Negative nitrogen balance due to protein catabolism.
Musculoskeletal: Aseptic necrosis of femoral and humeral heads, Charcot-like arthropathy, loss of muscle mass, muscle weakness, osteoporosis, pathologic fracture of long bones, postinjection flare (following intra-articular use), steroid myopathy, tendon rupture, vertebral compression fractures.
Neurologic/Psychiatric: Convulsions, depression, emotional instability, euphoria, headache, increased intracranial pressure with papilledema (pseudotumor cerebri) usually following discontinuation of treatment, insomnia, mood swings, neuritis, neuropathy, paresthesia, personality changes, psychic disorders, vertigo. Arachnoiditis, meningitis, paraparesis/paraplegia, and sensory disturbances have occurred after intrathecal administration (see WARNINGS, Neurologic).
Ophthalmic: Exophthalmos, glaucoma, increased intraocular pressure, posterior subcapsular cataracts, rare instances of blindness associated with periocular injections.
Other: Abnormal fat deposits, decreased resistance to infection, hiccups, increased or decreased motility and number of spermatozoa, injection site infections following non-sterile administration (see WARNINGS), malaise, moon face, weight gain.
- OVERDOSAGE
-
DOSAGE AND ADMINISTRATION
NOTE: Some of the SOLU-MEDROL formulations contain benzyl alcohol (see DESCRIPTION, WARNINGS and PRECAUTIONS, Pediatric Use)
Because of possible physical incompatibilities, SOLU-MEDROL should not be diluted or mixed with other solutions.
Use only the accompanying diluent or Bacteriostatic Water For Injection with Benzyl Alcohol when reconstituting SOLU-MEDROL (see DESCRIPTION).
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
This preparation may be administered by intravenous injection, by intravenous infusion, or by intramuscular injection, the preferred method for initial emergency use being intravenous injection. Following the initial emergency period, consideration should be given to employing a longer acting injectable preparation or an oral preparation.
There are reports of cardiac arrhythmias and/or cardiac arrest following the rapid administration of large intravenous doses of SOLU-MEDROL (greater than 0.5 gram administered over a period of less than 10 minutes). Bradycardia has been reported during or after the administration of large doses of methylprednisolone sodium succinate, and may be unrelated to the speed or duration of infusion. When high dose therapy is desired, the recommended dose of SOLU-MEDROL Sterile Powder is 30 mg/kg administered intravenously over at least 30 minutes. This dose may be repeated every 4 to 6 hours for 48 hours.
In general, high dose corticosteroid therapy should be continued only until the patient's condition has stabilized; usually not beyond 48 to 72 hours.
In other indications, initial dosage will vary from 10 to 40 mg of methylprednisolone depending on the specific disease entity being treated. However, in certain overwhelming, acute, life-threatening situations, administrations in dosages exceeding the usual dosages may be justified and may be in multiples of the oral dosages.
It Should Be Emphasized that Dosage Requirements are Variable and Must Be Individualized on the Basis of the Disease Under Treatment and the Response of the Patient. After a favorable response is noted, the proper maintenance dosage should be determined by decreasing the initial drug dosage in small decrements at appropriate time intervals until the lowest dosage which will maintain an adequate clinical response is reached. Situations which may make dosage adjustments necessary are changes in clinical status secondary to remissions or exacerbations in the disease process, the patient's individual drug responsiveness, and the effect of patient exposure to stressful situations not directly related to the disease entity under treatment. In this latter situation, it may be necessary to increase the dosage of the corticosteroid for a period of time consistent with the patient's condition. If after long-term therapy the drug is to be stopped, it is recommended that it be withdrawn gradually rather than abruptly.
SOLU-MEDROL may be administered by intravenous or intramuscular injection or by intravenous infusion, the preferred method for initial emergency use being intravenous injection. To administer by intravenous (or intramuscular) injection, prepare solution as directed. The desired dose may be administered intravenously over a period of several minutes. If desired, the medication may be administered in diluted solutions by adding Water for Injection or other suitable diluent (see below) to the Act-O-Vial and withdrawing the indicated dose.
To prepare solutions for intravenous infusion, first prepare the solution for injection as directed. This solution may then be added to indicated amounts of 5% dextrose in water, isotonic saline solution, or 5% dextrose in isotonic saline solution. From a microbiological point of view, unless the method of opening/reconstitution/dilution precludes the risk of microbial contamination, the product should be used immediately. If not used immediately, in-use storage times and conditions are the responsibility of the user. Chemical and physical stability of the further diluted product has been demonstrated within 4 hours of preparation if stored below 25°C or within 24 hours of preparation if stored at 2°C to 8°C.
In pediatric patients, the initial dose of methylprednisolone may vary depending on the specific disease entity being treated. The range of initial doses is 0.11 to 1.6 mg/kg/day in three or four divided doses (3.2 to 48 mg/m2bsa/day).
The National Heart, Lung, and Blood Institute (NHLBI) recommended dosing for systemic prednisone, prednisolone, or methylprednisolone in pediatric patients whose asthma is uncontrolled by inhaled corticosteroids and long-acting bronchodilators is 1–2 mg/kg/day in single or divided doses. It is further recommended that short course, or "burst" therapy, be continued until the patient achieves a peak expiratory flow rate of 80% of his or her personal best or until symptoms resolve. This usually requires 3 to 10 days of treatment, although it can take longer. There is no evidence that tapering the dose after improvement will prevent a relapse.
Dosage may be reduced for infants and children but should be governed more by the severity of the condition and response of the patient than by age or size. It should not be less than 0.5 mg per kg every 24 hours.
Dosage must be decreased or discontinued gradually when the drug has been administered for more than a few days. If a period of spontaneous remission occurs in a chronic condition, treatment should be discontinued. Routine laboratory studies, such as urinalysis, two-hour postprandial blood sugar, determination of blood pressure and body weight, and a chest X-ray should be made at regular intervals during prolonged therapy. Upper GI X-rays are desirable in patients with an ulcer history or significant dyspepsia.
In treatment of acute exacerbations of multiple sclerosis, daily doses of 160 mg of methylprednisolone for a week followed by 64 mg every other day for 1 month have been shown to be effective (see PRECAUTIONS, Neurologic-psychiatric).
For the purpose of comparison, the following is the equivalent milligram dosage of the various glucocorticoids:
Cortisone, 25
Triamcinolone, 4
Hydrocortisone, 20
Paramethasone, 2
Prednisolone, 5
Betamethasone, 0.75
Prednisone, 5
Dexamethasone, 0.75
Methylprednisolone, 4
These dose relationships apply only to oral or intravenous administration of these compounds. When these substances or their derivatives are injected intramuscularly or into joint spaces, their relative properties may be greatly altered.
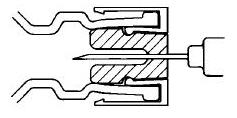
DIRECTIONS FOR USING THE ACT-O-VIAL SYSTEM
- 1.
- Press down on plastic activator to force diluent into the lower compartment.
- 2.
- Gently agitate to effect solution.
- 3.
- Remove plastic tab covering center of stopper.
- 4.
- Sterilize top of stopper with a suitable germicide.
- 5.
- Insert needle squarely through center of stopper until tip is just visible. Invert vial and withdraw dose.
-
STORAGE CONDITIONS
Protect from light.
Store unreconstituted product at controlled room temperature 20° to 25°C (68° to 77°F) [see USP].
Store reconstituted solution (not further diluted) at controlled room temperature 20° to 25°C (68° to 77°F) [see USP], and use it within 48 hours.
See section DOSAGE AND ADMINISTRATION, for storage of further diluted solution.
-
HOW SUPPLIED
SOLU-MEDROL Sterile Powder preservative-free is available in the following packages:
125 mg Act-O-Vial System (Single-Dose Vial)
Overbagged with 5 x 2 mL Act-O-Vial Systems in each bag, NDC 55154-3944-5This product's label may have been updated. For current full prescribing information, please visit www.pfizer.com.
- SPL UNCLASSIFIED SECTION
- Package/Label Display Panel
-
INGREDIENTS AND APPEARANCE
SOLU-MEDROL
methylprednisolone sodium succinate injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55154-3944(NDC:0009-0047) Route of Administration INTRAVENOUS, INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METHYLPREDNISOLONE SODIUM SUCCINATE (UNII: LEC9GKY20K) (METHYLPREDNISOLONE - UNII:X4W7ZR7023) METHYLPREDNISOLONE 125 mg in 2 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, MONOBASIC, ANHYDROUS (UNII: KH7I04HPUU) 1.6 mg in 2 mL SODIUM PHOSPHATE, DIBASIC, UNSPECIFIED FORM (UNII: GR686LBA74) 17.4 mg in 2 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55154-3944-5 5 in 1 BAG 04/02/1959 1 2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA011856 04/02/1959 Labeler - Cardinal Health 107, LLC (118546603)