Eptifibatide was studied in 3 placebo-controlled, randomized studies. PURSUIT evaluated patients with acute coronary syndromes: UA or NSTEMI. Two other studies, ESPRIT and IMPACT II, evaluated ...
Eptifibatide was studied in 3 placebo-controlled, randomized studies. PURSUIT evaluated patients with acute coronary syndromes: UA or NSTEMI. Two other studies, ESPRIT and IMPACT II, evaluated patients about to undergo a PCI. Patients underwent primarily balloon angioplasty in IMPACT II and intracoronary stent placement, with or without angioplasty, in ESPRIT.
14.1 Non-ST-Segment Elevation Acute Coronary Syndrome
Non-ST-segment elevation acute coronary syndrome is defined as prolonged (≥10 minutes) symptoms of cardiac ischemia within the previous 24 hours associated with either ST-segment changes (elevations between 0.6 mm and 1 mm or depression >0.5 mm), T-wave inversion (>1 mm), or positive CK-MB. This definition includes "unstable angina" and "NSTEMI" but excludes MI that is associated with Q waves or greater degrees of ST-segment elevation.
PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Eptifibatide Therapy)
PURSUIT was a 726-center, 27-country, double-blind, randomized, placebo-controlled study in 10,948 patients presenting with UA or NSTEMI. Patients could be enrolled only if they had experienced cardiac ischemia at rest (≥10 minutes) within the previous 24 hours and had either ST-segment changes (elevations between 0.6 mm and 1 mm or depression >0.5 mm), T-wave inversion (>1 mm), or increased CK-MB. Important exclusion criteria included a history of bleeding diathesis, evidence of abnormal bleeding within the previous 30 days, uncontrolled hypertension, major surgery within the previous 6 weeks, stroke within the previous 30 days, any history of hemorrhagic stroke, serum creatinine >2 mg/dL, dependency on renal dialysis, or platelet count <100,000/mm3.
Patients were randomized to placebo, to eptifibatide 180-mcg/kg bolus followed by a 2-mcg/kg/min infusion (180/2), or to eptifibatide 180-mcg/kg bolus followed by a 1.3-mcg/kg/min infusion (180/1.3). The infusion was continued for 72 hours, until hospital discharge, or until the time of CABG, whichever occurred first, except that if PCI was performed, the eptifibatide infusion was continued for 24 hours after the procedure, allowing for a duration of infusion up to 96 hours.
The lower-infusion-rate arm was stopped after the first interim analysis when the 2 active-treatment arms appeared to have the same incidence of bleeding.
Patient age ranged from 20 to 94 (mean 63) years, and 65% were male. The patients were 89% Caucasian, 6% Hispanic, and 5% Black, recruited in the United States and Canada (40%), Western Europe (39%), Eastern Europe (16%), and Latin America (5%).
This was a “real world” study; each patient was managed according to the usual standards of the investigational site; frequencies of angiography, PCI, and CABG therefore differed widely from site to site and from country to country. Of the patients in PURSUIT, 13% were managed with PCI during drug infusion, of whom 50% received intracoronary stents; 87% were managed medically (without PCI during drug infusion).
The majority of patients received aspirin (75-325 mg once daily). Heparin was administered intravenously or subcutaneously, at the physician's discretion, most commonly as an intravenous bolus of 5000 units followed by a continuous infusion of 1000 units/h. For patients weighing less than 70 kg, the recommended heparin bolus dose was 60 units/kg followed by a continuous infusion of 12 units/kg/h. A target aPTT of 50 to 70 seconds was recommended. A total of 1250 patients underwent PCI within 72 hours after randomization, in which case they received intravenous heparin to maintain an ACT of 300 to 350 seconds.
The primary endpoint of the study was the occurrence of death from any cause or new MI (evaluated by a blinded Clinical Endpoints Committee) within 30 days of randomization.
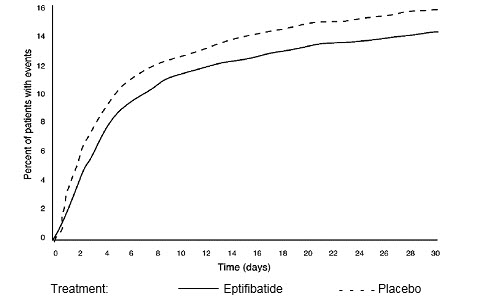
Compared to placebo, eptifibatide administered as a 180-mcg/kg bolus followed by a 2-mcg/kg/min infusion significantly (p=0.042) reduced the incidence of endpoint events (see Table 6). The reduction in the incidence of endpoint events in patients receiving eptifibatide was evident early during treatment, and this reduction was maintained through at least 30 days (see Figure 1). Table 5 also shows the incidence of the components of the primary endpoint, death (whether or not preceded by an MI) and new MI in surviving patients at 30 days.
Figure 1: Kaplan-Meier Plot of Time to Death or Myocardial Infarction Within 30 Days of Randomization
in the PURSUIT Study
Treatment with eptifibatide prior to determination of patient management strategy reduced clinical events regardless of whether patients ultimately underwent diagnostic catheterization, revascularization (i.e., PCI or CABG surgery) or continued to receive medical management alone. Table 6 shows the incidence of death or MI within 72 hours.
All of the effect of eptifibatide was established within 72 hours (during the period of drug infusion), regardless of management strategy. Moreover, for patients undergoing early PCI, a reduction in events was evident prior to the procedure.
An analysis of the results by sex suggests that women who would not routinely be expected to undergo PCI receive less benefit from eptifibatide (95% confidence limits for relative risk of 0.94 - 1.28) than do men (0.72 - 0.9). This difference may be a true treatment difference, the effect of other differences in these subgroups, or a statistical anomaly. No differential outcomes were seen between male and female patients undergoing PCI (see results for ESPRIT).
Follow-up data were available through 165 days for 10,611 patients enrolled in the PURSUIT trial (96.9% of the initial enrollment). This follow-up included 4566 patients who received eptifibatide at the 180/2 dose. As reported by the investigators, the occurrence of death from any cause or new MI for patients followed for at least 165 days was reduced from 13.6% with placebo to 12.1% with eptifibatide 180/2.
14.2 Percutaneous Coronary Intervention (PCI)
IMPACT II (Eptifibatide to Minimize Platelet Aggregation and Prevent Coronary Thrombosis II)
IMPACT II was a multicenter, double-blind, randomized, placebo-controlled study conducted in the United States in 4010 patients undergoing PCI. Major exclusion criteria included a history of bleeding diathesis, major surgery within 6 weeks of treatment, gastrointestinal bleeding within 30 days, any stroke or structural CNS abnormality, uncontrolled hypertension, PT >1.2 times control, hematocrit <30%, platelet count <100,000/mm3, and pregnancy.
Patient age ranged from 24 to 89 (mean 60) years, and 75% were male. The patients were 92% Caucasian, 5% Black, and 3% Hispanic. Forty-one percent of the patients underwent PCI for ongoing ACS. Patients were randomly assigned to 1 of 3 treatment regimens, each incorporating a bolus dose initiated immediately prior to PCI followed by a continuous infusion lasting 20 to 24 hours:
- 1)
- 135-mcg/kg bolus followed by a continuous infusion of 0.5 mcg/kg/min of eptifibatide (135/0.5);
- 2)
- 135-mcg/kg bolus followed by a continuous infusion of 0.75 mcg/kg/min of eptifibatide (135/0.75); or
- 3)
- a matching placebo bolus followed by a matching placebo continuous infusion.
Each patient received aspirin and an intravenous heparin bolus of 100 units/kg, with additional bolus infusions of up to 2000 additional units of heparin every 15 minutes to maintain an ACT of 300 to 350 seconds.
The primary endpoint was the composite of death, MI, or urgent revascularization, analyzed at 30 days after randomization in all patients who received at least 1 dose of study drug.
As shown in Table 7, each eptifibatide regimen reduced the rate of death, MI, or urgent intervention, although at 30 days, this finding was statistically significant only in the lower-dose eptifibatide group. As in the PURSUIT study, the effects of eptifibatide were seen early and persisted throughout the 30-day period.
ESPRIT (Enhanced Suppression of the Platelet IIb/IIIa Receptor with Eptifibatide Therapy)
The ESPRIT study was a multicenter, double-blind, randomized, placebo-controlled study conducted in the United States and Canada that enrolled 2064 patients undergoing elective or urgent PCI with intended intracoronary stent placement. Exclusion criteria included MI within the previous 24 hours, ongoing chest pain, administration of any oral antiplatelet or oral anticoagulant other than aspirin within 30 days of PCI (although loading doses of thienopyridine on the day of PCI were encouraged), planned PCI of a saphenous vein graft or subsequent “staged” PCI, prior stent placement in the target lesion, PCI within the previous 90 days, a history of bleeding diathesis, major surgery within 6 weeks of treatment, gastrointestinal bleeding within 30 days, any stroke or structural CNS abnormality, uncontrolled hypertension, PT >1.2 times control, hematocrit <30%, platelet count <100,000/mm3, and pregnancy.
Patient age ranged from 24 to 93 (mean 62) years, and 73% of patients were male. The study enrolled 90% Caucasian, 5% African American, 2% Hispanic, and 1% Asian patients. Patients received a wide variety of stents. Patients were randomized either to placebo or eptifibatide administered as an intravenous bolus of 180 mcg/kg followed immediately by a continuous infusion of 2 mcg/kg/min, and a second bolus of 180 mcg/kg administered 10 minutes later (180/2/180). Eptifibatide infusion was continued for 18 to 24 hours after PCI or until hospital discharge, whichever came first. Each patient received at least 1 dose of aspirin (162-325 mg) and 60 units/kg of heparin as a bolus (not to exceed 6000 units) if not already receiving a heparin infusion. Additional boluses of heparin (10-40 units/kg) could be administered in order to reach a target ACT between 200 and 300 seconds.
The primary endpoint of the ESPRIT study was the composite of death, MI, urgent target vessel revascularization (UTVR), and “bailout” to open-label eptifibatide due to a thrombotic complication of PCI (TBO) (e.g., visible thrombus, “no reflow,” or abrupt closure) at 48 hours. MI, UTVR, and TBO were evaluated by a blinded Clinical Events Committee.
As shown in Table 8, the incidence of the primary endpoint and selected secondary endpoints was significantly reduced in patients who received eptifibatide. A treatment benefit in patients who received eptifibatide was seen by 48 hours and at the end of the 30-day observation period.
The need for thrombotic “bailout” was significantly reduced with eptifibatide at 48 hours (2.1% for placebo, 1% for eptifibatide; p=0.029). Consistent with previous studies of GP IIb/IIIa inhibitors, most of the benefit achieved acutely with eptifibatide was in the reduction of MI. Eptifibatide reduced the occurrence of MI at 48 hours from 9% for placebo to 5.4% (p=0.0015) and maintained that effect with significance at 30 days.
There was no treatment difference with respect to sex in ESPRIT. Eptifibatide reduced the incidence of the primary endpoint in both men (95% confidence limits for relative risk: 0.54, 1.07) and women (0.24, 0.72) at 48 hours.
Follow-up (12-month) mortality data were available for 2024 patients (1017 on eptifibatide) enrolled in the ESPRIT trial (98.1% of the initial enrollment). Twelve-month clinical event data were available for 1964 patients (988 on eptifibatide), representing 95.2% of the initial enrollment. As shown in Table 9, the treatment effect of eptifibatide seen at 48 hours and 30 days appeared preserved at 6 months and 1 year. Most of the benefit was in reduction of MI.
Percentages are Kaplan-Meier event rates.
Close