Label: EMFLAZA- deflazacort tablet
EMFLAZA- deflazacort suspension











-
NDC Code(s):
52856-501-01,
52856-502-03,
52856-503-03,
52856-504-03, view more52856-505-22
- Packager: PTC Therapeutics, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated June 18, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use EMFLAZA® safely and effectively. See full prescribing information for EMFLAZA.
EMFLAZA® (deflazacort) tablets, for oral use
EMFLAZA® (deflazacort) oral suspension
Initial U.S. Approval: 2017
RECENT MAJOR CHANGES
Warnings and Precautions
Immunosuppression and Increased Risk of Infection (5.2) 06/2024
INDICATIONS AND USAGE
EMFLAZA is a corticosteroid indicated for the treatment of Duchenne muscular dystrophy (DMD) in patients 2 years of age and older (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Hypersensitivity to deflazacort or any of the inactive ingredients in EMFLAZA (4)
WARNINGS AND PRECAUTIONS
- Alterations in Endocrine Function: Hypothalamic-pituitary-adrenal axis suppression, Cushing’s syndrome, and hyperglycemia can occur; Monitor patients for these conditions with chronic use of EMFLAZA (2.3, 5.1)
- Immunosuppression and Increased Risk of Infection: Increased risk of new, exacerbation, dissemination, or reactivation of latent infections, which can be severe and at times fatal; Signs and symptoms of infection may be masked (5.2)
- Alterations in Cardiovascular/Renal Function: Monitor for elevated blood pressure and sodium, and for decreased potassium levels (5.3)
- Gastrointestinal Perforation: Increased risk in patients with certain GI disorders; Signs and symptoms may be masked (5.4)
- Behavioral and Mood Disturbances: May include euphoria, insomnia, mood swings, personality changes, severe depression, and psychosis (5.5)
- Effects on Bones: Monitor for decreases in bone mineral density with chronic use of EMFLAZA (5.6)
- Ophthalmic Effects: May include cataracts, infections, and glaucoma; Monitor intraocular pressure if EMFLAZA is continued for more than 6 weeks (5.7)
- Vaccination: Do not administer live or live attenuated vaccines to patients receiving immunosuppressive doses of corticosteroids. Administer live-attenuated or live vaccines at least 4 to 6 weeks prior to starting EMFLAZA (5.8)
- Serious Skin Rashes: Discontinue at the first sign of rash, unless the rash is clearly not drug related (5.9)
ADVERSE REACTIONS
The most common adverse reactions (≥ 10% for EMFLAZA and greater than placebo) are Cushingoid appearance, weight increased, increased appetite, upper respiratory tract infection, cough, pollakiuria, hirsutism, central obesity, and nasopharyngitis (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact PTC Therapeutics, Inc. at 1-866-562-4620 or Pharmacovigilance@PTCBio.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Assessments Prior to First Dose of EMFLAZA
2.2 Dosing Information
2.3 Discontinuation
2.4 Important Preparation and Administration Instructions
2.5 Dosage Modification for Use with CYP3A4 Inhibitors and Inducers
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Alterations in Endocrine Function
5.2 Immunosuppression and Increased Risk of Infection
5.3 Alterations in Cardiovascular/Renal Function
5.4 Gastrointestinal Perforation
5.5 Behavioral and Mood Disturbances
5.6 Effects on Bones
5.7 Ophthalmic Effects
5.8 Immunizations
5.9 Serious Skin Rashes
5.10 Effects on Growth and Development
5.11 Myopathy
5.12 Kaposi’s Sarcoma
5.13 Risk of Serious Adverse Reactions in Infants because of Benzyl Alcohol Preservative
5.14 Thromboembolic Events
5.15 Anaphylaxis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 CYP3A4 Inhibitors and Inducers
7.2 Neuromuscular Blockers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Assessments Prior to First Dose of EMFLAZA
Administer all immunizations according to immunization guidelines prior to starting EMFLAZA. Administer live-attenuated or live vaccines at least 4 to 6 weeks prior to starting EMFLAZA [see Warnings and Precautions (5.8)].
2.2 Dosing Information
The recommended oral dosage of EMFLAZA is approximately 0.9 mg/kg/day once daily. If tablets are used, round up to the nearest possible dose. Any combination of the four EMFLAZA tablet strengths can be used to achieve this dose. If the oral suspension is used, round up to the nearest tenth of a milliliter (mL).
2.3 Discontinuation
Dosage of EMFLAZA must be decreased gradually if the drug has been administered for more than a few days [see Warnings and Precautions (5.1)].
2.4 Important Preparation and Administration Instructions
EMFLAZA Tablets and Oral Suspension can be taken with or without food. Do not administer EMFLAZA with grapefruit juice [see Drug Interactions (7.1)]
EMFLAZA Tablets
EMFLAZA Tablets can be administered whole or crushed and taken immediately after mixing with applesauce.EMFLAZA Oral Suspension
Shake EMFLAZA Oral Suspension well before administration.Use only the oral dispenser provided with the product. After withdrawing the appropriate dose into the oral dispenser, slowly add the EMFLAZA Oral Suspension into 3 to 4 ounces of juice (except grapefruit juice) or milk and mix well. The dose should then be administered immediately.
Discard any unused EMFLAZA Oral Suspension remaining after 1 month of first opening the bottle.
2.5 Dosage Modification for Use with CYP3A4 Inhibitors and Inducers
CYP3A4 Inhibitors
Give one third of the recommended dosage when EMFLAZA is administered with moderate or strong CYP3A4 inhibitors. For example, a 36 mg per day dose would be reduced to a 12 mg per day dose when used with moderate or strong CYP3A4 inhibitors [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].CYP3A4 Inducers
Avoid use with moderate or strong CYP3A4 inducers with EMFLAZA [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]. - 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
EMFLAZA is contraindicated in patients with known hypersensitivity to deflazacort or to any of the inactive ingredients. Instances of hypersensitivity, including anaphylaxis, have occurred in patients receiving corticosteroid therapy [see Warnings and Precautions (5.15) and Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Alterations in Endocrine Function
Corticosteroids, such as EMFLAZA, can cause serious and life-threatening alterations in endocrine function, especially with chronic use. Monitor patients receiving EMFLAZA for Cushing’s syndrome, hyperglycemia, and adrenal insufficiency after EMFLAZA withdrawal. In addition, patients with hypopituitarism, primary adrenal insufficiency or congenital adrenal hyperplasia, altered thyroid function, or pheochromocytoma may be at increased risk for adverse endocrine events.
Risk of Adrenal Insufficiency Following Corticosteroid Withdrawal
Corticosteroids produce reversible hypothalamic-pituitary-adrenal (HPA) axis suppression, with the potential for the development of secondary adrenal insufficiency after withdrawal of corticosteroid treatment. Acute adrenal insufficiency can occur if corticosteroids are withdrawn abruptly, and can be fatal. The degree and duration of adrenocortical insufficiency produced is variable among patients and depends on the dose, frequency, and duration of corticosteroid therapy. The risk is reduced by gradually tapering the corticosteroid dose when withdrawing treatment. This insufficiency may persist, however, for months after discontinuation of prolonged therapy; therefore, in any situation of stress occurring during that period of discontinuation, corticosteroid therapy should be reinstituted. For patients already taking corticosteroids during times of stress, the dosage may need to be increased.A steroid “withdrawal syndrome”, seemingly unrelated to adrenocortical insufficiency, may also occur following abrupt discontinuance of corticosteroids. This syndrome includes symptoms such as anorexia, nausea, vomiting, lethargy, headache, fever, joint pain, desquamation, myalgia, and/or weight loss. These effects are thought to be due to the sudden change in corticosteroid concentration rather than to low corticosteroid levels.
Cushing’s Syndrome
Cushing’s syndrome (hypercortisolism) occurs with prolonged exposure to exogenous corticosteroids, including EMFLAZA. Symptoms include hypertension, truncal obesity and thinning of the limbs, purple striae, facial rounding, facial plethora, muscle weakness, easy and frequent bruising with thin fragile skin, posterior neck fat deposition, osteopenia, acne, amenorrhea, hirsutism and psychiatric abnormalities.Hyperglycemia
Corticosteroids can increase blood glucose, worsen pre-existing diabetes, predispose those on long-term therapy to diabetes mellitus, and may reduce the effect of anti-diabetic drugs. Monitor blood glucose at regular intervals. For patients with hyperglycemia, anti-diabetic treatment should be initiated or adjusted accordingly.Considerations for Use in Patients with Altered Thyroid Function
Metabolic clearance of corticosteroids is decreased in hypothyroid patients and increased in hyperthyroid patients. Changes in thyroid status of the patient may necessitate a dose adjustment of the corticosteroid. When concomitant administration of corticosteroids and levothyroxine is required, administration of corticosteroid should precede the initiation of levothyroxine therapy to reduce the risk of adrenal crisis.Pheochromocytoma Crisis
There have been reports of pheochromocytoma crisis, which can be fatal, after administration of systemic corticosteroids. In patients with suspected or identified pheochromocytoma, consider the risk of pheochromocytoma crisis prior to administering corticosteroids.5.2 Immunosuppression and Increased Risk of Infection
Corticosteroids, including EMFLAZA, suppress the immune system and increase the risk of infection with any pathogen, including viral, bacterial, fungal, protozoan, or helminthic pathogens. Corticosteroids can:
- Reduce resistance to new infections
- Exacerbate existing infections
- Increase the risk of disseminated infections
- Increase the risk of reactivation or exacerbation of latent infections
- Mask some signs of infections
Corticosteroid-associated infections can be mild but can be severe, and at times fatal. The rate of infectious complications increases with increasing corticosteroid dosages.
Monitor for the development of infection and consider EMFLAZA withdrawal or dosage reduction as needed.
Tuberculosis
If EMFLAZA is used to treat a condition in patients with latent tuberculosis or tuberculin reactivity, reactivation of tuberculosis may occur. Closely monitor such patients for reactivation. During prolonged EMFLAZA therapy, patients with latent tuberculosis or tuberculin reactivity should receive chemoprophylaxis.
Varicella Zoster and Measles Viral Infections
Varicella and measles can have a serious or even fatal course in non-immune patients taking corticosteroids, including EMFLAZA. In corticosteroid-treated patients who have not had these diseases or are non-immune, particular care should be taken to avoid exposure to varicella and measles.
- If an EMFLAZA-treated patient is exposed to varicella, prophylaxis with varicella zoster immunoglobulin may be indicated. If varicella develops, treatment with antiviral agents may be considered.
- If an EMFLAZA-treated patient is exposed to measles, prophylaxis with immunoglobulin may be indicated.
Hepatitis B Virus Reactivation
Hepatitis B virus reactivation can occur in patients who are hepatitis B carriers treated with immunosuppressive dosages of corticosteroids, including EMFLAZA. Reactivation can also occur infrequently in corticosteroid-treated patients who appear to have resolved hepatitis B infection.
Screen patients for hepatitis B infection before initiating immunosuppressive (e.g., prolonged) treatment with EMFLAZA. For patients who show evidence of hepatitis B infection, recommend consultation with physicians with expertise in managing hepatitis B regarding monitoring and consideration for hepatitis B antiviral therapy.
Fungal Infections
Corticosteroids, including EMFLAZA, may exacerbate systemic fungal infections; therefore, avoid EMFLAZA use in the presence of such infections unless EMFLAZA is needed to control drug reactions. For patients on chronic EMFLAZA therapy who develop systemic fungal infections, EMFLAZA withdrawal or dose reduction is recommended.
Amebiasis
Corticosteroids, including EMFLAZA, may activate latent amebiasis. Therefore, it is recommended that latent amebiasis or active amebiasis be ruled out before initiating EMFLAZA in patients who have spent time in the tropics or patients with unexplained diarrhea.
Strongyloides Infestation
Corticosteroids, including EMFLAZA, should be used with great care in patients with known or suspected Strongyloides (threadworm) infestation. In such patients, corticosteroid-induced immunosuppression may lead to Strongyloides hyperinfection and dissemination with widespread larval migration, often accompanied by severe enterocolitis and potentially fatal gram-negative septicemia.
Cerebral Malaria
Avoid corticosteroids, including EMFLAZA, in patients with cerebral malaria.
5.3 Alterations in Cardiovascular/Renal Function
Corticosteroids, including EMFLAZA, can cause elevation of blood pressure, salt, and water retention, and increased excretion of potassium and calcium. Monitor blood pressure and assess for signs and symptoms of volume overload. Monitor serum potassium levels. Dietary salt restriction and potassium supplementation may be necessary. EMFLAZA should be used with caution in patients with congestive heart failure, hypertension, or renal insufficiency.
Literature reports suggest an association between use of corticosteroids and left ventricular free wall rupture after a recent myocardial infarction; therefore, therapy with EMFLAZA should be used with great caution in these patients.
5.4 Gastrointestinal Perforation
There is an increased risk of gastrointestinal perforation during corticosteroid use in patients with certain gastrointestinal disorders such as active or latent peptic ulcers, diverticulitis, fresh intestinal anastomoses, and non-specific ulcerative colitis. Signs of gastrointestinal perforation, such as peritoneal irritation, may be masked in patients receiving corticosteroids.
Avoid corticosteroids if there is a probability of impending perforation, abscess, or other pyogenic infections; diverticulitis; fresh intestinal anastomoses; or active or latent peptic ulcer.
5.5 Behavioral and Mood Disturbances
Potentially severe psychiatric adverse reactions may occur with systemic corticosteroids, including EMFLAZA. Symptoms typically emerge within a few days or weeks of starting treatment and may be dose-related. These reactions may improve after either dose reduction or withdrawal, although pharmacologic treatment may be necessary. Psychiatric adverse reactions usually involve hypomanic or manic symptoms (e.g., euphoria, insomnia, mood swings) during treatment and depressive episodes after discontinuation of treatment. Inform patients or caregivers of the potential for behavioral and mood changes and encourage them to seek medical attention if psychiatric symptoms develop, especially if depressed mood or suicidal ideation is suspected.
5.6 Effects on Bones
Decreased Bone Mineral Density
Corticosteroids, including EMFLAZA, decrease bone formation and increase bone resorption both through their effect on calcium regulation (i.e., decreasing absorption and increasing excretion) and inhibition of osteoblast function. This, together with a decrease in the protein matrix of the bone secondary to an increase in protein catabolism and reduced sex hormone production, may lead to inhibition of bone growth in pediatric patients and the development of bone loss at any age. Bone loss can predispose patients to vertebral and long bone fractures. Consider a patient’s risk of osteoporosis before initiating corticosteroid therapy. Monitor bone mineral density in patients on long-term treatment with EMFLAZA.Avascular Necrosis
Corticosteroids, including EMFLAZA, may cause avascular necrosis.5.7 Ophthalmic Effects
Use of corticosteroids, including EMFLAZA, may produce posterior subcapsular cataracts. Corticosteroids may also cause glaucoma with possible damage to the optic nerves, and may increase the risk of secondary ocular infections caused by bacteria, fungi, or viruses. Corticosteroids are not recommended for patients with active ocular herpes simplex.
Intraocular pressure may become elevated in some patients taking corticosteroids. If treatment with EMFLAZA is continued for more than 6 weeks, monitor intraocular pressure.
5.8 Immunizations
Administer all immunizations according to immunization guidelines prior to starting EMFLAZA. Administer live-attenuated or live vaccines at least 4 to 6 weeks prior to starting EMFLAZA. Patients on EMFLAZA may receive concurrent vaccinations, except for live-attenuated or live vaccines.
5.9 Serious Skin Rashes
Toxic epidermal necrolysis has been reported with the use of deflazacort with symptoms beginning within 8 weeks of starting treatment. Discontinue at the first sign of rash, unless the rash is clearly not drug related.
5.10 Effects on Growth and Development
Long-term use of corticosteroids, including EMFLAZA, can have negative effects on growth and development in children.
5.11 Myopathy
Patients receiving corticosteroids, including EMFLAZA, and concomitant therapy with neuromuscular blocking agents (e.g., pancuronium) or patients with disorders of neuromuscular transmission (e.g., myasthenia gravis) may be at increased risk of developing acute myopathy. This acute myopathy is generalized, may involve ocular and respiratory muscles, and may result in quadriparesis. Elevation of creatine kinase may occur. Clinical improvement or recovery after stopping corticosteroids may require weeks to years.
5.12 Kaposi’s Sarcoma
Kaposi’s sarcoma has been reported to occur in patients receiving corticosteroid therapy, most often for chronic conditions. Discontinuation of corticosteroids may result in clinical improvement of Kaposi’s sarcoma.
5.13 Risk of Serious Adverse Reactions in Infants because of Benzyl Alcohol Preservative
EMFLAZA Oral Suspension contains benzyl alcohol and is not approved for use in pediatric patients less than 2 years of age. Serious and fatal adverse reactions including “gasping syndrome” can occur in neonates and low birth weight infants treated with benzyl alcohol-preserved drugs, including EMFLAZA. The “gasping syndrome” is characterized by central nervous system depression, metabolic acidosis, and gasping respirations.
When prescribing EMFLAZA Oral Suspension, consider the combined daily metabolic load of benzyl alcohol from all sources, including EMFLAZA Oral Suspension (EMFLAZA Oral Suspension contains 10.45 mg of benzyl alcohol per mL; EMFLAZA Tablets do not contain benzyl alcohol) and other drugs containing benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known. At the recommended dose of 0.9 mg/kg/day of EMFLAZA Oral Suspension, patients would receive approximately 0.4 mg/kg/day of benzyl alcohol [see Use in Specific Populations (8.4)].5.14 Thromboembolic Events
Observational studies have shown an increased risk of thromboembolism (including venous thromboembolism) particularly with higher cumulative doses of corticosteroids. It is unclear if risk differs by daily dose or duration of use. Use EMFLAZA with caution in patients who have or may be predisposed to thromboembolic disorders.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in more detail in other sections:
- Alterations in Endocrine Function [see Warnings and Precautions (5.1)]
- Immunosuppression and Increased Risk of Infection [see Warnings and Precautions (5.2)]
- Alterations in Cardiovascular/Renal Function [see Warnings and Precautions (5.3)]
- Gastrointestinal Perforation [see Warnings and Precautions (5.4)]
- Behavioral and Mood Disturbances [see Warnings and Precautions (5.5)]
- Effects on Bones [see Warnings and Precautions (5.6)]
- Ophthalmic Effects [see Warnings and Precautions (5.7)]
- Immunizations [see Warnings and Precautions (5.8)]
- Serious Skin Rashes [see Warnings and Precautions (5.9)]
- Effects on Growth and Development [see Warnings and Precautions (5.10)]
- Myopathy [see Warnings and Precautions (5.11)]
- Kaposi’s Sarcoma [see Warnings and Precautions (5.12)]
- Risk of Serious Adverse Reactions in Infants because of Benzyl Alcohol Preservative [see Warnings and Precautions (5.13)]
- Thromboembolic Events [see Warnings and Precautions (5.14)]
- Anaphylaxis [see Warnings and Precautions (5.15)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In Study 1 [see Clinical Studies (14)], the adverse reactions that were associated with deflazacort treatment discontinuation, in decreasing order of frequency, were weight increased, obesity, cataract, and sleep disorder.
Most Common Adverse Reactions in Clinical Studies
Table 1 lists the adverse reactions that occurred in ≥ 5% of patients in the 0.9 mg/kg/day deflazacort-treated group and that occurred more frequently than in placebo patients in Study 1, which included patients with DMD between the ages of 5 and 15 years.Table 1: Adverse Reactions that Occurred in ≥ 5% of Deflazacort-Treated Patients and Occurred More Frequently than in Placebo Patients with DMD (Study 1) 1 At 12 weeks placebo patients were re-randomized to receive either deflazacort or an active comparator.
Adverse Reaction Deflazacort
0.9 mg/kg/d (N=51)
% at 12 weeksPlacebo (N=50)
% at 12 weeks1Cushingoid appearance 33 12 Weight increased 20 6 Increased appetite 14 2 Upper respiratory tract infection 12 10 Cough 12 6 Pollakiuria 12 2 Nasopharyngitis 10 6 Hirsutism 10 2 Central obesity 10 4 Erythema 8 6 Irritability 8 4 Rhinorrhea 8 0 Abdominal discomfort 6 2 Common adverse reactions (≥ 5% of deflazacort-treated patients) that occurred over 52 weeks of exposure to deflazacort 0.9 mg/kg/day in Study 1 and at a higher rate than deflazacort 0.9 mg/kg/day in the 12-week placebo-controlled phase of the trial include Cushingoid appearance (60%), hirsutism (35%), weight increased (28%), erythema (28%), central obesity (25%), abdominal pain/abdominal pain upper (18% combined), pollakiuria (15%), constipation (10%), irritability (10%), abnormal behavior (9%), pyrexia (9%), back pain (7%), rash (7%), contusion (6%), nausea (6%), psychomotor hyperactivity (6%), epistaxis (6%), and skin striae (6%).
Study 1 also evaluated a higher dosage of deflazacort (1.2 mg/kg/day). Compared with the 0.9 mg/kg/day dosage, deflazacort 1.2 mg/kg/day over 52 weeks was associated with a higher incidence of certain adverse reactions, including Cushingoid appearance (69%), erythema (49%), hirsutism (37%), headache (34%), weight increased (32%), constipation (15%), abdominal pain upper (14%), skin striae (11%), acne (11%), and abdominal discomfort (8%). As there was no additional benefit with the 1.2 mg/kg/day dose of deflazacort, use of EMFLAZA 1.2 mg/kg/day is not recommended for the treatment of DMD [see Dosage and Administration (2.2)].
In an additional clinical study of two years duration with extended follow-up (Study 2), many of the same adverse reactions were observed. In addition, musculoskeletal events associated with long-term steroid use were also observed, including muscle weakness, tendon disorder, and osteopenia.
Less Common Adverse Reactions Observed in Clinical Studies
Other adverse reactions (≥ 1% frequency in any deflazacort treatment group and greater than placebo) that were observed during the 12-week placebo-controlled phase of Study 1 are shown below.Eye Disorders: Lacrimation increased
Gastrointestinal Disorders: Dyspepsia, nausea, gastrointestinal disorder
General Disorders and Administration Site Conditions: Thirst
Infections: Hordeolum, impetigo, influenza, otitis externa, pharyngitis, tooth abscess, urinary tract infection, viral infection
Injury, Poisoning and Procedural Complications: Back injury, contusion, face injury, fibula fracture, greenstick fracture, heat exhaustion
Investigations: Glucose urine present, heart rate irregular
Musculoskeletal and Connective Tissue Disorders: Back pain, muscle spasms, myalgia, neck mass, neck pain, pain in extremity
Nervous System Disorders: Dizziness, psychomotor hyperactivity
Psychiatric Disorders: Affect lability, aggression, depression, emotional disorder, middle insomnia, mood altered, mood swings, sleep disorder
Renal and Urinary Disorders: Chromaturia, dysuria, hypertonic bladder
Reproductive System and Breast Disorders: Testicular pain
Respiratory, Thoracic, and Mediastinal Disorders: Hypoventilation, rhinorrhea
Skin and Subcutaneous Tissue Disorders: Acne, alopecia, dermatitis acneiform
Vascular Disorders: Hot flush
6.2 Postmarketing Experience
The following adverse reactions have been reported during post-approval use of deflazacort worldwide or during post-approval use of other corticosteroids. These reactions are reported voluntarily from a population of uncertain size; therefore, it is not always possible to estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: Leukocytosis
Cardiac Disorder: Heart failure
Eye Disorders: Chorioretinopathy, corneal or scleral thinning
Gastrointestinal Disorders: Acute pancreatitis (especially in children), hemorrhage, peptic ulceration, perforation of peptic ulcer
General Disorders and Administration Site Conditions: Edema, impaired healing
Immune System Disorders: Hypersensitivity including anaphylaxis
Metabolism and Nutrition Disorders: Impaired carbohydrate tolerance with increased requirement for anti-diabetic therapy, negative protein and calcium balance, potassium loss and hypokalemic alkalosis when co-administered with beta 2-agonist and xanthines
Musculoskeletal and Connective Tissue Disorders: Avascular necrosis, muscle wasting, negative nitrogen balance, tendonitis and tendon rupture when co-administered with quinolones, vertebral and long bone fractures
Nervous System Disorders: Aggravation of epilepsy, increased intra-cranial pressure with papilledema in children (pseudotumor cerebri) usually after treatment withdrawal, vertigo
Psychiatric Disorders: Anxiety, cognitive dysfunction including confusion and amnesia, delusions, hallucinations, mania, suicidal thoughts
Skin and Subcutaneous Tissue Disorders: Toxic epidermal necrolysis
Vascular Disorders: Thromboembolism, in particular in patients with underlying conditions associated with increased thrombotic tendency, benign intracranial hypertension
-
7 DRUG INTERACTIONS
7.1 CYP3A4 Inhibitors and Inducers
Moderate or Strong CYP3A4 Inhibitors
The active metabolite of deflazacort, 21-desDFZ, is a substrate of CYP3A4 [see Clinical Pharmacology (12.3)]. Co-administration of deflazacort with clarithromycin, a strong CYP3A4 inhibitor, increased total exposure to 21-desDFZ by about 3-fold. Therefore, give one third the recommended dosage of EMFLAZA when moderate or strong CYP3A4 inhibitors (e.g., clarithromycin, fluconazole, diltiazem, verapamil, grapefruit juice) are used concomitantly with EMFLAZA [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].Moderate or Strong CYP3A4 Inducers
Co-administration of deflazacort with rifampin, a strong CYP3A4 inducer, significantly decreased the exposure of 21-desDFZ. Avoid concomitant use of strong (e.g., efavirenz) or moderate (e.g., carbamazepine, phenytoin) CYP3A4 inducers with EMFLAZA [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].7.2 Neuromuscular Blockers
Patients receiving corticosteroids, including EMFLAZA, and concomitant therapy with neuromuscular blocking drugs (e.g., pancuronium) may be at increased risk of developing an acute myopathy [see Warnings and Precautions (5.11)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Corticosteroids should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Infants born to mothers who have received substantial doses of corticosteroids during pregnancy should be carefully observed for signs of hypoadrenalism. There are no adequate and well-controlled studies with EMFLAZA in pregnant women to inform drug-associated risks.Corticosteroids, including EMFLAZA, readily cross the placenta. Adverse developmental outcomes, including orofacial clefts (cleft lip, with or without cleft palate) and intrauterine growth restriction, and decreased birth weight, have been reported with maternal use of corticosteroids, including EMFLAZA, during pregnancy. Some epidemiologic studies report an increased risk of orofacial clefts from about 1 per 1000 infants to 3 to 5 per 1000 infants; however, a risk for orofacial clefts has not been observed in all studies. Intrauterine growth restriction and decreased birth weight appear to be dose-related; however, the underlying maternal condition may also contribute to these risks (see Data). The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Animal reproduction studies have not been conducted with deflazacort. Animal reproduction studies conducted with other corticosteroids in pregnant mice, rats, hamsters, and rabbits using clinically relevant doses have shown an increased incidence of cleft palate. An increase in embryofetal death, intrauterine growth retardation, and constriction of the ductus arteriosus were observed in some animal species.
Data
Human Data
Multiple cohort and case-controlled studies in humans suggest that maternal corticosteroid use during the first trimester increases the rate of cleft lip, with or without cleft palate, from about 1/1000 infants to 3-5/1000 infants. Two prospective case-controlled studies showed decreased birth weight in infants exposed to maternal corticosteroids in utero.8.2 Lactation
Risk Summary
Systemically administered corticosteroids appear in human milk and could suppress growth, interfere with endogenous corticosteroid production, or cause other untoward effects. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for EMFLAZA and any potential adverse effects on the breastfed infant from EMFLAZA. There are no data on the effects on milk production.8.4 Pediatric Use
The safety and effectiveness of EMFLAZA for the treatment of DMD have been established in patients 2 years of age and older. Use of EMFLAZA in pediatric patients is supported by a multicenter, randomized, double-blind, placebo- and active-controlled study in 196 males 5 to 15 years of age [see Clinical Studies (14)]. Use of EMFLAZA in patients 2 years to less than 5 years of age is supported by the findings of efficacy and safety in patients 5 years and older with DMD.
Safety and effectiveness in pediatric patients below the age of 2 years have not been established.
EMFLAZA Oral Suspension contains benzyl alcohol and is not approved for use in pediatric patients less than 2 years of age. Serious adverse reactions including fatal reactions and “gasping syndrome” occurred in premature neonates and low birth weight infants in the neonatal intensive care unit who received drugs containing benzyl alcohol as a preservative. In these cases, benzyl alcohol dosages of 99 to 234 mg/kg/day produced high levels of benzyl alcohol and its metabolites in the blood and urine (blood levels of benzyl alcohol were 0.61 to 1.378 mmol/L). Additional adverse reactions included gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Preterm, low-birth weight infants may be more likely to develop these reactions because they may be less able to metabolize benzyl alcohol.
When prescribing EMFLAZA Oral Suspension consider the combined daily metabolic load of benzyl alcohol from all sources including EMFLAZA Oral Suspension (EMFLAZA Oral Suspension contains 10.45 mg of benzyl alcohol per mL; EMFLAZA Tablets do not contain benzyl alcohol) and other drugs containing benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known. At the recommended dose of 0.9 mg/kg/day of EMFLAZA Oral Suspension, patients would receive approximately 0.4 mg/kg/day of benzyl alcohol [see Warnings and Precautions (5.13)].
Juvenile Animal Toxicity Data
Oral administration of deflazacort (0, 0.1, 0.3, and 1.0 mg/kg/day) to juvenile rats from postnatal day (PND) 21 to 80 resulted in decreased body weight gain and adverse effects on skeletal development (including decreased cellularity of growth plate and altered bone distribution) and on lymphoid tissue (decreased cellularity). A no-effect dose was not identified. In addition, neurological and neurobehavioral abnormalities were observed at the mid and/or high dose. Plasma 21-desDFZ exposure (AUC) at the lowest dose tested (0.1 mg/kg/day) was lower than that in humans at the recommended human dose of EMFLAZA (0.9 mg/kg/day).8.5 Geriatric Use
DMD is largely a disease of children and young adults; therefore, there is no geriatric experience with EMFLAZA.
8.6 Renal Impairment
No dose adjustment is required in patients with mild, moderate or severe renal impairment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is required in patients with mild or moderate hepatic impairment [see Clinical Pharmacology (12.3)]. There is no clinical experience in patients with severe hepatic impairment, and a dosing recommendation cannot be provided for patients with severe hepatic impairment.
-
10 OVERDOSAGE
Treatment of acute overdosage is by immediate gastric lavage or emesis followed by supportive and symptomatic therapy. For chronic overdosage in the face of severe disease requiring continuous steroid therapy, the dosage of EMFLAZA may be reduced temporarily, or alternate day treatment may be introduced.
-
11 DESCRIPTION
The active ingredient in EMFLAZA is deflazacort (a corticosteroid). Corticosteroids are adrenocortical steroids, both naturally occurring and synthetic. The molecular formula for deflazacort is C25H31NO6. The chemical name for deflazacort is (11β,16β)-21-(acetyloxy)-11-hydroxy-2'-methyl-5'H-pregna-1,4-dieno[17,16-d]oxazole-3,20-dione, and the structure is:
![The structural formula for EMFLAZA is deflazacort (a corticosteroid). Corticosteroids are adrenocortical steroids, both naturally occurring and synthetic. The molecular formula for deflazacort is C25H31NO6. The chemical name for deflazacort is (11β,16β)-21-(acetyloxy)-11-hydroxy-2'-methyl-5'H-pregna-1,4-dieno[17,16-d]oxazole-3,20-dione.](/dailymed/image.cfm?name=emflaza-01.jpg&setid=31b347d2-f156-4055-9d8f-7cf0df420296)
Deflazacort is a white to off white, odorless fine powder and has a molecular weight of 441.517. Deflazacort is freely soluble in acetic acid and dichloromethane and soluble in methanol and acetone.
EMFLAZA for oral administration is available as an immediate-release tablet in strengths of 6, 18, 30 and 36 mg and an immediate-release oral suspension in a strength of 22.75 mg/mL. Each tablet contains deflazacort and the following inactive ingredients: colloidal silicon dioxide, lactose monohydrate, magnesium stearate, and pre-gelatinized corn starch. The oral suspension contains deflazacort and the following inactive ingredients: acetic acid, aluminum magnesium silicate, benzyl alcohol, carboxymethylcellulose sodium, polysorbate 80, purified water, and sorbitol.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Deflazacort is a corticosteroid prodrug, whose active metabolite, 21-desDFZ, acts through the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. The precise mechanism by which deflazacort exerts its therapeutic effects in patients with DMD is unknown.
12.3 Pharmacokinetics
Absorption
After oral administration in the fasted state, the median Tmax with deflazacort tablets or suspension is about 1 hour (range 0.25 to 2 hours).Food Effect: Co-administration of deflazacort tablets with a high-fat meal reduced Cmax by about 30% and delayed Tmax by one hour, relative to administration under fasting conditions, but there was no effect on the overall systemic absorption as measured by AUC. The bioavailability of deflazacort tablets was similar to that of the oral suspension. The administration of deflazacort with food or crushed in applesauce did not affect the absorption and bioavailability of deflazacort.
Distribution
The protein binding of the active metabolite of deflazacort is about 40%.Elimination
Metabolism
Deflazacort is rapidly converted to the active metabolite 21-desDFZ by esterases after oral administration. 21-desDFZ is further metabolized by CYP3A4 to several other inactive metabolites, including 6β-hydroxy-21-desacetyl deflazacort.Excretion
Urinary excretion is the predominant route of deflazacort elimination (about 68% of the dose), and the elimination is almost completed by 24 hours post dose. 21-desDFZ accounts for 18% of the eliminated drug in the urine.Specific Populations
Pediatric Patients
The Cmax values (Geometric mean, %CV) of 21-desDFZ in children (ages 4-11, N=16) and adolescents (ages 12-16, N=8) was 206 ng/mL (95.6%) and 381 ng/mL (37.7%), respectively, on Day 1 after administration of 0.9 mg/kg deflazacort. The AUCinf (Geometric mean, %CV) of 21-desDFZ in children (ages 4-11, N=16) and adolescents (ages 12-16, N=8) was 400 ng•h/mL (87.5%) and 655 ng•h/mL (58.1%) on Day 1 after administration of 0.9 mg/kg deflazacort.Male and Female Patients
There are no differences in the pharmacokinetics of 21-desDFZ between males and females.Racial or Ethnic Groups
There are no differences in the pharmacokinetics of 21-desDFZ between Caucasians and non-Caucasians.Patients with Renal Impairment
In a study (N=16) comparing subjects with end stage renal disease (creatinine clearance less than 15 mL/min) with healthy matched controls, 21-desDFZ exposure was similar between the groups.Patients with Hepatic Impairment
In a study (N=16) comparing subjects with moderate hepatic impairment (Child-Pugh Class B) with healthy matched controls, 21-desDFZ exposure was similar between the groups. There is no experience in patients with severe hepatic impairment.Drug Interaction Studies
In Vivo Assessment of Drug Interactions
Compared to administration of deflazacort alone, administration of deflazacort following multiple doses of a strong CYP3A4 and Pgp inhibitor (clarithromycin) resulted in markedly higher Cmax, AUClast, and AUCinf values of 21-desDFZ. Geometric mean exposure (Cmax, AUClast, and AUCinf) of 21-desDFZ ranged from 2.3-fold to 3.4-fold higher following administration of clarithromycin [see Dosage and Administration (2.5)].
Compared to administration of deflazacort alone, administration of deflazacort following multiple doses of a strong CYP3A4 inducer (rifampicin) resulted in markedly lower Cmax, AUClast, and AUCinf values of 21-desDFZ. Geometric mean exposures (Cmax, AUClast, and AUCinf) of 21-desDFZ were approximately 95% lower following administration of rifampin [see Drug Interactions (7.1)].
6β-Hydroxy-21-desacetyl deflazacort, a secondary and inactive metabolite, is not expected to cause any clinically meaningful interactions with the CYP enzymes or transporters.
In Vitro Assessment of Drug Interactions
Drug-Metabolizing Enzyme Inhibition
21-desDFZ at clinically relevant concentrations did not inhibit CYP1A2, 2C9, 2C19, 3A4, UGT1A1, UGT1A4, UGT1A6, UGT1A9, or UGT2B7 and exhibited weak and not likely clinically meaningful inhibition for 2B6, 2C8, 2D6, and 3A4, UGT1A3 and UGT2B15.6β-Hydroxy-21-desacetyl deflazacort at clinically relevant concentrations did not significantly inhibit CYP2C19, 3A4 1A2, 2B6, 2C8, 2C9, or 2D6.
Drug-Metabolizing Enzyme Induction
21-desDFZ and 6β-hydroxy-21-desacetyl deflazacort at clinically relevant concentrations did not significantly induce CYP1A2, 2B6, or 3A4.Transporters
Both deflazacort and 21-desDFZ are substrates of Pgp. 21-desDFZ is not a substrate for BCRP. Neither deflazacort nor 21-desDFZ inhibited Pgp or BCRP in vitro. 21-desDFZ was not a substrate for SLC transporters OATP1B1 or OATP1B3, and did not inhibit SLC transporters OATP1B1, OATP1B3, OAT1, OAT3, or OCT2.6β-Hydroxy-21-desacetyl deflazacort at clinically relevant concentrations did not significantly inhibit BCRP, OAT1, OAT3, Pgp, OATP1B1, OATP1B3 MATE1, MATE2-K, OCT1, OCT2, or BSEP transporters.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a published 2-year carcinogenicity study in rats, oral administration of deflazacort (0, 0.03, 0.06, 0.12, 0.25, 0.50, or 1.0 mg/kg/day) resulted in bone tumors (osteosarcoma and osteoma) of the head at 0.25 mg/kg/day, the highest evaluable dose. Doses higher than 0.25 mg/kg/day could not be evaluated for tumors because of a marked decrease in survival.In a 6-month carcinogenicity study in transgenic (Tg.RasH2) mice, oral administration of deflazacort (0, 2, 5, or 20 mg/kg/day in males; 0, 0.5, 2, or 5 mg/kg/day in females) resulted in an increase in stomach tumors (adenoma) at the highest dose tested in males and females.
Mutagenesis
Deflazacort and 21-desDFZ were negative in in vitro (bacterial reverse mutation and human lymphocyte chromosomal aberration) assays and deflazacort was negative in an in vivo (rat micronucleus) assay.Impairment of Fertility
Fertility studies in animals were not conducted with deflazacort. No effects on the male reproductive system were observed following oral administration of deflazacort to monkeys (0, 1.0, 3.0, or 6.0 mg/kg/day) for 39 weeks or rats (0, 0.05, 0.15, or 0.5 mg/kg/day) for 26 weeks. Plasma 21-desDFZ exposures (AUC) at the highest doses tested in monkey and rat were 4 and 2 times, respectively, that in humans at the recommended human dose of EMFLAZA (0.9 mg/kg/day). -
14 CLINICAL STUDIES
The effectiveness of EMFLAZA for the treatment of DMD was established in Study 1, a multicenter, randomized, double-blind, placebo-controlled, 52-week study conducted in the US and Canada. The study population consisted of 196 male pediatric patients 5 to 15 years of age with documented mutation of the dystrophin gene, onset of weakness before 5 years of age, and serum creatinine kinase activity at least 10 times the upper limit of normal (ULN) at some stage in their illness. Patients were randomized to therapy with deflazacort (0.9 or 1.2 mg/kg/day), an active comparator, or placebo. A comparison to placebo was made after 12 weeks of treatment. After 12 weeks, placebo patients were re-randomized to receive either deflazacort or the active comparator; all patients continued treatment for an additional 40 weeks. Baseline characteristics were comparable between the treatment arms.
In Study 1, efficacy was evaluated by assessing the change between Baseline and Week 12 in average strength of 18 muscle groups. Individual muscle strength was graded using a modified Medical Research Council (MRC) 11-point scale, with higher scores representing greater strength.
The change in average muscle strength score between Baseline and Week 12 was significantly greater for the deflazacort 0.9 mg/kg/day dose group than for the placebo group (see Table 2).
Table 2: Analysis of Change from Baseline at Week 12 in Average Muscle Strength Score (Study 1) Treatment N Change from
Baseline
LS Mean (95% CI)P-value Deflazacort 0.9 mg/kg/day 51 0.15 (0.01, 0.28) 0.017 Placebo 50 -0.10 (-0.23, 0.03) Compared with the deflazacort 0.9 mg/kg/day group, the deflazacort 1.2 mg/kg/day group demonstrated a small additional benefit compared to placebo at Week 12, but had a greater incidence of adverse reactions. Therefore, use of a 1.2 mg/kg/day dosage of EMFLAZA is not recommended [see Dosage and Administration (2.2)].
Although not a pre-specified statistical analysis, compared with placebo, the deflazacort 0.9 mg/kg/day dose group demonstrated at Week 52 the persistence of the treatment effect observed at Week 12 and the small advantage of the 1.2 mg/kg/day dose that was observed at Week 12 was no longer present. Also not statistically controlled for multiple comparisons, results on several timed measures of patient function (i.e., time to stand from supine, time to climb 4 stairs, and time to walk or run 30 feet) numerically favored deflazacort 0.9 mg/kg/day at Week 12, in comparison with placebo.
An additional randomized, double-blind, placebo-controlled, 104-week clinical trial evaluated deflazacort in comparison to placebo (Study 2). The study population consisted of 29 male children 6 to 12 years of age with a DMD diagnosis confirmed by the documented presence of abnormal dystrophin or a confirmed mutation of the dystrophin gene. The results of the analysis of the primary endpoint of average muscle strength scores in Study 2 (graded on a 0-5 scale) at 2 years were not statistically significant, possibly because of a limited number of patients remaining in the placebo arm (subjects were discontinued from the trial when they lost ambulation). Although not statistically controlled for multiple comparisons, average muscle strength scores at Months 6 and 12, as well as the average time to loss of ambulation, numerically favored deflazacort in comparison with placebo.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
EMFLAZA Tablets
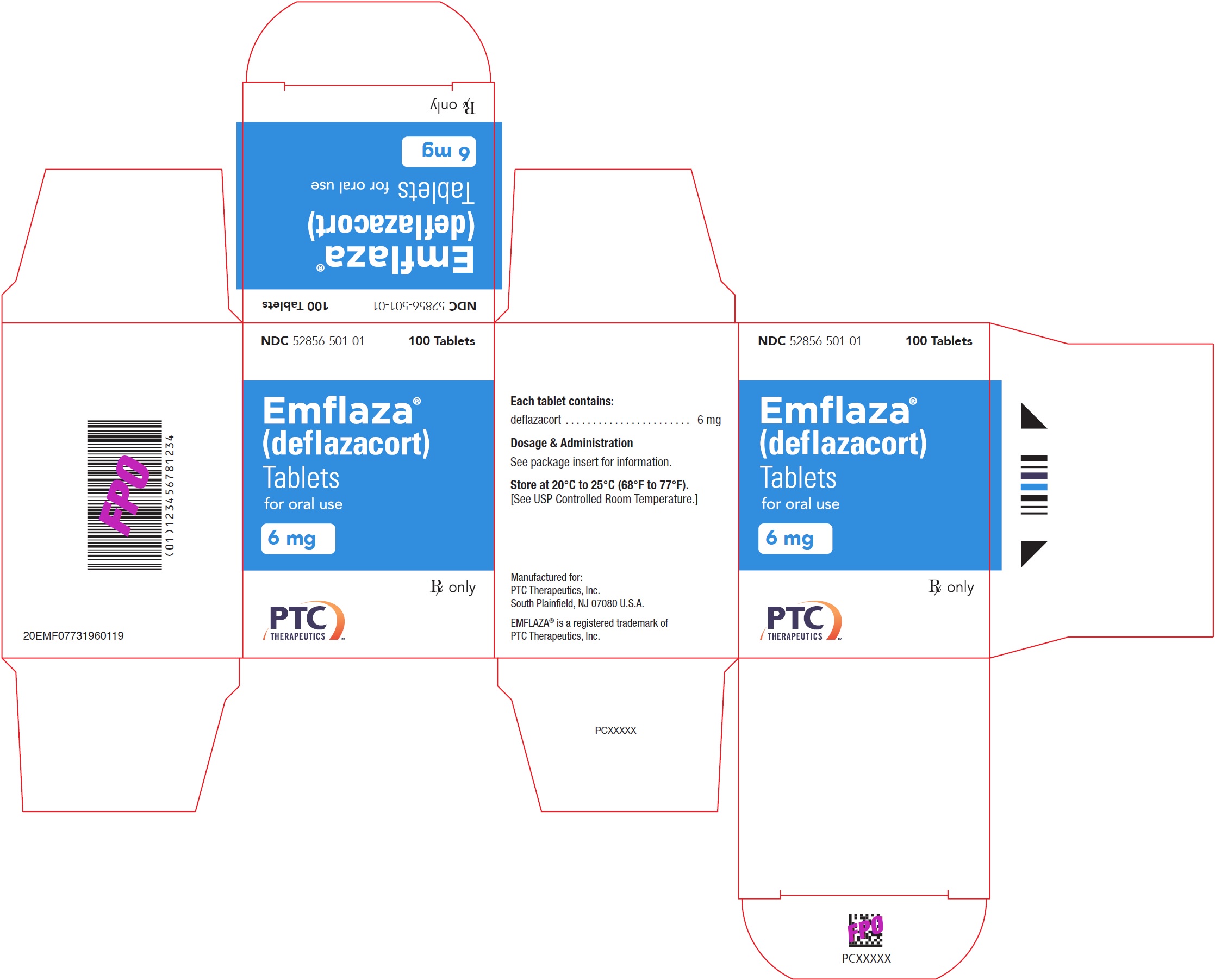
- 6 mg are white, round with “6” debossed on one side. They are supplied as follows:
NDC 52856-501-01 Bottle of 100 tablets
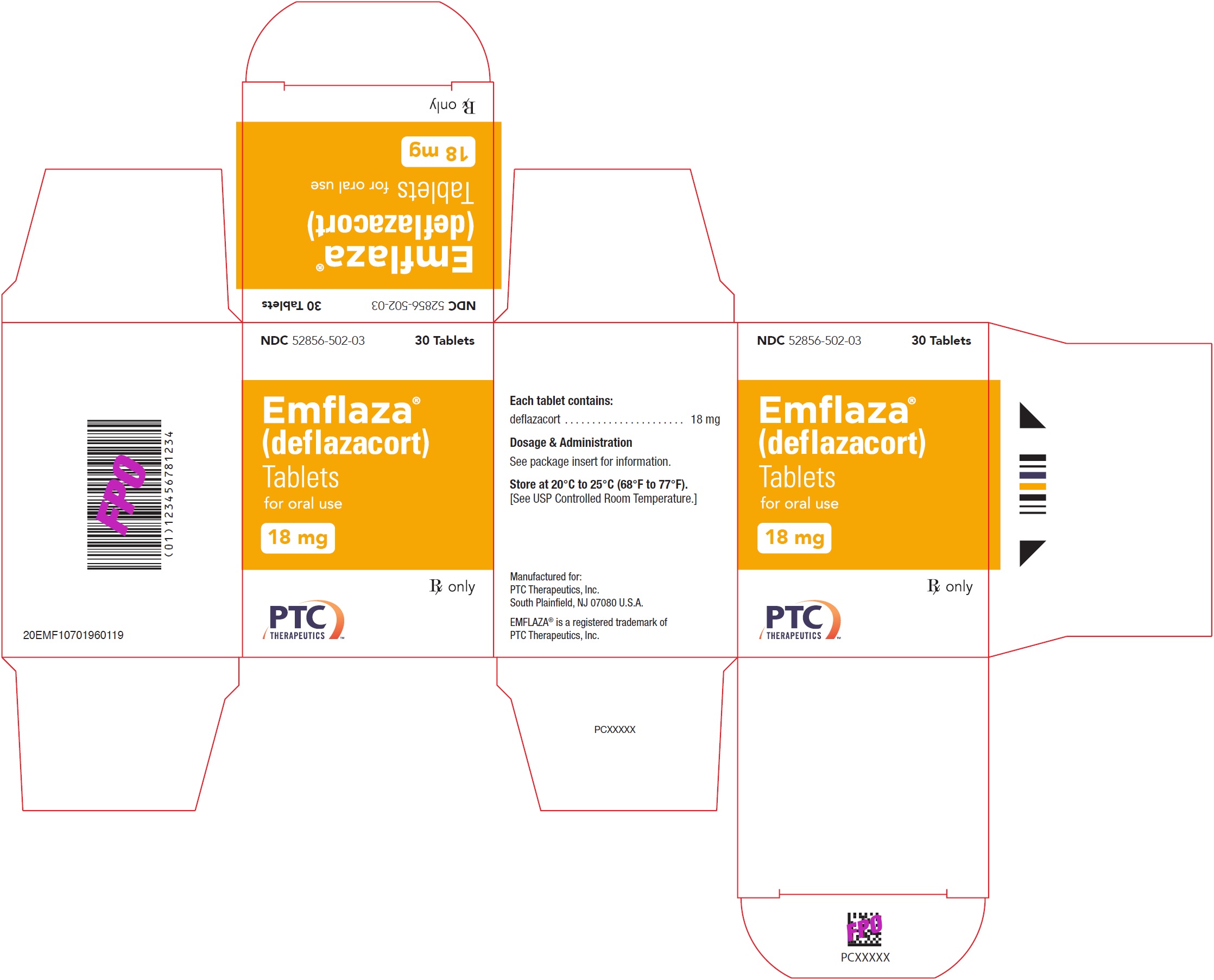
- 18 mg are white, round with “18” debossed on one side. They are supplied as follows:
NDC 52856-502-03 Bottle of 30 tablets
- 30 mg are white, oval with “30” debossed on one side. They are supplied as follows:
NDC 52856-503-03 Bottle of 30 tablets
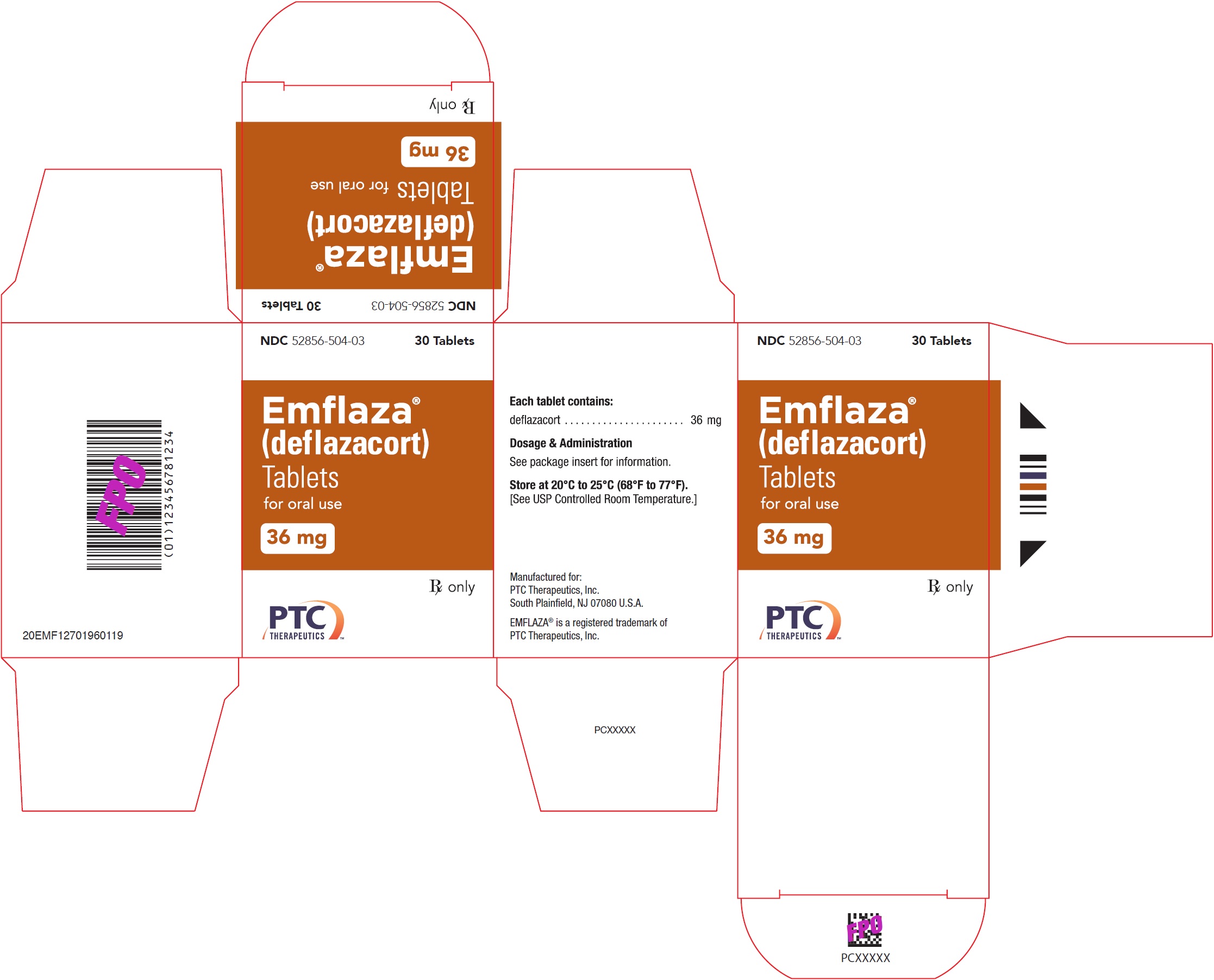
- 36 mg are white, oval with “36” debossed on one side. They are supplied as follows:
NDC 52856-504-03 Bottle of 30 tablets
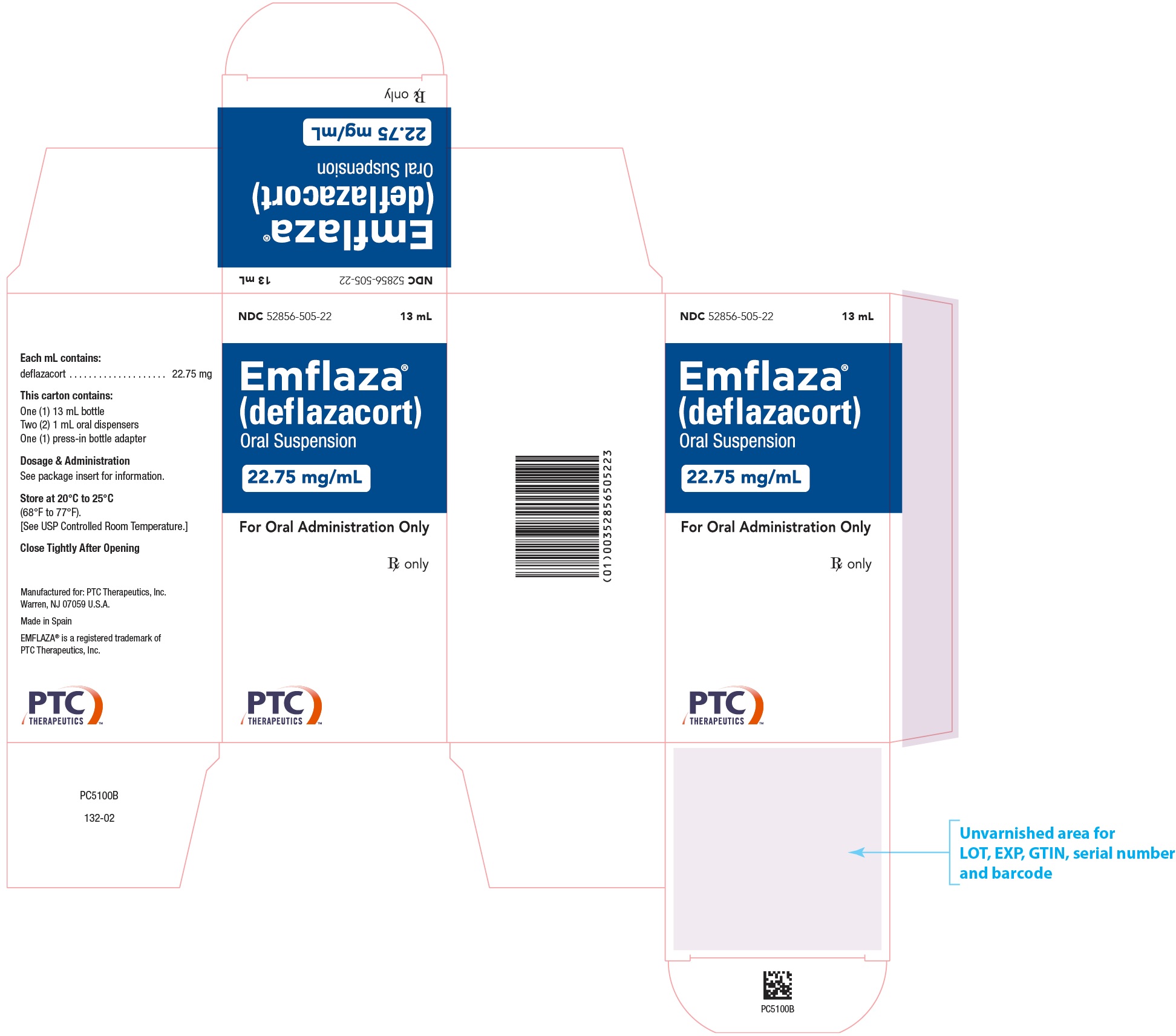
EMFLAZA Oral Suspension
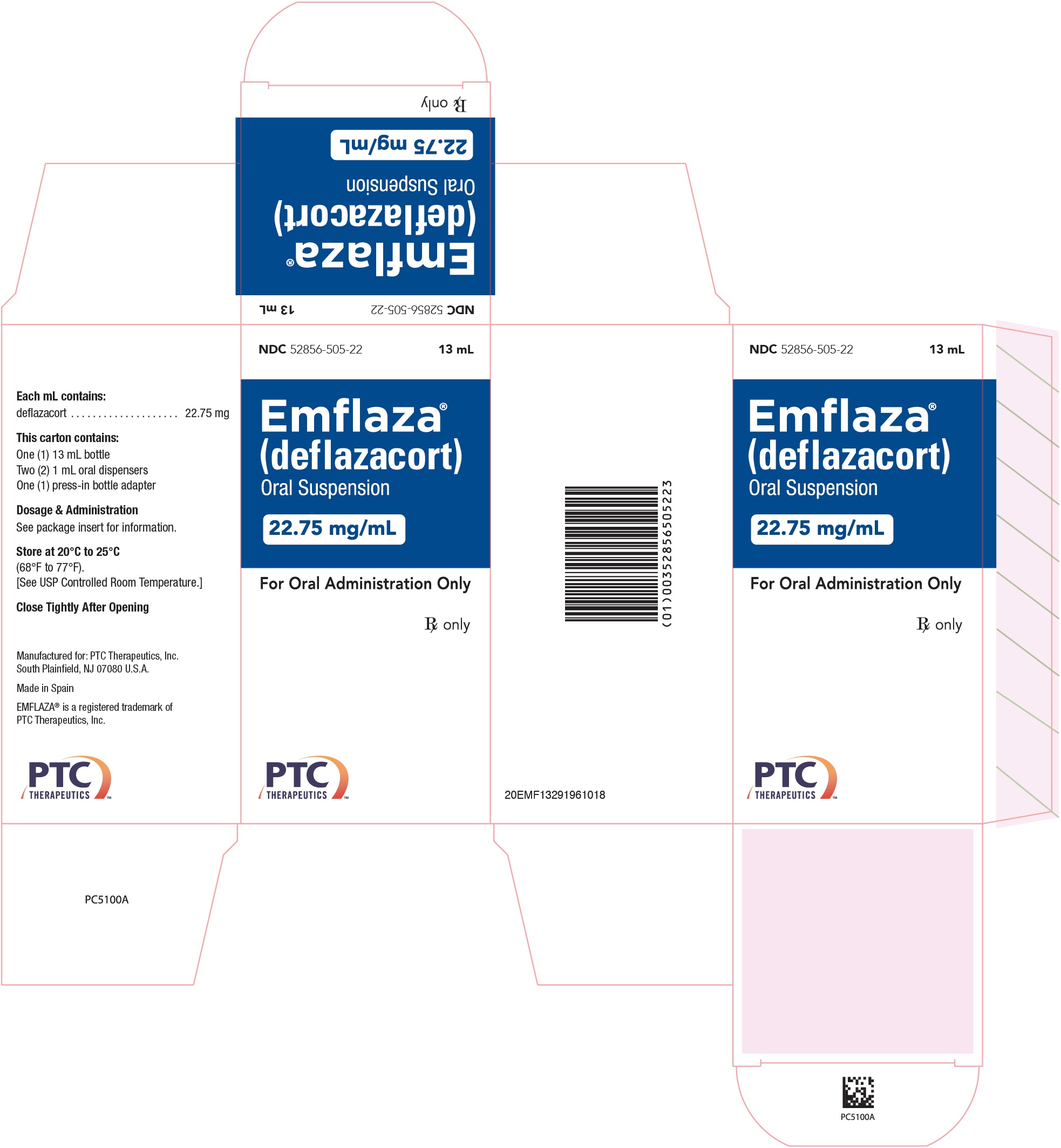
- 22.75 mg/mL is a whitish colored suspension. Supplied as 13 mL in a 30 mL bottle packaged with one bottle adapter and two 1 mL oral dispensers.
NDC 52856-505-22
- 6 mg are white, round with “6” debossed on one side. They are supplied as follows:
-
17 PATIENT COUNSELING INFORMATION
Advise the patients and/or caregivers to read the FDA-approved patient labeling if EMFLAZA Oral Suspension is prescribed (Instructions for Use).
Administration
- Warn patients and/or caregivers to not stop taking EMFLAZA abruptly or without first checking with their healthcare providers as there may be a need for gradual dose reduction to decrease the risk of adrenal insufficiency [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
- EMFLAZA may be taken with or without food. Do not take EMFLAZA with grapefruit juice.
Tablets
- EMFLAZA Tablets may be taken whole or crushed and taken immediately after mixing with applesauce.
Oral Suspension
- EMFLAZA Oral Suspension must be shaken well prior to measuring out each dose with the enclosed oral dispenser.
- The EMFLAZA Oral Suspension dose may be placed in 3-4 ounces of juice (except grapefruit juice) or milk, mixed thoroughly, and immediately administered.
- Discard any unused EMFLAZA Oral Suspension remaining after 1 month of first opening the bottle.
Increased Risk of Infection
Tell patients and/or caregivers to inform their healthcare provider if the patient has had recent or ongoing infections or if they have recently received a vaccine. Medical advice should be sought immediately if the patient develops fever or other signs of infection. Patients and/or caregivers should be made aware that some infections can potentially be severe and fatal.Warn patients who are on corticosteroids to avoid exposure to chickenpox or measles and to alert their healthcare provider immediately if they are exposed [see Warnings and Precautions (5.2)].
Alterations in Cardiovascular/Renal Function
Inform patients and/or caregivers that EMFLAZA can cause an increase in blood pressure and water retention. If this occurs, dietary salt restriction and potassium supplementation may be needed [see Warnings and Precautions (5.3)].Behavioral and Mood Disturbances
Advise patients and/or caregivers about the potential for severe behavioral and mood changes with EMFLAZA and encourage them to seek medical attention if psychiatric symptoms develop [see Warnings and Precautions (5.5)].Decreases in Bone Mineral Density
Advise patients and/or caregivers about the risk of osteoporosis with prolonged use of EMFLAZA, which can predispose the patient to vertebral and long bone fractures [see Warnings and Precautions (5.6)].Ophthalmic Effects
Inform patients and/or caregivers that EMFLAZA may cause cataracts or glaucoma and advise monitoring if corticosteroid therapy is continued for more than 6 weeks [see Warnings and Precautions (5.7)].Vaccination
Advise patients and/or caregivers to bring immunizations up-to-date according to immunization guidelines prior to starting therapy with EMFLAZA. Live-attenuated or live vaccines should be administered at least 4 to 6 weeks prior to starting EMFLAZA. Inform patients and/or caregivers that they may receive concurrent vaccinations with use of EMFLAZA, except for live-attenuated or live vaccines. [see Warnings and Precautions (5.8)].Serious Skin Rashes
Instruct patients and/or caregivers to seek medical attention at the first sign of a rash [see Warnings and Precautions (5.9)].Drug Interactions
Certain medications can cause an interaction with EMFLAZA. Advise patients and/or caregivers to inform their healthcare provider of all the medicines the patient is taking, including over-the-counter medicines (such as insulin, aspirin or other NSAIDS), dietary supplements, and herbal products. Inform patients and/or caregivers that alternate therapy, dosage adjustment, and/or special test(s) may be needed during the treatment.
PTC Therapeutics
Manufactured for:
PTC Therapeutics, Inc.
Warren, NJ 07059 U.S.A.EMFLAZA Oral Suspension made in Spain
EMFLAZA® is a trademark of PTC Therapeutics, Inc.
-
INSTRUCTIONS FOR USE
Instructions for Use
EMFLAZA™ (em fla' zah)
(deflazacort)
oral suspensionRead this Instructions for Use before you start using EMFLAZA oral suspension and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment.
Important information before you use EMFLAZA oral suspension:
- Only use the oral dispenser (see Figure A below) that comes in your EMFLAZA carton when using this medicine.
- EMFLAZA oral suspension can be taken with or without food.
- Take your dose of EMFLAZA oral suspension with juice or milk. Do not take EMFLAZA with grapefruit juice.
- Throw away (discard) any unused EMFLAZA oral suspension after 1 month of first opening the bottle.
- Do not stop taking EMFLAZA oral suspension without talking to your healthcare provider first.
Preparing for your EMFLAZA oral suspension dose:
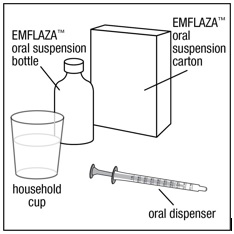
You will need the following supplies: See Figure A.
- 1 EMFLAZA oral suspension bottle
- 1 oral dispenser (2 oral dispensers are included in the EMFLAZA oral suspension carton. 1 is to be used to give (administer) the product and 1 extra oral dispenser is included as a spare, if needed.)
- 1 household cup filled with 3 to 4 ounces of juice or milk
- 1 spoon
Figure A
How to prepare your dose of EMFLAZA oral suspension:
Step 1. Remove the EMFLAZA oral suspension bottle and 1 of the oral dispensers from the carton.
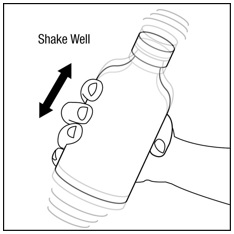
Step 2. Make sure the bottle cap is put on tightly and shake well before each use. See Figure B.
Figure B
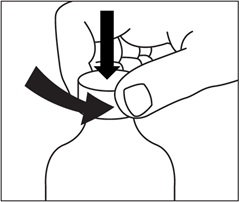
Step 3. Remove the cap from the bottle by pushing down firmly on the cap and turning the cap in a counter-clockwise direction (to the left). See Figure C. Place the open bottle upright on a flat surface.
Figure C
First time use of bottle only:
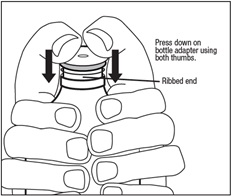
Unwrap the bottle adapter.
Place the ribbed end of the bottle adapter in the bottle top.
Wrap both hands around the bottle. Press down on the bottle adapter using both thumbs. Push the ribbed end of the bottle adapter firmly into the neck of the bottle until the adapter top is even with the bottle top. See Figure D.
Do not remove the bottle adapter from the bottle after it is inserted.
Write the date that you first open the bottle on the bottle label.Figure D
Step 4. Unwrap the oral dispenser if it is the first time you are using the oral dispenser.
Check your dose in milliliters (mL) as prescribed by your healthcare provider. Find this number on the barrel of the oral dispenser. See Figure E.Figure E
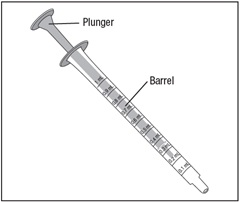
The oral dispenser only holds 1 mL of medicine at a time. If your dose is more than 1 mL, you will need to repeat Steps 5 through 8 using the same oral dispenser until your entire dose has been drawn from the bottle.
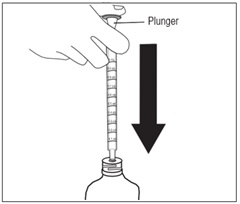
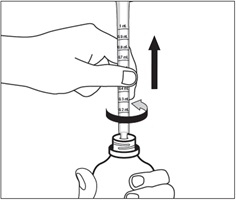
Step 5. Push the plunger of the oral dispenser all the way down. Insert the oral dispenser into the bottle adapter. See Figure F.
Figure F
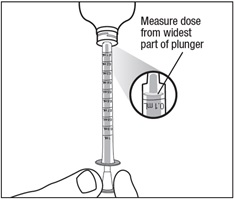
Step 6. With the oral dispenser in the bottle adapter, carefully turn the bottle upside down. Slowly pull back on the plunger until the widest part of the plunger is at the line marking on the oral dispenser of the number of mL needed for your dose. See Figure G. Do not use the narrow tip on the end of the plunger to measure the dose.
If you see air bubbles in the oral suspension, fully push in the plunger so the oral suspension flows back into the bottle. Then withdraw the dose of oral suspension that you need.
Figure G
Step 7. Leave the oral dispenser in the bottle adapter and turn the bottle to an upright position. Place the bottle onto a flat surface. Remove the oral dispenser from the bottle adapter by gently twisting the oral dispenser while pulling it straight up. See Figure H.
Figure H
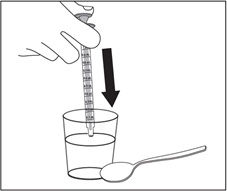
Step 8. Slowly push all the way down on the plunger of the oral dispenser to add the EMFLAZA oral suspension dose to a household cup filled with 3 to 4 ounces of juice or milk. See Figure I.
Figure I
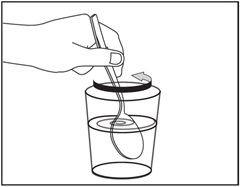
Step 9. Stir the EMFLAZA oral suspension and juice or milk with a spoon to mix well. See Figure J.
Figure J
Step 10. Immediately drink the juice or milk that is mixed with EMFLAZA oral suspension.
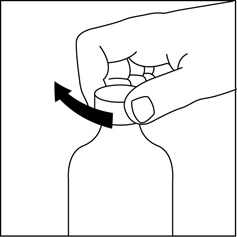
Step 11. Replace the cap tightly on the bottle by turning the cap in a clockwise direction (to the right). See Figure K.
Figure K
Step 12. Wash the oral dispenser after each use. The oral dispenser should be taken apart by pulling back on the plunger and removing it from the barrel of the oral dispenser. See Figure L.
Figure L
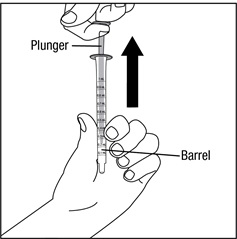
Step 13. After the barrel and plunger are dry, put the oral dispenser back together by pushing the plunger back into the barrel.
How should I store EMFLAZA oral suspension?
- Store the bottle upright at room temperature between 68°F to 77°F (20°C to 25°C).
- Throw away (discard) any unused EMFLAZA oral suspension after 1 month of first opening the bottle.
What are the ingredients in EMFLAZA oral suspension?
Active ingredient: deflazacort
Inactive ingredients: acetic acid, aluminum magnesium silicate, benzyl alcohol, carboxymethylcellulose sodium, polysorbate 80, purified water, and sorbitol.
Manufactured for: PTC Therapeutics, Inc., Warren, NJ 07059, U.S.A.
This Instructions for Use has been approved by the U.S. Food and Drug Administration. EMFLAZA™ is a trademark of PTC Therapeutics, Inc.
20EMF13891961118
Revised: 06/2024
- PRINCIPAL DISPLAY PANEL - NDC: 52856-501-01 - 6 mg Tablet 100-Count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-501-01 - 6 mg Tablet 100-Count Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-502-03 - 18 mg Tablet 30-Count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-502-03 - 18 mg Tablet 30-Count Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-503-03 - 30 mg Tablet 30-Count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-503-03 - 30 mg Tablet 30-Count Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-504-03 - 36 mg Tablet 30-Count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-504-03 - 36 mg Tablet 30-Count Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-505-22 - 22.75 mg/mL Oral Suspension 13 mL Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-505-22 - 22.75 mg/mL Oral Suspension 13 mL Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 52856-505-22 - 22.75 mg/mL Oral Suspension 13 mL Carton Label
-
INGREDIENTS AND APPEARANCE
EMFLAZA
deflazacort tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52856-501 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEFLAZACORT (UNII: KR5YZ6AE4B) (DEFLAZACORT - UNII:KR5YZ6AE4B) DEFLAZACORT 6 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color WHITE Score no score Shape ROUND Size 6mm Flavor Imprint Code 6 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52856-501-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208684 09/10/2018 EMFLAZA
deflazacort tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52856-502 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEFLAZACORT (UNII: KR5YZ6AE4B) (DEFLAZACORT - UNII:KR5YZ6AE4B) DEFLAZACORT 18 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color WHITE Score no score Shape ROUND Size 10mm Flavor Imprint Code 18 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52856-502-03 30 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208684 09/10/2018 EMFLAZA
deflazacort tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52856-503 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEFLAZACORT (UNII: KR5YZ6AE4B) (DEFLAZACORT - UNII:KR5YZ6AE4B) DEFLAZACORT 30 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color WHITE Score no score Shape OVAL Size 15mm Flavor Imprint Code 30 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52856-503-03 30 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208684 09/10/2018 EMFLAZA
deflazacort tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52856-504 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEFLAZACORT (UNII: KR5YZ6AE4B) (DEFLAZACORT - UNII:KR5YZ6AE4B) DEFLAZACORT 36 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color WHITE Score no score Shape OVAL Size 17mm Flavor Imprint Code 36 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52856-504-03 30 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208684 09/10/2018 EMFLAZA
deflazacort suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52856-505 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEFLAZACORT (UNII: KR5YZ6AE4B) (DEFLAZACORT - UNII:KR5YZ6AE4B) DEFLAZACORT 22.75 mg in 1 mL Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) MAGNESIUM ALUMINUM SILICATE (UNII: 6M3P64V0NC) BENZYL ALCOHOL (UNII: LKG8494WBH) CARBOXYMETHYLCELLULOSE SODIUM (UNII: K679OBS311) POLYSORBATE 80 (UNII: 6OZP39ZG8H) WATER (UNII: 059QF0KO0R) SORBITOL (UNII: 506T60A25R) Product Characteristics Color WHITE Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52856-505-22 13 mL in 1 BOTTLE; Type 0: Not a Combination Product 08/30/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208685 09/10/2018 Labeler - PTC Therapeutics, Inc. (124371951)