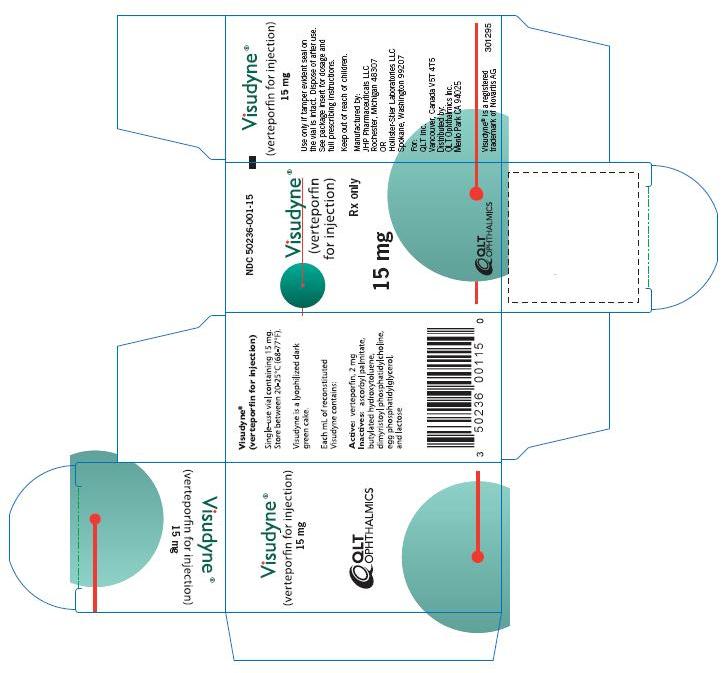
Label: VISUDYNE- verteporfin for injection injection, powder, lyophilized, for solution

-
Contains inactivated NDC Code(s)
NDC Code(s): 50236-001-15 - Packager: QLT Ophthalmics, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated April 7, 2010
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
Visudyne® (verteporfin for injection) is a light activated drug used in photodynamic therapy. The finished drug product is a lyophilized dark green cake. Verteporfin is a 1:1 mixture of two regioisomers (I and II), represented by the following structures:
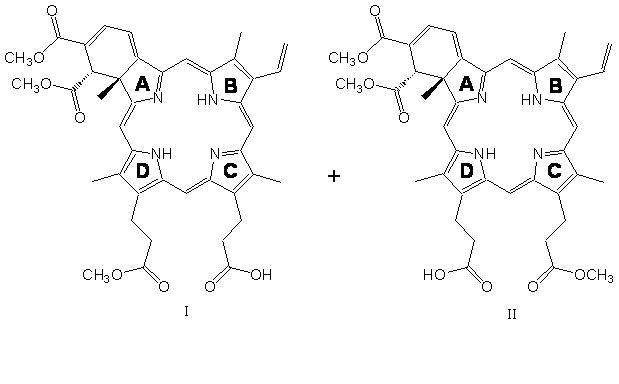
The chemical names for the verteporfin regioisomers are:
9-methyl (I) and 13-methyl (II) trans-(±)-18-ethenyl-4,4a-dihydro-3,4-bis(methoxycarbonyl)-4a,8,14,19-tetramethyl-23H, 25H-benzo[b]porphine-9,13-dipropanoate
The molecular formula is C41H42N4O8 with a molecular weight of approximately 718.8.
Each mL of reconstituted VISUDYNE contains:
ACTIVE: Verteporfin, 2 mg
INACTIVES: ascorbyl palmitate, butylated hydroxytoluene, dimyristoyl phosphatidylcholine, egg phosphatidylglycerol and lactose
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Visudyne® (verteporfin for injection) therapy is a two-stage process requiring administration of both verteporfin for injection and nonthermal red light.
Verteporfin is transported in the plasma primarily by lipoproteins. Once verteporfin is activated by light in the presence of oxygen, highly reactive, short-lived singlet oxygen and reactive oxygen radicals are generated. Light activation of verteporfin results in local damage to neovascular endothelium, resulting in vessel occlusion. Damaged endothelium is known to release procoagulant and vasoactive factors through the lipo-oxygenase (leukotriene) and cyclo-oxygenase (eicosanoids such as thromboxane) pathways, resulting in platelet aggregation, fibrin clot formation and vasoconstriction. Verteporfin appears to somewhat preferentially accumulate in neovasculature, including choroidal neovasculature. However, animal models indicate that the drug is also present in the retina. Therefore, there may be collateral damage to retinal structures following photoactivation including the retinal pigmented epithelium and outer nuclear layer of the retina. The temporary occlusion of choroidal neovascularization (CNV) following Visudyne therapy has been confirmed in humans by fluorescein angiography.
Pharmacokinetics
Following intravenous infusion, verteporfin exhibits a bi-exponential elimination with a terminal elimination half-life of approximately 5-6 hours. The extent of exposure and the maximal plasma concentration are proportional to the dose between 6 and 20 mg/m2. At the intended dose, pharmacokinetic parameters are not significantly affected by gender.
Verteporfin is metabolized to a small extent to its diacid metabolite by liver and plasma esterases. NADPH-dependent liver enzyme systems (including the cytochrome P450 isozymes) do not appear to play a role in the metabolism of verteporfin. Elimination is by the fecal route, with less than 0.01% of the dose recovered in urine.
In a study of patients with mild hepatic insufficiency (defined as having two abnormal hepatic function tests at enrollment), AUC and Cmax were not significantly different from the control group; half-life, however, was significantly increased by approximately 20%.
Clinical Studies
Age-Related Macular Degeneration (AMD)
Two adequate and well-controlled, double-masked, placebo-controlled, randomized studies were conducted in patients with classic-containing subfoveal CNV secondary to age-related macular degeneration. A total of 609 patients (Visudyne 402, placebo 207) were enrolled in these two studies. During these studies, retreatment was allowed every 3 months if fluorescein angiograms showed any recurrence or persistence of leakage. The placebo control (sham treatment) consisted of intravenous administration of Dextrose 5% in Water, followed by light application identical to that used for Visudyne therapy.
The difference between treatment groups statistically favored Visudyne at the 1-year and 2-year analyses for visual acuity endpoints.
The subgroup of patients with predominantly classic CNV lesions was more likely to exhibit a treatment benefit (N=242; Visudyne 159, placebo 83). Predominantly classic CNV lesions were defined as those in which the classic component comprised 50% or more of the area of the entire lesion. For the primary efficacy endpoint (percentage of patients who lost <3 lines of visual acuity), these patients showed a difference of approximately 28% between treatment groups at both Months 12 and 24 (67% for Visudyne patients compared to 40% for placebo patients, at Month 12; and 59% for Visudyne patients compared to 31% for placebo patients, at Month 24). Severe vision loss (≥6 lines of visual acuity from baseline) was experienced by 12% of Visudyne-treated patients compared to 34% of placebo-treated patients at Month 12, and by 15% of Visudyne-treated patients compared to 36% of placebo-treated patients at Month 24.
Patients with predominantly classic CNV lesions that did not contain occult CNV exhibited the greatest benefit (N=134; Visudyne 90, placebo 44). At 1 year, these patients demonstrated a 49% difference between treatment groups when assessed by the <3 lines-lost definition (77% vs. 27%).
Older patients (≥75 years), patients with dark irides, patients with occult lesions or patients with less than 50% classic CNV were less likely to benefit from Visudyne therapy.
The safety and efficacy of Visudyne beyond 2 years have not been demonstrated.
Based on the TAP extension study, the average number of treatments per year were 3.5 in the first year after diagnosis, 2.4 in the second, 1.3 in the third, 0.4 in the fourth and 0.1 in the fifth year.
Pathologic Myopia
One adequate and well-controlled, double-masked, placebo-controlled, randomized study was conducted in patients with subfoveal CNV secondary to pathologic myopia. A total of 120 patients (Visudyne 81, placebo 39) were enrolled in the study. The treatment dosing and retreatments were the same as in the AMD studies. The difference between treatment groups statistically favored Visudyne at the 1-year analysis but not at the 2-year analysis for visual acuity endpoints. For the primary efficacy endpoint (percentage of patients who lost <3 lines of visual acuity), patients at the 1-year timepoint showed a difference of approximately 19% between treatment groups (86% for Visudyne patients compared to 67% for placebo patients). However, by the 2-year timepoint, the effect was no longer statistically significant (79% for Visudyne patients compared to 72% for placebo patients).
Based on the VIP-PM extension study in pathologic myopia, the average number of treatments per year were 3.5 in the first year after diagnosis, 1.8 in the second, 0.4 in the third, 0.2 in the fourth and 0.1 in the fifth.
Presumed Ocular Histoplasmosis
One open-label study was conducted in patients with subfoveal CNV secondary to presumed ocular histoplasmosis. A total of 26 patients were treated with Visudyne in the study. The treatment dosing and retreatments for Visudyne were the same as in the AMD studies. Visudyne-treated patients compare favorably with historical control data demonstrating a reduction in the number of episodes of severe visual acuity loss (>6 lines of loss).
Based on the VOH extension study in presumed ocular histoplasmosis, the average number of treatments per year were 2.9 in the first year after diagnosis, 1.2 in the second, 0.2 in the third and 0.1 in the fourth.
-
INDICATIONS AND USAGE
Visudyne® (verteporfin for injection) therapy is indicated for the treatment of patients with predominantly classic subfoveal choroidal neovascularization due to age-related macular degeneration, pathologic myopia or presumed ocular histoplasmosis.
There is insufficient evidence to indicate Visudyne for the treatment of predominantly occult subfoveal choroidal neovascularization.
- CONTRAINDICATIONS
-
WARNINGS
Following injection with Visudyne® (verteporfin for injection), care should be taken to avoid exposure of skin or eyes to direct sunlight or bright indoor light for 5 days. In the event of extravasation during infusion, the extravasation area must be thoroughly protected from direct light until the swelling and discoloration have faded in order to prevent the occurrence of a local burn which could be severe. If emergency surgery is necessary within 48 hours after treatment, as much of the internal tissue as possible should be protected from intense light.
Patients who experience severe decrease of vision of ≥4 lines within 1 week after treatment should not be retreated, at least until their vision completely recovers to pretreatment levels and the potential benefits and risks of subsequent treatment are carefully considered by the treating physician.
Use of incompatible lasers that do not provide the required characteristics of light for the photoactivation of Visudyne could result in incomplete treatment due to partial photoactivation of Visudyne , overtreatment due to overactivation of Visudyne , or damage to surrounding normal tissue.
-
PRECAUTIONS
General
Standard precautions should be taken during infusion of Visudyne® (verteporfin for injection) to avoid extravasation. Examples of standard precautions include, but are not limited to:
- A free-flowing intravenous (IV) line should be established before starting Visudyne infusion and the line should be carefully monitored.
- Due to the possible fragility of vein walls of some elderly patients, it is strongly recommended that the largest arm vein possible, preferably antecutibal, be used for injection.
- Small veins in the back of the hand should be avoided.
Extravasation of Visudyne, especially if the affected area is exposed to light, can cause severe pain, inflammation, swelling or discoloration at the injection site.
If extravasation does occur, the infusion should be stopped immediately. The extravasation area must be thoroughly protected from direct light until swelling and discoloration have faded in order to prevent the occurrence of a local burn, which could be severe. Cold compresses should be applied to the injection site. (see Warnings). Oral medications for pain relief may be administered.
Visudyne therapy should be considered carefully in patients with moderate to severe hepatic impairment or biliary obstruction since there is no clinical experience with verteporfin in such patients.
There is no clinical data related to the use of Visudyne in anesthetized patients. At a >10-fold higher dose given by bolus injection to sedated or anesthetized pigs, verteporfin caused severe hemodynamic effects, including death, probably as a result of complement activation. These effects were diminished or abolished by pretreatment with antihistamine and they were not seen in conscious nonsedated pigs. VISUDYNE resulted in a concentration-dependent increase in complement activation in human blood in vitro. At 10 µg/mL (approximately 5 times the expected plasma concentration in human patients), there was mild to moderate complement activation. At ≥100 µg/mL, there was significant complement activation. Signs (chest pain, syncope, dyspnea, and flushing) consistent with complement activation have been observed in <1% of patients administered Visudyne. Patients should be supervised during Visudyne infusion.
Information for Patients
Patients who receive Visudyne will become temporarily photosensitive after the infusion. Patients should wear a wristband to remind them to avoid direct sunlight for 5 days. During that period, patients should avoid exposure of unprotected skin, eyes or other body organs to direct sunlight or bright indoor light. Sources of bright light include, but are not limited to, tanning salons, bright halogen lighting and high power lighting used in surgical operating rooms or dental offices. Prolonged exposure to light from light-emitting medical devices such as pulse oximeters should also be avoided for 5 days following Visudyne administration.
If treated patients must go outdoors in daylight during the first 5 days after treatment, they should protect all parts of their skin and their eyes by wearing protective clothing and dark sunglasses. UV sunscreens are not effective in protecting against photosensitivity reactions because photoactivation of the residual drug in the skin can be caused by visible light.
Patients should not stay in the dark and should be encouraged to expose their skin to ambient indoor light, as it will help inactivate the drug in the skin through a process called photobleaching.
Following Visudyne treatment, patients may develop visual disturbances such as abnormal vision, vision decrease, or visual field defects that may interfere with their ability to drive or use machines. Patients should not drive or use machines as long as these symptoms persist.
Drug Interactions
Drug interaction studies in humans have not been conducted with Visudyne.
Verteporfin is rapidly eliminated by the liver, mainly as unchanged drug. Metabolism is limited and occurs by liver and plasma esterases. Microsomal cytochrome P450 does not appear to play a role in verteporfin metabolism.
Based on the mechanism of action of verteporfin, many drugs used concomitantly could influence the effect of Visudyne therapy. Possible examples include the following:
Calcium channel blockers, polymyxin B or radiation therapy could enhance the rate of Visudyne uptake by the vascular endothelium. Other photosensitizing agents (e.g., tetracyclines, sulfonamides, phenothiazines, sulfonylurea hypoglycemic agents, thiazide diuretics and griseofulvin) could increase the potential for skin photosensitivity reactions. Compounds that quench active oxygen species or scavenge radicals, such as dimethyl sulfoxide, β-carotene, ethanol, formate and mannitol, would be expected to decrease Visudyne activity. Drugs that decrease clotting, vasoconstriction or platelet aggregation, e.g., thromboxane A2 inhibitors, could also decrease the efficacy of Visudyne therapy.
Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been conducted to evaluate the carcinogenic potential of verteporfin.
Photodynamic therapy (PDT) as a class has been reported to result in DNA damage including DNA strand breaks, alkali-labile sites, DNA degradation, and DNA-protein cross links which may result in chromosomal aberrations, sister chromatid exchanges (SCE), and mutations. In addition, other photodynamic therapeutic agents have been shown to increase the incidence of SCE in Chinese hamster ovary (CHO) cells irradiated with visible light and in Chinese hamster lung fibroblasts irradiated with near UV light, increase mutations and DNA-protein cross-linking in mouse L5178 cells, and increase DNA-strand breaks in malignant human cervical carcinoma cells, but not in normal cells. Verteporfin was not evaluated in these latter systems. It is not known how the potential for DNA damage with PDT agents translates into human risk.
No effect on male or female fertility has been observed in rats following intravenous administration of verteporfin for injection up to 10 mg/kg/day (approximately 60- and 40-fold human exposure at 6 mg/m2 based on AUCinf in male and female rats, respectively).
Pregnancy
Teratogenic Effects: Pregnancy Category C
Rat fetuses of dams administered verteporfin for injection intravenously at ≥10 mg/kg/day during organogenesis (approximately 40-fold human exposure at 6 mg/m2 based on AUCinf in female rats) exhibited an increase in the incidence of anophthalmia/microphthalmia. Rat fetuses of dams administered 25 mg/kg/day (approximately 125 fold the human exposure at 6 mg/m2 based on AUCinf in female rats) had an increased incidence of wavy ribs and anophthalmia/microphthalmia.
In pregnant rabbits, a decrease in body weight gain and food consumption was observed in animals that received verteporfin for injection intravenously at ≥10 mg/kg/day during organogenesis. The no observed adverse effect level (NOAEL) for maternal toxicity was 3 mg/kg/day (approximately 7-fold human exposure at 6 mg/m2 based on body surface area). There were no teratogenic effects observed in rabbits at doses up to 10 mg/kg/day.
There are no adequate and well-controlled studies in pregnant women. Visudyne should be used during pregnancy only if the benefit justifies the potential risk to the fetus.
Nursing Mothers
Verteporfin and its diacid metabolite have been found in the breast milk of one woman after a 6 mg/m2 infusion. The verteporfin breast milk levels were up to 66% of the corresponding plasma levels and declined below the limit of quantification (2 ng/mL) within 24 hours. The diacid metabolite had lower peak concentrations but persisted up to at least 48 hours.
Because of the potential for serious adverse reactions in nursing infants from Visudyne, a decision should be made whether to discontinue nursing or postpone treatment, taking into account the importance of the drug to the mother.
-
ADVERSE REACTIONS
Severe chest pain, vasovagal and hypersensitivity reactions have been reported. Vasovagal and hypersensitivity reactions on rare occasions can be severe. These reactions may include syncope, sweating, dizziness, rash, dyspnea, flushing and changes in blood pressure and heart rate. General symptoms can include headache, malaise, urticaria, and pruritus.
The most frequently reported adverse events to Visudyne® (verteporfin for injection) are injection site reactions (including pain, edema, inflammation, extravasation, rashes, hemorrhage and discoloration) and visual disturbances (including blurred vision, flashes of light, decreased visual acuity and visual field defects, including scotoma). These events occurred in approximately 10%-30% of patients. The following events, listed by Body System, were reported more frequently with Visudyne therapy than with placebo therapy and occurred in 1%-10% of patients:
Ocular Treatment Site: Blepharitis, cataracts, conjunctivitis/conjunctival injection, dry eyes, ocular itching, severe vision decrease with or without subretinal/retinal or vitreous hemorrhage
Body as a Whole: Asthenia, fever, flu syndrome, infusion related pain primarily presenting as back pain, photosensitivity reactions
Cardiovascular: Atrial fibrillation, hypertension, peripheral vascular disorder, varicose veins
Dermatologic: Eczema
Digestive: Constipation, gastrointestinal cancers, nausea
Hemic and Lymphatic: Anemia, white blood cell count decreased, white blood cell count increased
Hepatic: Elevated liver function tests
Metabolic/Nutritional: Albuminuria, creatinine increased
Musculoskeletal: Arthralgia, arthrosis, myasthenia
Nervous System: Hypesthesia, sleep disorder, vertigo
Respiratory: Cough, pharyngitis, pneumonia
Special Senses: Cataracts, decreased hearing, diplopia, lacrimation disorder
Urogenital: Prostatic disorder
Severe vision decrease, equivalent of ≥4 lines, within 7 days after treatment has been reported in 1%-5% of patients. Partial recovery of vision was observed in some patients. Photosensitivity reactions usually occurred in the form of skin sunburn following exposure to sunlight. The higher incidence of back pain in the Visudyne group occurred primarily during infusion.
The following adverse events have occurred either at low incidence (<1%) during clinical trials or have been reported during the use of Visudyne in clinical practice where these events were reported voluntarily from a population of unknown size and frequency of occurrence cannot be determined precisely. They have been chosen for inclusion based on factors such as seriousness, frequency of reporting, possible causal connection to Visudyne, or a combination of these factors:
Ocular Treatment Site: Retinal detachment (nonrhegmatogenous), retinal or choroidal vessel nonperfusion, retinal pigment epithelial tear
Non-ocular Events: Chest pain and other musculoskeletal pain during infusion
-
OVERDOSAGE
Overdose of drug and/or light in the treated eye may result in nonperfusion of normal retinal vessels with the possibility of severe decrease in vision that could be permanent. An overdose of drug will also result in the prolongation of the period during which the patient remains photosensitive to bright light. In such cases, it is recommended to extend the photosensitivity precautions for a time proportional to the overdose.
-
DOSAGE AND ADMINISTRATION
A course of Visudyne® (verteporfin for injection) therapy is a two-step process requiring administration of both drug and light.
The first step is the intravenous infusion of Visudyne. The second step is the activation of Visudyne with light from a nonthermal diode laser.
The physician should reevaluate the patient every 3 months and if choroidal neovascular leakage is detected on fluorescein angiography, therapy should be repeated.
Lesion Size Determination
The greatest linear dimension (GLD) of the lesion is estimated by fluorescein angiography and color fundus photography. All classic and occult CNV, blood and/or blocked fluorescence, and any serous detachments of the retinal pigment epithelium should be included for this measurement. Fundus cameras with magnification within the range of 2.4-2.6X are recommended. The GLD of the lesion on the fluorescein angiogram must be corrected for the magnification of the fundus camera to obtain the GLD of the lesion on the retina.
Spot Size Determination
The treatment spot size should be 1000 microns larger than the GLD of the lesion on the retina to allow a 500 micron border, ensuring full coverage of the lesion. The maximum spot size used in the clinical trials was 6400 microns.
The nasal edge of the treatment spot must be positioned at least 200 microns from the temporal edge of the optic disc, even if this will result in lack of photoactivation of CNV within 200 microns of the optic nerve.
Visudyne® Administration
Reconstitute each vial of Visudyne with 7 mL of sterile Water for Injection to provide 7.5 mL containing 2 mg/mL. Reconstituted Visudyne must be protected from light and used within 4 hours. It is recommended that reconstituted Visudyne be inspected visually for particulate matter and discoloration prior to administration. Reconstituted Visudyne is an opaque dark green solution. Visudyne may precipitate in saline solutions. Do not use normal saline or other parenteral solutions, except 5% Dextrose for Injection, for dilution of the reconstituted Visudyne. Do not mix Visudyne in the same solution with other drugs.
The volume of reconstituted Visudyne required to achieve the desired dose of 6 mg/m2 body surface area is withdrawn from the vial and diluted with 5% Dextrose for Injection to a total infusion volume of 30 mL. After dilution, protect from light and use within a maximum of 4 hours. The full infusion volume is administered intravenously over 10 minutes at a rate of 3 mL/minute, using an appropriate syringe pump and in-line filter. The clinical studies were conducted using a standard infusion line filter of 1.2 microns.
Precautions should be taken to prevent extravasation at the injection site. If extravasation occurs, protect the site from light (See PRECAUTIONS).
Light Administration
Initiate 689 nm wavelength laser light delivery to the patient 15 minutes after the start of the 10-minute infusion with Visudyne.
Photoactivation of Visudyne is controlled by the total light dose delivered. In the treatment of choroidal neovascularization, the recommended light dose is 50 J/cm2 of neovascular lesion administered at an intensity of 600 mW/cm2. This dose is administered over 83 seconds.
Light dose, light intensity, ophthalmic lens magnification factor and zoom lens setting are important parameters for the appropriate delivery of light to the predetermined treatment spot. Follow the laser system manuals for procedure set up and operation.
The laser system must deliver a stable power output at a wavelength of 689±3 nm. Light is delivered to the retina as a single circular spot via a fiber optic and a slit lamp, using a suitable ophthalmic magnification lens.
The following laser systems have been tested for compatibility with Visudyne and are approved for delivery of a stable power output at a wavelength of 689±3 nm:
Coherent Opal Photoactivator laser console and modified Coherent LaserLink adapter, manufactured by Lumenis, Inc., 2400 Condensa Street, Santa Clara, CA 95051-0901,
Zeiss VISULAS 690s laser and VISULINK® PDT adapter manufactured by Carl Zeiss Meditec Inc., 5160 Hacienda Drive, Dublin, CA 94568,
Ceralas™ I laser system and Ceralink™ Slit Lamp Adapter manufactured by Biolitec Inc., 515 Shaker Road, East Longmeadow, MA 01028,
Quantel Activis laser console and the ZSL30 ACT™, ZSL120 ACT™ and HSBMBQ ACT™ slit lamp adapters distributed by Quantel Medical, 601 Haggerty Lane, Bozeman, MT 59715.
Concurrent Bilateral Treatment
The controlled trials only allowed treatment of one eye per patient. In patients who present with eligible lesions in both eyes, physicians should evaluate the potential benefits and risks of treating both eyes concurrently. If the patient has already received previous Visudyne therapy in one eye with an acceptable safety profile, both eyes can be treated concurrently after a single administration of Visudyne. The more aggressive lesion should be treated first, at 15 minutes after the start of infusion. Immediately at the end of light application to the first eye, the laser settings should be adjusted to introduce the treatment parameters for the second eye, with the same light dose and intensity as for the first eye, starting no later than 20 minutes from the start of infusion.
In patients who present for the first time with eligible lesions in both eyes without prior Visudyne therapy, it is prudent to treat only one eye (the most aggressive lesion) at the first course. One week after the first course, if no significant safety issues are identified, the second eye can be treated using the same treatment regimen after a second Visudyne infusion. Approximately 3 months later, both eyes can be evaluated and concurrent treatment following a new Visudyne infusion can be started if both lesions still show evidence of leakage.
-
HOW SUPPLIED
Visudyne (verteporfin for injection) is supplied in a single use glass vial with a gray bromobutyl stopper and aluminum flip-off cap. It contains a lyophilized dark green cake with 15 mg verteporfin. The product is intended for intravenous injection only.
Spills and Disposal
Spills of Visudyne should be wiped up with a damp cloth. Skin and eye contact should be avoided due to the potential for photosensitivity reactions upon exposure to light. Use of rubber gloves and eye protection is recommended. All materials should be disposed of properly.
Accidental Exposure
Because of the potential to induce photosensitivity reactions, it is important to avoid contact with the eyes and skin during preparation and administration of Visudyne. Any exposed person must be protected from bright light (See WARNINGS).
NDC 50236-001-15
Store Visudyne between 20°C and 25°C (68°F-77°F).
REV: JANUARY 2010
301293 CC# 100020
QLT Inc.
Vancouver, Canada V5T 4T5
Distributed by:
QLT Ophthalmics Inc.
Menlo Park CA 94025
Visudyne® is a registered trademark of Novartis AG
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
VISUDYNE
verteporfin for injection injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50236-001 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VERTEPORFIN (UNII: 0X9PA28K43) (VERTEPORFIN - UNII:0X9PA28K43) VERTEPORFIN 15 mg Inactive Ingredients Ingredient Name Strength ASCORBYL PALMITATE (UNII: QN83US2B0N) BUTYLATED HYDROXYTOLUENE (UNII: 1P9D0Z171K) LACTOSE (UNII: J2B2A4N98G) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50236-001-15 1 in 1 CARTON Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021119 04/12/2000 Labeler - QLT Ophthalmics, Inc. (832856848)