Label: AMILORIDE HYDROCHLORIDE tablet

- NDC Code(s): 63629-2114-1
- Packager: Bryant Ranch Prepack
- This is a repackaged label.
- Source NDC Code(s): 49884-117
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated November 28, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
Amiloride HCl, an antikaliuretic-diuretic agent, is a pyrazine-carbonyl-guanidine that is unrelated chemically to other known antikaliuretic or diuretic agents. It is the salt of a moderately strong base (pKa 8.7). It is designated chemically as 3,5-diamino-6-chloro-N-(diaminomethylene) pyrazinecarboxamide monohydrochloride, dihydrate and has a molecular weight of 302.12. Its empirical formula is C 6H8CIN7O•HCl•2H2O and its structural formula is:
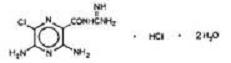
Each tablet for oral administration contains 5 mg of Amiloride HCI, calculated on the anhydrous basis. Each tablet contains the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, D&C yellow #10 lake, dibasic calcium phosphate dihydrate, FD&C yellow #6 lake, magnesium stearate, microcrystalline cellulose and silicon dioxide.
-
CLINICAL PHARMACOLOGY
Amiloride HCl is a potassium-conserving (antikaliuretic) drug that possesses weak (compared with thiazide diuretics) natriuretic, diuretic, and antihypertensive activity. These effects have been partially additive to the effects of thiazide diuretics in some clinical studies. When administered with a thiazide or loop diuretic, amiloride has been shown to decrease the enhanced urinary excretion of magnesium which occurs when a thiazide or loop diuretic is used alone. Amiloride has potassium-conserving activity in patients receiving kaliureticdiuretic agents.
Amiloride HCl is not an aldosterone antagonist and its effects are seen even in the absence of aldosterone.
Amiloride exerts its potassium sparing effect through the inhibition of sodium reabsorption at the distal convoluted tubule, cortical collecting tubule and collecting duct; this decreases the net negative potential of the tubular lumen and reduces both potassium and hydrogen secretion and their subsequent excretion. This mechanism accounts in large part for the potassium sparing action of amiloride.
Amiloride usually begins to act within 2 hours after an oral dose. Its effect on electrolyte excretion reaches a peak between 6 and 10 hours and lasts about 24 hours. Peak plasma levels are obtained in 3 to 4 hours and the plasma half-life varies from 6 to 9 hours. Effects on electrolytes increase with single doses of amiloride HCl up to approximately 15 mg.
Amiloride HCl is not metabolized by the liver but is excreted unchanged by the kidneys. About 50 percent of a 20 mg dose of amiloride HCl is excreted in the urine and 40 percent in the stool within 72 hours. Amiloride has little effect on glomerular filtration rate or renal blood flow. Because amiloride HCl is not metabolized by the liver, drug accumulation is not anticipated in patients with hepatic dysfunction, but accumulation can occur if the hepatorenal syndrome develops.
-
INDICATIONS AND USAGE
Amiloride HCl is indicated as adjunctive treatment with thiazide diuretics or other kaliureticdiuretic agents in congestive heart failure or hypertension to:
- help restore normal serum potassium levels in patients who develop hypokalemia on the kaliuretic diuretic.
- prevent development of hypokalemia in patients who would be exposed to particular risk if hypokalemia were to develop, e.g., digitalized patients or patients with significant cardiac arrhythmias.
The use of potassium-conserving agents is often unnecessary in patients receiving diuretics for uncomplicated essential hypertension when such patients have a normal diet. Amiloride HCl has little additive diuretic or antihypertensive effect when added to a thiazide diuretic.
Amiloride HCl should rarely be used alone. It has weak (compared with thiazides) diuretic and antihypertensive effects. Used as single agents, potassium sparing diuretics, including amiloride HCl, result in an increased risk of hyperkalemia (approximately 10% with amiloride). Amiloride HCl should be used alone only when persistent hypokalemia has been documented and only with careful titration of the dose and close monitoring of serum electrolytes.
-
CONTRAINDICATIONS
Hyperkalemia
Amiloride HCl should not be used in the presence of elevated serum potassium levels (greater than 5.5 mEq per liter).
Antikaliuretic Therapy or Potassium Supplementation
Amiloride HCl should not be given to patients receiving other potassium-conserving agents, such as spironolactone or triamterene. Potassium supplementation in the form of medication, potassium-containing salt substitutes or a potassium-rich diet should not be used with amiloride HCl except in severe and/or refractory cases of hypokalemia. Such concomitant therapy can be associated with rapid increases in serum potassium levels. If potassium supplementation is used, careful monitoring of the serum potassium level is necessary.
Impaired Renal Function
Anuria, acute or chronic renal insufficiency, and evidence of diabetic nephropathy are contraindications to the use of amiloride HCl. Patients with evidence of renal functional impairment (blood urea nitrogen [BUN] levels over 30 mg per 100 mL or serum creatinine levels over 1.5 mg per 100 mL) or diabetes mellitus should not receive the drug without careful, frequent and continuing monitoring of serum electrolytes, creatinine, and BUN levels. Potassium retention associated with the use of an antikaliuretic agent is accentuated in the presence of renal impairment and may result in the rapid development of hyperkalemia.
Hypersensitivity
Amiloride HCl is contraindicated in patients who are hypersensitive to this product.
-
WARNINGS
Hyperkalemia
Like other potassium-conserving agents, amiloride may cause hyperkalemia (serum potassium levels greater than 5.5 mEq per liter) which, if uncorrected, is potentially fatal. Hyperkalemia occurs commonly (about 10%) when amiloride is used without a kaliuretic diuretic. This incidence is greater in patients with renal impairment, diabetes mellitus (with or without recognized renal insufficiency), and in the elderly. When amiloride is used concomitantly with a thiazide diuretic in patients without these complications, the risk of hyperkalemia is reduced to about 1-2%. It is thus essential to monitor serum potassium levels carefully in any patient receiving amiloride, particularly when it is first introduced, at the time of diuretic dosage adjustments, and during any illness that could affect renal function.
The risk of hyperkalemia may be increased when potassium-conserving agents, including amiloride HCl, are administered concomitantly with an angiotensin-converting enzyme inhibitor, an angiotensin II receptor antagonist, cyclosporine or tacrolimus. (See PRECAUTIONS, Drug Interactions.) Warning signs or symptoms of hyperkalemia include paresthesias, muscular weakness, fatigue, flaccid paralysis of the extremities, bradycardia, shock, and ECG abnormalities. Monitoring of the serum potassium level is essential because mild hyperkalemia is not usually associated with an abnormal ECG.
When abnormal, the ECG in hyperkalemia is characterized primarily by tall, peaked T waves or elevations from previous tracings. There may also be lowering of the R wave and increased depth of the S wave, widening and even disappearance of the P wave, progressive widening of the QRS complex, prolongation of the PR interval, and ST depression.
Treatment of hyperkalemia:If hyperkalemia occurs in patients taking amiloride HCl, the drug should be discontinued immediately. If the serum potassium level exceeds 6.5 mEq per liter, active measures should be taken to reduce it. Such measures include the intravenous administration of sodium bicarbonate solution or oral or parenteral glucose with a rapid-acting insulin preparation. If needed, a cation exchange resin such as sodium polystyrene sulfonate may be given orally or by enema. Patients with persistent hyperkalemia may require dialysis.
Diabetes Mellitus
In diabetic patients, hyperkalemia has been reported with the use of all potassium-conserving diuretics, including amiloride HCl, even in patients without evidence of diabetic nephropathy. Therefore, amiloride HCl should be avoided, if possible, in diabetic patients and, if it is used, serum electrolytes and renal function must be monitored frequently.
Amiloride HCl should be discontinued at least 3 days before glucose tolerance testing.
Metabolic or Respiratory Acidosis
Antikaliuretic therapy should be instituted only with caution in severely ill patients in whom respiratory or metabolic acidosis may occur, such as patients with cardiopulmonary disease or poorly controlled diabetes. If amiloride HCl is given to these patients, frequent monitoring of acid-base balance is necessary. Shifts in acid-base balance alter the ratio of extracellular/intracellular potassium, and the development of acidosis may be associated with rapid increases in serum potassium levels.
-
GENERAL PRECAUTIONS
Electrolyte Imbalance and BUN Increases
Hyponatremia and hypochloremia may occur when amiloride HCl is used with other diuretics and increases in BUN levels have been reported. These increases usually have accompanied vigorous fluid elimination, especially when diuretic therapy was used in seriously ill patients, such as those who had hepatic cirrhosis with ascites and metabolic alkalosis, or those with resistant edema. Therefore, when amiloride HCl is given with other diuretics to such patients, careful monitoring of serum electrolytes and BUN levels is important. In patients with pre-existing severe liver disease, hepatic encephalopathy, manifested by tremors, confusion, and coma, and increased jaundice, have been reported in association with diuretics, including amiloride HCl.
Drug Interactions
When amiloride HCl is administered concomitantly with an angiotensin-converting enzyme inhibitor, an angiotensin II receptor antagonist, cyclosporine or tacrolimus, the risk of hyperkalemia may be increased. Therefore, if concomitant use of these agents is indicated because of demonstrated hypokalemia, they should be used with caution and with frequent monitoring of serum potassium. (See WARNINGS).
Lithium generally should not be given with diuretics because they reduce its renal clearance and add a high risk of lithium toxicity. Read circulars for lithium preparations before use of such concomitant therapy.
In some patients, the administration of a non-steroidal anti-inflammatory agent can reduce the diuretic, natriuretic, and antihypertensive effects of loop, potassium-sparing and thiazide diuretics. Therefore, when amiloride HCl and non-steroidal anti-inflammatory agents are used concomitantly, the patient should be observed closely to determine if the desired effect of the diuretic is obtained. Since indomethacin and potassium-sparing diuretics, including amiloride HCl, may each be associated with increased serum potassium levels, the potential effects on potassium kinetics and renal function should be considered when these agents are administered concurrently.
Carcinogenesis and Mutagenesis and Impairment of Fertility
There was no evidence of a tumorigenic effect when amiloride HCl was administered for 92 weeks to mice at doses up to 10 mg/kg/day (25 times the maximum daily human dose). Amiloride HCl has also been administered for 104 weeks to male and female rats at doses up to 6 and 8 mg/kg/day (15 and 20 times the maximum daily dose for humans, respectively) and showed no evidence of carcinogenicity.
Amiloride HCl was devoid of mutagenic activity in various strains of Salmonella typhimurium with or without a mammalian liver microsomal activation system (Ames test).
Pregnancy
Pregnancy Category B. Teratogenicity studies with amiloride HCl in rabbits and mice given 20 and 25 times the maximum human dose, respectively, revealed no evidence of harm to the fetus, although studies showed that the drug crossed the placenta in modest amounts. Reproduction studies in rats at 20 times the expected maximum daily dose for humans showed no evidence of impaired fertility. At approximately 5 or more times the expected maximum daily dose for humans, some toxicity was seen in adult rats and rabbits and a decrease in rat pup growth and survival occurred.
There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Studies in rats have shown that amiloride is excreted in milk in concentrations higher than those found in blood, but it is not known whether amiloride is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from amiloride HCl, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of amiloride HCI did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. (See CONTRAINDICATIONS, Impaired Renal Function.)
-
ADVERSE REACTIONS
Amiloride HCl is usually well tolerated and, except for hyperkalemia (serum potassium levels greater than 5.5 mEq liter - see WARNINGS), significant adverse effects have been reported infrequently. Minor adverse reactions were reported relatively frequently (about 20%) but the relationship of many of the reports to amiloride HCl is uncertain and the overall frequency was similar in hydrochlorothiazide treated groups. Nausea/anorexia, abdominal pain, flatulence, and mild skin rash have been reported and probably are related to amiloride. Other adverse experiences that have been reported with amiloride are generally those known to be associated with diuresis, or with the underlying disease being treated.
The adverse reactions for amiloride HCl listed in the following table have been arranged into two groups: (1) incidence greater than one percent; and (2) incidence one percent or less. The incidence for group (1) was determined from clinical studies conducted in the United States (837 patients treated with amiloride HCl). The adverse effects listed in group (2) include reports from the same clinical studies and voluntary reports since marketing. The probability of a causal relationship exists between amiloride HCl and these adverse reactions, some of which have been reported only rarely.
*Reactions occurring in 3% to 8% of patients treated with amiloride HCl. (Those reactions occurring in less than 3% of the patients are unmarked.) **See WARNINGS. Incidence > 1%
Incidence ≤ 1%
Body as a Whole
Headache*
Back pain
Weakness
Chest pain
Fatigability
Neck/shoulder ache
Pain, extremities
Cardiovascular
None
Angina pectoris
Orthostatic hypotension
Arrhythmia
Palpitation
Digestive
Nausea/anorexia*
Jaundice
Diarrhea*
GI bleeding
Vomiting*
Abdominal fullness
Abdominal pain
GI disturbance
Gas pain
Thirst
Appetite changes
Heartburn
Constipation
Flatulence
Dyspepsia
Metabolic Elevated serum potassium levels (>5.5 mEq per liter)**
None
Skin
None
Skin rash
Itching
Dryness of mouth
Pruritus
Alopecia
Musculoskeletal
Muscle cramps
Joint pain
Leg ache
Nervous
Dizziness
Paresthesia
Encephalopathy
Tremors
Vertigo
Psychiatric
None
Nervousness
Mental confusion
Insomnia
Decreased libido
Depression
Somnolence
Respiratory
Cough
Shortness of Breath
Dyspnea
Special Senses
None
Visual disturbances
Nasal congestion
Tinnitus
Increased intraocular pressure
Urogenital
Impotence
Polyuria
Dysuria
Urinary frequency
Bladder spasms
Gynecomastia
Causal Relationship Unknown
Other reactions have been reported but occurred under circumstances where a causal relationship could not be established. However, in these rarely reported events, that possibility cannot be excluded. Therefore, these observations are listed to serve as alerting information to physicians.
Activation of probable pre-existing peptic ulcer
Aplastic anemia
Neutropenia
Abnormal liver function
-
OVERDOSAGE
No data are available in regard to overdosage in humans.
The oral LD 50 of amiloride HCl (calculated as the base) is 56 mg/kg in mice and 36 to 85 mg/kg in rats, depending on the strain.
It is not known whether the drug is dialyzable.
The most likely signs and symptoms to be expected with overdosage are dehydration and electrolyte imbalance. These can be treated by established procedures. Therapy with amiloride HCl should be discontinued and the patient observed closely. There is no specific antidote.Emesis should be induced or gastric lavage performed.Treatment is symptomatic and supportive. If hyperkalemia occurs, active measures should be taken to reduce the serum potassium levels.
-
DOSAGE AND ADMINISTRATION
Amiloride HCl should be administered with food.
Amiloride HCl, one 5 mg tablet daily, should be added to the usual antihypertensive or diuretic dosage of a kaliuretic diuretic. The dosage may be increased to 10 mg per day, if necessary. More than two 5 mg tablets of amiloride HCl daily usually are not needed, and there is little controlled experience with such doses. If persistent hypokalemia is documented with 10 mg, the dose can be increased to 15 mg, then 20 mg, with careful monitoring of electrolytes.
In treating patients with congestive heart failure after an initial diuresis has been achieved, potassium loss may also decrease and the need for amiloride HCl should be re-evaluated. Dosage adjustment may be necessary. Maintenance therapy may be on an intermittent basis.
If it is necessary to use amiloride HCl alone (see INDICATIONS), the starting dosage should be one 5 mg tablet daily. This dosage may be increased to 10 mg per day, if necessary. More than two 5 mg tablets usually are not needed, and there is little controlled experience with such doses. If persistent hypokalemia is documented with 10 mg, the dose can be increased to 15 mg, then 20 mg, with careful monitoring of electrolytes.
- HOW SUPPLIED
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
AMILORIDE HYDROCHLORIDE
amiloride hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63629-2114(NDC:49884-117) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AMILORIDE HYDROCHLORIDE ANHYDROUS (UNII: 7M458Q65S3) (AMILORIDE - UNII:7DZO8EB0Z3) AMILORIDE HYDROCHLORIDE ANHYDROUS 5 mg Inactive Ingredients Ingredient Name Strength D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) DIBASIC CALCIUM PHOSPHATE DIHYDRATE (UNII: O7TSZ97GEP) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Product Characteristics Color yellow Score no score Shape ROUND Size 7mm Flavor Imprint Code Par;117 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63629-2114-1 1000 in 1 BOTTLE; Type 0: Not a Combination Product 05/12/1986 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA070346 01/22/1986 Labeler - Bryant Ranch Prepack (171714327) Registrant - Bryant Ranch Prepack (171714327) Establishment Name Address ID/FEI Business Operations Bryant Ranch Prepack 171714327 REPACK(63629-2114) , RELABEL(63629-2114)
