Label: SPIRONOLACTONE tablet, film coated

- NDC Code(s): 63629-1095-1
- Packager: Bryant Ranch Prepack
- This is a repackaged label.
- Source NDC Code(s): 53489-329
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated February 13, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SPIRONOLACTONE TABLETS safely and effectively.
See full prescribing information for SPIRONOLACTONE TABLETS.
SPIRONOLACTONE tablets for oral use
Initial U.S. Approval: 1960INDICATIONS AND USAGE
Spironolactone tablets are an aldosterone antagonist indicated for:
- The treatment of NYHA Class III-IV heart failure and reduced ejection fraction to increase survival, manage edema, and to reduce the need for hospitalization for heart failure ( 1.1).
- Use as an add-on therapy for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions ( 1.2).
- The management of edema in adult patients who are cirrhotic when edema is not responsive to fluid and sodium restrictions and in the setting of nephrotic syndrome when treatment of the underlying disease, restriction of fluid and sodium intake, and the use of other diuretics produce an inadequate response ( 1.3).
- Treatment of primary hyperaldosternism for: (
1.4).
- Short-term preoperative treatment
- Long-term maintenance for patients with discrete aldosterone-producing adrenal adenomas who are not candidates for surgery and patients with bilateral micro or macronodular adrenal hyperplasia
DOSAGE AND ADMINISTRATION
- Heart Failure: Initiate treatment at 25 mg once daily ( 2.2).
- Hypertension: Initiate treatment at 25 to 100 mg daily in either single or divided doses ( 2.3).
- Edema: Initiate therapy in a hospital setting and titrate slowly. The recommended initial daily dose is 100 mg in single or divided doses ( 2.4).
- Primary hyperaldosteronism: Initiate treatment at 100 to 400 mg in preparation for surgery. In patients unsuitable for surgery use the lowest effective dosage determined for the individual patient ( 2.5).
DOSAGE FORMS AND STRENGTHS
Tablets: 25 mg, 50 mg, and 100 mg ( 3)
CONTRAINDICATIONS
Spironolactone Tablets are contraindicated in patients with ( 4):
- Hyperkalemia
- Addison’s disease
- Concomitant use of eplerenone
WARNINGS AND PRECAUTIONS
- Hyperkalemia: Monitor serum potassium within one week of initiation and regularly thereafter ( 5.1).
- Hypotension and Worsening Renal Function: Monitor volume status and renal function periodically ( 5.2).
- Electrolyte and Metabolic Abnormalities: Monitor serum electrolytes, uric acid and blood glucose periodically ( 5.3).
- Gynecomastia: Spironolactone tablets can cause gynecomastia ( 5.4).
ADVERSE REACTIONS
The most common adverse reaction with spironolactone tablets treatment is gynecomastia ( 5.4, 6).
To report SUSPECTED ADVERSE REACTIONS, contact Sun Pharmaceutical Industries, Inc. at 1-800-406-7984 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Agents increasing serum potassium: Concomitant administration can lead to hyperkalemia ( 5.1, 7.1).
- Lithium: Increased risk of lithium toxicity ( 7.2).
- NSAIDs: May reduce the diuretic, natriuretic and antihypertensive effect of spironolactone tablets ( 7.3).
- Digoxin: Spironolactone tablets can interfere with radioimmunologic assays of digoxin exposure ( 7.4).
- Cholestyramine: Hyperkalemic metabolic acidosis has been reported with concomitant use ( 7.5).
- Acetylsalicylic Acid (ASA): ASA may reduce the efficacy of spironolactone tablets ( 7.6).
USE IN SPECIFIC POPULATIONS
- Pregnancy: Based on animal data, spironolactone tablets may affect sex differentiation of the male during embryogenesis ( 8.1).
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Heart Failure
1.2 Hypertension
1.3 Edema Associated with Hepatic Cirrhosis or Nephrotic Syndrome
1.4 Primary Hyperaldosteronism
2 DOSAGE AND ADMINISTRATION
2.1 General Considerations
2.2 Treatment of Heart Failure
2.3 Treatment of Essential Hypertension
2.4 Treatment of Edema
2.5 Treatment of Primary Hyperaldosteronism
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hyperkalemia
5.2 Hypotension and Worsening Renal Function
5.3 Electrolyte and Metabolic Abnormalities
5.4 Gynecomastia
6 ADVERSE REACTIONS
7 DRUG INTERACTIONS
7.1 Drugs and Supplements Increasing Serum Potassium
7.2 Lithium
7.3 Nonsteroidal anti-inflammatory drugs (NSAIDs)
7.4 Digoxin
7.5 Cholestyramine
7.6 Acetylsalicylic Acid
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Use in Renal Impairment
8.7 Use in Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Heart Failure
14.2 Hypertension
16 HOW SUPPLIED
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Heart Failure
Spironolactone tablets are indicated for treatment of NYHA Class III-IV heart failure and reduced ejection fraction to increase survival, manage edema, and reduce the need for hospitalization for heart failure.
Spironolactone tablets are usually administered in conjunction with other heart failure therapies.
1.2 Hypertension
Spironolactone tablets are indicated as add-on therapy for the treatment of hypertension, to lower blood pressure in patients who are not adequately controlled on other agents. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
1.3 Edema Associated with Hepatic Cirrhosis or Nephrotic Syndrome
Spironolactone tablets are indicated for the management of edema in the following settings:
- Cirrhosis of the liver when edema is not responsive to fluid and sodium restriction.
- Nephrotic syndrome when treatment of the underlying disease, restriction of fluid and sodium intake, and the use of other diuretics produce an inadequate response.
Because it increases serum potassium, spironolactone tablets may be useful for treating edema when administration of other diuretics has caused hypokalemia.
1.4 Primary Hyperaldosteronism
Spironolactone tablets are indicated in the following settings:
- Short-term preoperative treatment of patients with primary hyperaldosteronism.
- Long-term maintenance therapy for patients with discrete aldosterone-producing adrenal adenomas who are not candidates for surgery.
- Long-term maintenance therapy for patients with bilateral micro or macronodular adrenal hyperplasia (idiopathic hyperaldosteronism).
-
2 DOSAGE AND ADMINISTRATION
2.1 General Considerations
Spironolactone tablets can be taken with or without food, but should be taken consistently with respect to food [ see Clinical Pharmacology (12.3)] .
2.2 Treatment of Heart Failure
In patients with serum potassium ≤5.0 mEq/L and eGFR >50 mL/min/1.73 m², initiate treatment at 25 mg once daily. Patients who tolerate 25 mg once daily may have their dosage increased to 50 mg once daily as clinically indicated. Patients who develop hyperkalemia on 25 mg once daily may have their dosage reduced to 25 mg every other day [ see Warnings and Precautions (5.1)] . In patients with an eGFR between 30 and 50 mL/min/1.73 m 2, consider initiating therapy at 25 mg every other day because of the risk of hyperkalemia [ see Use in Specific Populations (8.6)].
2.3 Treatment of Essential Hypertension
The recommended initial daily dose is 25 to 100 mg of spironolactone tablets administered in either single or divided doses is recommended. Dosage can be titrated at two-week intervals. Doses greater than 100 mg/day generally do not provide additional reductions in blood pressure.
2.4 Treatment of Edema
In patients with cirrhosis, initiate therapy in a hospital setting and titrate slowly [ see Use in Specific Populations (8.7)] . The recommended initial daily dosage is 100 mg of spironolactone tablets administered in either single or divided doses, but may range from 25 to 200 mg daily. When given as the sole agent for diuresis, administer for at least five days before increasing dose to obtain desired effect.
2.5 Treatment of Primary Hyperaldosteronism
Administer spironolactone tablets in doses of 100 to 400 mg daily in preparation for surgery. For patients who are considered unsuitable for surgery, spironolactone tablets can be used as long-term maintenance therapy at the lowest effective dosage determined for the individual patient.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hyperkalemia
Spironolactone tablets can cause hyperkalemia. This risk is increased by impaired renal function or concomitant potassium supplementation, potassium-containing salt substitutes or drugs that increase potassium, such as angiotensin converting enzyme inhibitors and angiotensin receptor blockers [see Drug Interactions (7.1)] .
Monitor serum potassium within 1 week of initiation or titration of spironolactone tablets and regularly thereafter. More frequent monitoring may be needed when spironolactone tablets are given with other drugs that cause hyperkalemia or in patients with impaired renal function.
If hyperkalemia occurs, decrease the dose or discontinue spironolactone tablets and treat hyperkalemia.
5.2 Hypotension and Worsening Renal Function
Excessive diuresis may cause symptomatic dehydration, hypotension and worsening renal function, particularly in salt-depleted patients or those taking angiotensin converting enzyme inhibitors and angiotensin II receptor blockers. Worsening of renal function can also occur with concomitant use of nephrotoxic drugs (e.g., aminoglycosides, cisplatin, and NSAIDs). Monitor volume status and renal function periodically.
5.3 Electrolyte and Metabolic Abnormalities
In addition to causing hyperkalemia, spironolactone tablets can cause hyponatremia, hypomagnesemia, hypocalcemia, hypochloremic alkalosis, and hyperglycemia. Asymptomatic hyperuricemia can occur and rarely gout is precipitated. Monitor serum electrolytes, uric acid and blood glucose periodically.
5.4 Gynecomastia
Spironolactone tablets can cause gynecomastia. In the Randomized Spironolactone Evaluation Study, patients with heart failure treated with a mean dose of 26 mg of spironolactone once daily, about 9% of the male subjects developed gynecomastia. The risk of gynecomastia increases in a dose-dependent manner with an onset that varies widely from 1-2 months to over a year. Gynecomastia is usually reversible.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hyperkalemia [see Warnings and Precautions ( 5.1)]
- Hypotension and Worsening Renal Function [see Warnings and Precautions ( 5.2)]
- Electrolyte and Metabolic Abnormalities [see Warnings and Precautions ( 5.3)]
- Gynecomastia [see Warnings and Precautions ( 5.4)]
- Impaired neurological function/ coma in patients with hepatic impairment, cirrhosis and ascites [ see Use in Specific Populations (8.7)]
The following adverse reactions associated with the use of spironolactone were identified in clinical trials or postmarketing reports. Because these reactions were reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency, reliably, or to establish a causal relationship to drug exposure.
Digestive: Gastric bleeding, ulceration, gastritis, diarrhea and cramping, nausea, vomiting.
Reproductive: Decreased libido, inability to achieve or maintain erection, irregular menses or amenorrhea, postmenopausal bleeding, breast and nipple pain.
Hematologic: Leukopenia (including agranulocytosis), thrombocytopenia.
Hypersensitivity: Fever, urticaria, maculopapular or erythematous cutaneous eruptions, anaphylactic reactions, vasculitis.
Metabolism: Hyperkalemia, electrolyte disturbances [ see Warnings and Precautions (5.1, 5.3)] , hyponatremia, hypovolemia.
Musculoskeletal: Leg cramps.
Nervous system/psychiatric: Lethargy, mental confusion, ataxia, dizziness, headache, drowsiness.
Liver/biliary: A very few cases of mixed cholestatic/hepatocellular toxicity, with one reported fatality, have been reported with spironolactone administration.
Renal: Renal dysfunction (including renal failure).
Skin: Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN), drug rash with eosinophilia and systemic symptoms (DRESS), alopecia, pruritis.
-
7 DRUG INTERACTIONS
7.1 Drugs and Supplements Increasing Serum Potassium
Concomitant administration of spironolactone tablets with potassium supplementation or drugs that can increase potassium may lead to severe hyperkalemia. In general, discontinue potassium supplementation in heart failure patients who start spironolactone tablets [ see Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)] .
Check serum potassium levels when ACE inhibitor or ARB therapy is altered in patients receiving spironolactone tablets.
Examples of drugs that can increase potassium include:
- ACE inhibitors
- angiotensin receptor blockers
- non-steroidal anti-inflammatory drugs (NSAIDs)
- heparin and low molecular weight heparin
- trimethoprim
7.2 Lithium
Like other diuretics, spironolactone tablets reduces the renal clearance of lithium, thus increasing the risk of lithium toxicity. Monitor lithium levels periodically when spironolactone tablets are coadministered [ see Clinical Pharmacology (12.3)] .
7.3 Nonsteroidal anti-inflammatory drugs (NSAIDs)
In some patients, the administration of an NSAID can reduce the diuretic, natriuretic, and antihypertensive effect of diuretics. Therefore, when spironolactone tablets and NSAIDs are used concomitantly, monitor closely to determine if the desired effect of the diuretic is obtained [ see Clinical Pharmacology (12.3)] .
7.4 Digoxin
Spironolactone and its metabolites interfere with radioimmunoassays for digoxin and increase the apparent exposure to digoxin. It is unknown to what extent, if any, spironolactone may increase actual digoxin exposure. In patients taking concomitant digoxin, use an assay that does not interact with spironolactone.
7.5 Cholestyramine
Hyperkalemic metabolic acidosis has been reported in patients given spironolactone tablets concurrently with cholestyramine.
7.6 Acetylsalicylic Acid
Acetylsalicylic acid may reduce the efficacy of spironolactone. Therefore, when spironolactone tablets and acetylsalicylic acid are used concomitantly, spironolactone tablets may need to be titrated to higher maintenance dose and the patient should be observed closely to determine if the desired effect is obtained [ see Clinical Pharmacology (12.3)] .
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on mechanism of action and findings in animal studies, spironolactone may affect sex differentiation of the male during embryogenesis (see Data). Rat embryofetal studies report feminization of male fetuses and endocrine dysfunction in females exposed to spironolactone in utero. Limited available data from published case reports and case series did not demonstrate an association of major malformations or other adverse pregnancy outcomes with spironolactone . There are risks to the mother and fetus associated with heart failure, cirrhosis and poorly controlled hypertension during pregnancy ( see Clinical Considerations) . Because of the potential risk to the male fetus due to anti-androgenic properties of spironolactone and animal data, avoid spironolactone in pregnant women or advise a pregnant woman of the potential risk to a male fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnant women with congestive heart failure are at increased risk for preterm birth. Stroke volume and heart rate increase during pregnancy, increasing cardiac output, especially during the first trimester. Clinical classification of heart disease may worsen with pregnancy and lead to maternal death. Closely monitor pregnant patients for destabilization of their heart failure.
Pregnant women with symptomatic cirrhosis generally have poor outcomes including hepatic failure, variceal hemorrhage, preterm delivery, fetal growth restriction and maternal death. Outcomes are worse with coexisting esophageal varices. Pregnant women with cirrhosis of the liver should be carefully monitored and managed accordingly.
Hypertension in pregnancy increases the maternal risk for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage). Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death.
Data
Animal Data
Teratology studies with spironolactone tablets have been carried out in mice and rabbits at doses of up to
20 mg/kg/day. On a body surface area basis, this dose in the mouse is substantially below the maximum recommended human dose and, in the rabbit, approximates the maximum recommended human dose. No teratogenic or other embryotoxic effects were observed in mice, but the 20 mg/kg dose caused an increased rate of resorption and a lower number of live fetuses in rabbits. Because of its antiandrogenic activity and the requirement of testosterone for male morphogenesis, spironolactone tablets may have the potential for adversely affecting sex differentiation of the male during embryogenesis. When administered to rats at 200 mg/kg/day between gestation days 13 and 21 (late embryogenesis and fetal development), feminization of male fetuses was observed. Offspring exposed during late pregnancy to 50 and 100 mg/kg/day doses of spironolactone tablets exhibited changes in the reproductive tract including dose-dependent decreases in weights of the ventral prostate and seminal vesicle in males, ovaries and uteri that were enlarged in females, and other indications of endocrine dysfunction, that persisted into adulthood. Spironolactone tablets has known endocrine effects in animals including progestational and antiandrogenic effects.
8.2 Lactation
Risk Summary
Spironolactone is not present in breastmilk; however, limited data from a lactating woman at 17 days postpartum reports the presence of the active metabolite, canrenone, in human breast milk in low amounts that are expected to be clinically inconsequential. In this case, there were no adverse effects reported for the breastfed infant after short term exposure to spironolactone; however, long term effects on a breastfed infant are unknown. There are no data on spironolactone effects on milk production. Consider the developmental and health benefits of breastfeeding along with the mother’s clinical need for spironolactone and any potential adverse effects on the breastfed child from spironolactone or from the underlying maternal condition.
8.5 Geriatric Use
Spironolactone tablets are substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, monitor renal function.
8.6 Use in Renal Impairment
Spironolactone tablets are substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Patients with renal impairment are at increased risk of hyperkalemia. Monitor potassium closely.
8.7 Use in Hepatic Impairment
Spironolactone tablets can cause sudden alterations of fluid and electrolyte balance which may precipitate impaired neurological function, worsening hepatic encephalopathy and coma in patients with hepatic disease with cirrhosis and ascites. In these patients, initiate spironolactone tablets in the hospital [ see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)] .
Clearance of spironolactone and its metabolites is reduced in patients with cirrhosis. In patients with cirrhosis, start with lowest initial dose and titrate slowly [ see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)] .
-
10 OVERDOSAGE
The oral LD 50 of spironolactone tablets is greater than 1000 mg/kg in mice, rats, and rabbits.
Acute overdosage of spironolactone tablets may be manifested by drowsiness, mental confusion, maculopapular or erythematous rash, nausea, vomiting, dizziness, or diarrhea. Rarely, instances of hyponatremia, hyperkalemia, or hepatic coma may occur in patients with severe liver disease, but these are unlikely due to acute overdosage. Hyperkalemia may occur, especially in patients with impaired renal function.
Treatment: Induce vomiting or evacuate the stomach by lavage. There is no specific antidote. Treatment is supportive to maintain hydration, electrolyte balance, and vital functions. Patients who have renal impairment may develop hyperkalemia. In such cases, discontinue spironolactone tablets.
-
11 DESCRIPTION
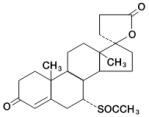
Spironolactone oral tablets contain 25 mg, 50 mg, or 100 mg of the aldosterone antagonist spironolactone, 17‑ hydroxy-7α-mercapto-3-oxo-17α-pregn-4-ene-21-carboxylic acid γ-lactone acetate, which has the following structural formula:
Spironolactone is practically insoluble in water, soluble in alcohol, and freely soluble in benzene and in chloroform.
Spironolactone tablets, 25 mg contain the following inactive ingredients: anhydrous lactose, colloidal silicon dioxide, docusate sodium 85%/sodium benzoate 15%, entrapped peppermint flavor, magnesium stearate, microcrystalline cellulose, povidone, and sodium starch glycolate.
Spironolactone tablets, 50 mg and 100 mg contain the following inactive ingredients: anhydrous lactose, carnauba wax, colloidal silicon dioxide, docusate sodium 85%/sodium benzoate 15%, entrapped peppermint flavor, hypromellose, magnesium stearate, microcrystalline cellulose, polydextrose, polyethylene glycol, povidone, sodium starch glycolate, titanium dioxide, and triacetin.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Spironolactone and its active metabolites are specific pharmacologic antagonists of aldosterone, acting primarily through competitive binding of receptors at the aldosterone-dependent sodium-potassium exchange site in the distal convoluted renal tubule. Spironolactone causes increased amounts of sodium and water to be excreted, while potassium is retained. Spironolactone acts both as a diuretic and as an antihypertensive drug by this mechanism. It may be given alone or with other diuretic agents that act more proximally in the renal tubule.
12.2 Pharmacodynamics
Aldosterone antagonist activity: Increased levels of the mineralocorticoid, aldosterone, are present in primary and secondary hyperaldosteronism. Edematous states in which secondary aldosteronism is usually involved include congestive heart failure, hepatic cirrhosis, and nephrotic syndrome. By competing with aldosterone for receptor sites, spironolactone provides effective therapy for the edema and ascites in those conditions.
Spironolactone counteracts secondary aldosteronism induced by the volume depletion and associated sodium loss caused by active diuretic therapy.
12.3 Pharmacokinetics
Absorption
The mean time to reach peak plasma concentration of spironolactone and the active metabolite, canrenone, in healthy volunteers is 2.6 and 4.3 hours, respectively.
Effect of food: Food increased the bioavailability of spironolactone (as measured by AUC) by approximately 95.4%. Patients should establish a routine pattern for taking spironolactone tablets with regard to meals [ see Dosage and Administration (2.1)] .
Distribution
Spironolactone and its metabolites are more than 90% bound to plasma proteins.
Elimination
The mean half-life of spironolactone is 1.4 hour. The mean half-life values of its metabolites including canrenone, 7-α-(thiomethyl) spirolactone (TMS), and 6-ß-hydroxy-7-α-(thiomethyl) spirolactone (HTMS) are 16.5, 13.8, and 15 hours, respectively.
Metabolism: Spironolactone is rapidly and extensively metabolized. Metabolites can be divided into two main categories: those in which sulfur of the parent molecule is removed (e.g., canrenone) and those in which the sulfur is retained (e.g., TMS and HTMS). In humans, the potencies of TMS and 7-α-thiospirolactone in reversing the effects of the synthetic mineralocorticoid, fludrocortisone, on urinary electrolyte composition were approximately a third relative to spironolactone. However, since the serum concentrations of these steroids were not determined, their incomplete absorption and/or first-pass metabolism could not be ruled out as a reason for their reduced in vivo activities.
Excretion: The metabolites are excreted primarily in the urine and secondarily in bile.
Specific Populations
The impact of age, sex, race/ethnicity, and renal impairment on the pharmacokinetics of spironolactone have not been specifically studied.
Patients with Hepatic Impairment: The terminal half-life of spironolactone has been reported to be increased in patients with cirrhotic ascites [ see Use in Specific Populations (8.7)] .
Drug Interaction Studies:
Drugs and Supplements Increasing Serum Potassium: Concomitant administration of spironolactone tablets with potassium supplementation, salt substitutes containing potassium, a diet rich in potassium, or drugs that can increase potassium, including ACE inhibitors, angiotensin II antagonists, non-steroidal anti-inflammatory drugs (NSAIDs), heparin and low molecular weight heparin, may lead to severe hyperkalemia [ see Warnings and Precautions (5.1) and Drug Interactions (7.1)] .
Lithium: Spironolactone tablets reduces the renal clearance of lithium, inducing a high risk of lithium toxicity [see Drug Interactions (7.2)].
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs): In some patients, the administration of an NSAID can reduce the diuretic, natriuretic, and antihypertensive effect of loop, potassium-sparing, and thiazide diuretics [ see Drug Interactions (7.3)] .
Acetylsalicylic acid: A single dose of 600 mg of acetylsalicylic acid inhibited the natriuretic effect of spironolactone, which was hypothesized be due to inhibition of tubular secretion of canrenone, causing decreased effectiveness of spironolactone [ see Drug Interactions (7.6)] .
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Orally administered spironolactone tablets has been shown to be a tumorigen in dietary administration studies performed in rats, with its proliferative effects manifested on endocrine organs and the liver. In an 18-month study using doses of about 50, 150, and 500 mg/kg/day, there were statistically significant increases in benign adenomas of the thyroid and testes and, in male rats, a dose-related increase in proliferative changes in the liver (including hepatocytomegaly and hyperplastic nodules). In a 24-month study in which the same strain of rat was administered doses of about 10, 30, and 100 mg spironolactone tablets/kg/day, the range of proliferative effects included significant increases in hepatocellular adenomas and testicular interstitial cell tumors in males, and significant increases in thyroid follicular cell adenomas and carcinomas in both sexes. There was also a statistically significant, but not dose-related, increase in benign uterine endometrial stromal polyps in females. No increased tumors were seen at doses of 100 mg/kg/day. This dose represents about 5X the human recommended daily dose of 200 mg/day, when based on body surface area.
Mutagenesis
Neither spironolactone tablets nor potassium canrenoate produced mutagenic effects in tests using bacteria or yeast. In the absence of metabolic activation, neither spironolactone tablets nor potassium canrenoate has been shown to be mutagenic in mammalian tests in vitro. In the presence of metabolic activation, spironolactone tablets has been reported to be negative in some mammalian mutagenicity tests in vitro and inconclusive (but slightly positive) for mutagenicity in other mammalian tests in vitro. In the presence of metabolic activation, potassium canrenoate has been reported to test positive for mutagenicity in some mammalian tests in vitro, inconclusive in others, and negative in still others.
Impairment of Fertility
In a three-litter reproduction study in which female rats received dietary doses of 15 and 50 mg spironolactone tablets/kg/day, there were no effects on mating and fertility, but there was a small increase in incidence of stillborn pups at 50 mg/kg/day. When injected into female rats (100 mg/kg/day for 7 days, i.p.), spironolactone tablets were found to increase the length of the estrous cycle by prolonging diestrus during treatment and inducing constant diestrus during a two-week post-treatment observation period. These effects were associated with retarded ovarian follicle development and a reduction in circulating estrogen levels, which would be expected to impair mating, fertility, and fecundity. Spironolactone tablets (100 mg/kg/day), administered i.p. to female mice during a two-week cohabitation period with untreated males, decreased the number of mated mice that conceived (effect shown to be caused by an inhibition of ovulation) and decreased the number of implanted embryos in those that became pregnant (effect shown to be caused by an inhibition of implantation), and at 200 mg/kg, also increased the latency period to mating.
-
14 CLINICAL STUDIES
14.1 Heart Failure
The Randomized Spironolactone Evaluation Study was a placebo controlled, double-blind study of the effect of spironolactone on mortality in patients with highly symptomatic heart failure and reduced ejection fraction. To be eligible to participate patients had to have an ejection fraction of ≤ 35%, NYHA class III-IV symptoms, and a history of NYHA class IV symptoms within the last 6 months before enrollment. Patients with a baseline serum creatinine of >2.5 mg/dL or a recent increase of 25% or with a baseline serum potassium of >5.0 mEq/L were excluded.
Follow-up visits and laboratory measurements (including serum potassium and creatinine) were performed every four weeks for the first 12 weeks, then every 3 months for the first year, and then every 6 months thereafter.
The initial dose of spironolactone was 25 mg once daily. Patients who were intolerant of the initial dosage regimen had their dose decreased to one 25 mg tablet every other day at one to four weeks. Patients who were tolerant of one tablet daily at 8 weeks may have had their dose increased to 50 mg daily at the discretion of the investigator. The mean daily dose at study end for patients randomized to spironolactone was 26 mg.
1663 patients were randomized 1:1 to spironolactone or placebo. 87% of patients were white, 7% black, 2% Asian. 73% were male and median age was 67. The median ejection fraction was 26%. 70% were NYHA class III and 29% class IV. The etiology of heart failure was ischemic in 55%, and non-ischemic in 45%. There was a history of myocardial infarction in 28%, of hypertension in 24%, and of diabetes in 22%. The median baseline serum creatinine was 1.2 mg/dL and the median baseline creatinine clearance was 57 mL/min.
At baseline 100% of patients were taking loop diuretic and 95% were taking an ACE inhibitor. Other medications used at any time during the study included digoxin (78%), anticoagulants (58%), aspirin (43%), and beta-blockers (15%).
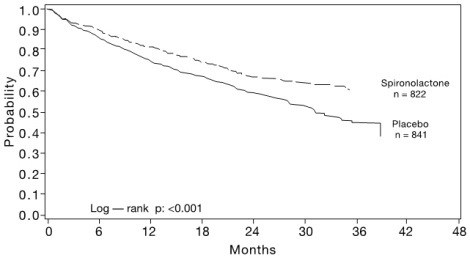
The primary endpoint for the Randomized Spironolactone Evaluation Study was time to all-cause mortality. The Randomized Spironolactone Evaluation Study was terminated early because of significant mortality benefit demonstrated during a planned interim analysis. Compared to placebo, spironolactone reduced the risk of death by 30% (p<0.001; 95% confidence interval 18% to 40%).
Spironolactone also reduced the risk of hospitalization for cardiac causes (defined as worsening heart failure, angina, ventricular arrhythmias, or myocardial infarction) by 30% (p <0.001; 95% confidence interval 18% to 41%).
The survival curves by treatment group are shown in Figure 1.
Figure 1. Survival by Treatment Group in the Randomized Spironolactone Evaluation Study
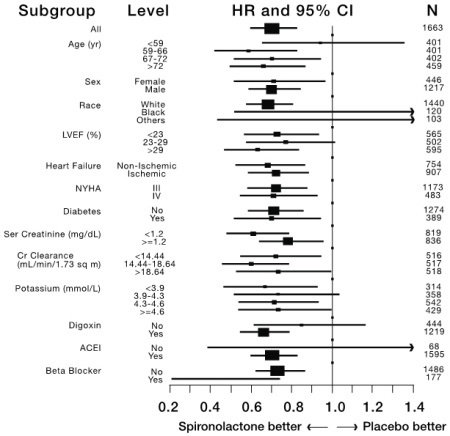
Mortality hazard ratios for some subgroups are shown in Figure 2. The favorable effect of spironolactone on mortality appeared similar for both genders and all age groups except patients younger than 55. There were too few non-whites in the Randomized Spironolactone Evaluation Study to evaluate if the effects differ by race. Spironolactone’s benefit appeared greater in patients with low baseline serum potassium levels and less in patients with ejection fractions <0.2. These subgroup analyses must be interpreted cautiously.
Figure 2. Hazard Ratios of All-Cause Mortality by Subgroup in the Randomized Spironolactone Evaluation Study
Figure 2: The size of each box is proportional to the sample size as well as the event rate. LVEF denotes left ventricular ejection fraction, Ser Creatinine denotes serum creatinine, Cr Clearance denotes creatinine clearance, and ACEI denotes angiotensin-converting enzyme inhibitor.
14.2 Hypertension
The dose response of spironolactone for hypertension has not been well characterized. In patients with hypertension, decreases in systolic blood pressure have been observed at doses ranging from 25 to 100 mg/day. Doses greater than 100 mg/day generally do not provide additional reductions in blood pressure [ see Dosage and Administration (2.3)] .
- 16 HOW SUPPLIED
- 17 PATIENT COUNSELING INFORMATION
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SPIRONOLACTONE
spironolactone tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63629-1095(NDC:53489-329) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SPIRONOLACTONE (UNII: 27O7W4T232) (SPIRONOLACTONE - UNII:27O7W4T232) SPIRONOLACTONE 100 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) CARNAUBA WAX (UNII: R12CBM0EIZ) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) DOCUSATE SODIUM (UNII: F05Q2T2JA0) SODIUM BENZOATE (UNII: OJ245FE5EU) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYDEXTROSE (UNII: VH2XOU12IE) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) Product Characteristics Color white Score 2 pieces Shape OVAL Size 19mm Flavor PEPPERMINT Imprint Code MP;303 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63629-1095-1 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 07/23/1986 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA089424 07/23/1986 Labeler - Bryant Ranch Prepack (171714327) Registrant - Bryant Ranch Prepack (171714327) Establishment Name Address ID/FEI Business Operations Bryant Ranch Prepack 171714327 REPACK(63629-1095) , RELABEL(63629-1095)