Label: LOPRESSOR- metoprolol tartrate tablet


- NDC Code(s): 30698-458-01, 30698-459-01
- Packager: Validus Pharmaceuticals LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated September 14, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LOPRESSOR safely and effectively. See full prescribing information for LOPRESSOR.
LOPRESSOR (metoprolol tartrate) tablets, for oral use
Initial U.S. Approval: 1992
INDICATIONS AND USAGE
LOPRESSOR is a beta-adrenergic blocker indicated for the treatment of:
- Hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and non-fatal cardiovascular events, primarily strokes and myocardial infarctions. (1.1)
- Angina Pectoris. (1.2)
- Myocardial Infarction, to reduce the risk of cardiovascular mortality when used in conjunction with intravenous metoprolol therapy in patients with definite or suspected acute myocardial infarction in hemodynamically stable patients. (1.3)
DOSAGE AND ADMINISTRATION
- Administer once daily with food or after a meal. Titrate at weekly or longer intervals as needed and tolerated. (2)
- Hypertension: Recommended starting dosage is 100 mg daily, in single or divided doses. (2.1)
- Angina Pectoris: Recommended starting dosage is 100 mg daily, given as two divided doses. (2.2)
- Myocardial Infarction: The starting dosage depends upon tolerance of intravenous metoprolol, see full prescribing information. (2.3)
DOSAGE FORMS AND STRENGTHS
- LOPRESSOR (metoprolol tartrate) tablets: 50 mg and 100 mg. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Abrupt cessation may exacerbate myocardial ischemia. (5.1)
- Heart Failure: Worsening cardiac failure may occur. (5.2)
- Bronchospastic Disease: Avoid beta-blockers. (5.3)
- Pheochromocytoma: Initiate therapy with an alpha blocker. (5.4)
- Major Surgery: Avoid initiation of high-dose extended-release metoprolol in patients undergoing non-cardiac surgery. Do not routinely withdraw chronic beta-blocker therapy prior to surgery. (5.5, 6.1)
- Diabetes: May mask symptoms of hypoglycemia. (5.6)
- Thyrotoxicosis: Abrupt withdrawal in patients with thyrotoxicosis might precipitate a thyroid storm. (5.7)
- Peripheral Vascular Disease: Can aggravate symptoms of arterial insufficiency. (5.9)
ADVERSE REACTIONS
- Most common adverse reactions: tiredness, dizziness, depression, shortness of breath, bradycardia, hypotension, diarrhea, pruritus, rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Validus Pharmaceuticals LLC at 1-866-982-5438 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- Catecholamine-depleting drugs may have an additive effect when given with beta-blocking agents. (7.1)
- Patients may be unresponsive to the usual doses of epinephrine used to treat allergic reaction. (7.2)
- CYP2D6 Inhibitors are likely to increase metoprolol concentration. (7.3)
- Concomitant use of glycosides, clonidine, and diltiazem and verapamil with beta-blockers can increase the risk of bradycardia. (7.4)
- Beta-blockers including metoprolol, may exacerbate the rebound hypertension that can follow the withdrawal of clonidine. (7.4)
USE IN SPECIFIC POPULATIONS
- Hepatic Impairment: Consider initiating LOPRESSOR therapy at low doses and gradually increase dosage to optimize therapy, while monitoring closely for adverse events. (8.6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2023
- Hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and non-fatal cardiovascular events, primarily strokes and myocardial infarctions. (1.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Hypertension
1.2 Angina Pectoris
1.3 Myocardial Infarction
2 DOSAGE AND ADMINISTRATION
2.1 Hypertension
2.2 Angina Pectoris
2.3 Myocardial Infarction
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Abrupt Cessation of Therapy
5.2 Heart Failure
5.3 Bronchospastic Disease
5.4 Pheochromocytoma
5.5 Major Surgery
5.6 Hypoglycemia
5.7 Thyrotoxicosis
5.8 Risk of Anaphylactic Reactions
5.9 Peripheral Vascular Disease
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Catecholamine Depleting Drugs
7.2 Epinephrine
7.3 CYP2D6 Inhibitors
7.4 Negative Chronotropes
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Hypertension
14.2 Angina Pectoris
14.3 Myocardial Infarction
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Hypertension
LOPRESSOR is indicated for the treatment of hypertension in adult patients, to lower blood pressure. Lowering blood pressure lowers the risk of fatal and non-fatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including metoprolol.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than 1 drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
LOPRESSOR may be administered with other antihypertensive agents.
-
2 DOSAGE AND ADMINISTRATION
2.1 Hypertension
Individualize the dosage of Lopressor tablets. Lopressor tablets should be taken with or immediately following meals.
The usual initial dosage is 100 mg daily in single or divided doses. Adjust dosage at weekly (or longer) intervals until optimum blood pressure reduction is achieved. In general, the maximum effect of any given dosage level will be apparent after 1 week of therapy. The effective dosage range of Lopressor tablets is 100 mg to 450 mg per day. Dosages above 450 mg per day have not been studied. While once-daily dosing can maintain a reduction in blood pressure throughout the day, lower doses (especially 100 mg) may not maintain a full effect at the end of the 24-hour period. Larger or more frequent daily doses may be required. Measure blood pressure near the end of the dosing interval to determine whether satisfactory control is being maintained throughout the day.
2.2 Angina Pectoris
The dosage of Lopressor tablets should be individualized. Lopressor tablets should be taken with or immediately following meals.
The usual initial dosage is 100 mg daily, given in two divided doses. Gradually increase the dosage at weekly intervals until optimum clinical response has been obtained or there is a pronounced slowing of the heart rate. The effective dosage range of Lopressor tablets is 100 to 400 mg per day. Dosages above 400 mg per day have not been studied. If treatment is to be discontinued, reduce the dosage gradually over a period of 1 - 2 weeks [see Warnings and Precautions (5.1)].
2.3 Myocardial Infarction
See prescribing information of intravenous metoprolol for dosage instructions for intravenous therapy.
In patients who tolerate the full intravenous dose, initiate Lopressor tablets, 50 mg every 6 hours, 15 minutes after the last intravenous dose of metoprolol and continue for 48 hours. In the case of intolerance, reduce dose to 25 mg and administer for 48 hours. Titrate, based on tolerability, to a maintenance dosage of 100 mg twice daily. Continue therapy for at least 3 months. Although the efficacy of Lopressor beyond 3 months has not been conclusively established, data from studies with other beta-blockers suggest that treatment should be continued for 1 to 3 years.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
LOPRESSOR is contraindicated in severe bradycardia, second- or third-degree heart block, cardiogenic shock, systolic blood pressure <100, decompensated heart failure, sick sinus syndrome (unless a permanent pacemaker is in place), and in patients who are hypersensitive to any component of this product.
-
5 WARNINGS AND PRECAUTIONS
5.1 Abrupt Cessation of Therapy
Following abrupt cessation of therapy with certain beta-blocking agents, exacerbations of angina pectoris and, in some cases, myocardial infarction have occurred. When discontinuing chronically administered LOPRESSOR, particularly in patients with ischemic heart disease, gradually reduce the dosage over a period of 1 to 2 weeks and monitor the patient. If angina markedly worsens or acute coronary ischemia develops, promptly reinstate LOPRESSOR, and take measures appropriate for the management of unstable angina. Warn patients not to interrupt therapy without their physician’s advice. Because coronary artery disease is common and may be unrecognized, avoid abruptly discontinuing LOPRESSOR in patients treated only for hypertension.
5.2 Heart Failure
Worsening cardiac failure may occur during up-titration of LOPRESSOR. If such symptoms occur, increase diuretics and restore clinical stability before advancing the dose of LOPRESSOR [see Dosage and Administration (2)]. It may be necessary to lower the dose of LOPRESSOR or temporarily discontinue it. Such episodes do not preclude subsequent successful titration of LOPRESSOR.
5.3 Bronchospastic Disease
Patients with bronchospastic disease, should in general, not receive beta-blockers, including Lopressor. Because of its relative beta1 cardio-selectivity, however, LOPRESSOR may be used in patients with bronchospastic disease who do not respond to, or cannot tolerate, other antihypertensive treatment. Because beta1-selectivity is not absolute, use the lowest possible dose of LOPRESSOR. Bronchodilators, including beta2-agonists, should be readily available or administered concomitantly [see Dosage and Administration (2)].
5.4 Pheochromocytoma
If LOPRESSOR is used in the setting of pheochromocytoma, it should be given in combination with an alpha blocker, and only after the alpha blocker has been initiated. Administration of beta-blockers alone in the setting of pheochromocytoma has been associated with a paradoxical increase in blood pressure due to the attenuation of beta-mediated vasodilatation in skeletal muscle.
5.5 Major Surgery
Avoid initiation of a high-dose regimen of beta-blocker therapy in patients undergoing non-cardiac surgery, since such use in patients with cardiovascular risk factors has been associated with bradycardia, hypotension, stroke and death.
Chronically administered beta-blocking therapy should not be routinely withdrawn prior to major surgery, however, the impaired ability of the heart to respond to reflex adrenergic stimuli may augment the risks of general anesthesia and surgical procedures.
5.6 Hypoglycemia
Beta-blockers may prevent early warning signs of hypoglycemia, such as tachycardia, and increase the risk for severe or prolonged hypoglycemia at any time during treatment, especially in patients with diabetes mellitus or children and patients who are fasting (i.e., surgery, not eating regularly, or are vomiting). If severe hypoglycemia occurs, patients should be instructed to seek emergency treatment. Beta-blockers may ask some of the manifestations of hypoglycemia, particularly tachycardia.
5.7 Thyrotoxicosis
Beta-adrenergic blockade may mask certain clinical signs of hyperthyroidism, such as tachycardia. Abrupt withdrawal of beta-blockade may precipitate a thyroid storm.
5.8 Risk of Anaphylactic Reactions
While taking beta-blockers, patients with a history of severe anaphylactic reaction to a variety of allergens may be more reactive to repeated challenge, either accidental, diagnostic, or therapeutic. Such patients may be unresponsive to the usual doses of epinephrine used to treat allergic reaction.
-
6 ADVERSE REACTIONS
The following adverse reactions are described elsewhere in labeling:
- Worsening angina or myocardial infarction [see Warnings and Precautions (5)]
- Worsening heart failure [see Warnings and Precautions (5)].
- Worsening AV block [see Contraindications (4)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Hypertension and Angina
Most adverse effects have been mild and transient.
Central Nervous System: Tiredness and dizziness have occurred in about 10% of patients. Depression has been reported in about 5 of 100 patients. Mental confusion and short-term memory loss have been reported. Headache, nightmares, and insomnia have also been reported.
Cardiovascular: Shortness of breath and bradycardia have occurred in approximately 3% of patients. Cold extremities; arterial insufficiency, usually of the Raynaud type; palpitations; heart failure exacerbations; peripheral edema; and hypotension have been reported in about 1% of patients. Gangrene in patients with pre-existing severe peripheral circulatory disorders has also been reported. [see Contraindications (4) and Warnings and Precautions (5.2)].
Respiratory: Wheezing (bronchospasm) and dyspnea have been reported in about 1% of patients [see Warnings and Precautions (5.3)]. Rhinitis has also been reported.
Gastrointestinal: Diarrhea has occurred in about 5% of patients. Nausea, dry mouth, gastric pain, constipation, flatulence, and heartburn have been reported in about 1% of patients. Vomiting was a common occurrence.
Hypersensitive Reactions: Pruritus or rash have occurred in about 5% of patients. Photosensitivity and worsening of psoriasis has been reported.
Miscellaneous: Peyronie’s disease, musculoskeletal pain, blurred vision, and tinnitus has been reported.
Myocardial Infarction
In general, the adverse reactions observed in trials with metoprolol in MI are consistent with the hypertension and angina experience.
In a randomized comparison of Lopressor and placebo in the setting of acute MI the following adverse reactions were reported:
Lopressor® Placebo Hypotension (systolic BP < 90 mm Hg) 27.4% 23.2% Bradycardia (heart rate < 40 beats/min) 15.9% 6.7% Second- or third-degree heart block 4.7% 4.7% First-degree heart block (P-R ≥ 0.26 sec) 5.3% 1.9% Heart failure 27.5% 29.6% 6.2 Post-Marketing Experience
The following adverse reactions have been identified during post approval use of LOPRESSOR. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Central Nervous System: Reversible mental depression progressing to catatonia; an acute reversible syndrome characterized by disorientation for time and place, short-term memory loss, emotional lability, slightly clouded sensorium, and decreased performance on neuropsychometrics.
Cardiovascular: Intensification of AV block [see Contraindications (4)].
Hematologic: Agranulocytosis, nonthrombocytopenic purpura and thrombocytopenic purpura.
Hypersensitive Reactions: Fever combined with aching and sore throat, laryngospasm and respiratory distress.
Laboratory Findings:
Increase in blood triglycerides, elevated transaminase and decrease in High Density Lipoprotein (HDL)
- Worsening angina or myocardial infarction [see Warnings and Precautions (5)]
-
7 DRUG INTERACTIONS
7.1 Catecholamine Depleting Drugs
Catecholamine depleting drugs (e.g., reserpine, monoamine oxidase (MAO) inhibitors) may have an additive effect when given with beta-blocking agents. Observe patients treated with LOPRESSOR plus a catecholamine depletor for evidence of hypotension or marked bradycardia, which may produce vertigo, syncope, or postural hypotension.
7.2 Epinephrine
While taking beta-blockers, patients with a history of severe anaphylactic reactions to a variety of allergens may be more reactive to repeated challenge and may be unresponsive to the usual doses of epinephrine used to treat an allergic reaction.
7.3 CYP2D6 Inhibitors
Drugs that are strong inhibitors of CYP2D6 such as quinidine, fluoxetine, paroxetine, and propafenone were shown to double metoprolol concentrations. While there is no information about moderate or weak inhibitors, these too are likely to increase metoprolol concentration. Increases in plasma concentration decrease the cardioselectivity of metoprolol [see Clinical Pharmacology (12.3)]. Monitor patients closely, when the combination cannot be avoided.
7.4 Negative Chronotropes
Digitalis glycosides, clonidine, diltiazem and verapamil slow atrioventricular conduction and decrease heart rate. Concomitant use with beta-blockers can increase the risk of bradycardia.
If clonidine and a beta-blocker, such as metoprolol are coadministered, withdraw the beta-blocker several days before the gradual withdrawal of clonidine because beta-blockers may exacerbate the rebound hypertension that can follow the withdrawal of clonidine. If replacing clonidine by beta-blocker therapy, delay the introduction of beta-blockers for several days after clonidine administration has stopped.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published observational studies have not demonstrated an association of adverse developmental outcomes with maternal use of metoprolol during pregnancy (see Data). Untreated hypertension and myocardial infarction during pregnancy can lead to adverse outcomes for the mother and the fetus (see Clinical Considerations). In animal reproduction studies, metoprolol has been shown to increase post-implantation loss and decrease neonatal survival in rats at oral dosages of 500 mg/kg/day, approximately 11 times the daily dose of 450 mg in a 60-kg patient on a mg/m2 basis.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical consideration
Disease-associated maternal and/or embryo/fetal risk
Hypertension in pregnancy increases the maternal risk for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage). Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death. Pregnant women with hypertension should be carefully monitored and managed accordingly.
Fetal/Neonatal adverse reactions
Metoprolol crosses the placenta. Neonates born to mothers who are receiving metoprolol during pregnancy, may be at risk for hypotension, hypoglycemia, bradycardia, and respiratory depression. Observe neonates and manage accordingly.
Data
Human Data
Data from published observational studies did not demonstrate an association of major congenital malformations and use of metoprolol in pregnancy. The published literature has reported inconsistent findings of intrauterine growth retardation, preterm birth and perinatal mortality with maternal use of metoprolol during pregnancy; however, these studies have methodological limitations hindering interpretation. Methodological limitations include retrospective design, concomitant use of other medications, and other unadjusted confounders that may account for the study findings including the underlying disease in the mother. These observational studies cannot definitely establish or exclude any drug-associated risk during pregnancy.
Animal Data
Metoprolol has been shown to increase post-implantation loss and decrease neonatal survival in rats at oral dosages of 500 mg/kg/day, i.e., 11 times, on a mg/m2 basis, the daily dose of 450 mg in a 60-kg patient.
No fetal abnormalities were observed when pregnant rats received metoprolol orally up to a dose of 200 mg/kg/day, i.e., 4 times, the daily dose of 400mg in a 60-kg patient.
8.2 Lactation
Risk Summary
Limited available data from published literature report that metoprolol is present in human milk. The estimated daily infant dose of metoprolol received from breastmilk ranges from 0.05 mg to less than 1 mg. The estimated relative infant dosage was 0.5% to 2% of the mother's weight-adjusted dosage (see Data). No adverse reactions of metoprolol on the breastfed infant have been identified. There is no information regarding the effects of metoprolol on milk production.
Clinical consideration
Monitoring for adverse reactions
For a lactating woman who is a slow metabolizer of metoprolol, monitor the breastfed infant for bradycardia and other symptoms of beta-blockade such as dry mouth, skin or eyes, diarrhea or constipation. In a report of 6 mothers taking metoprolol, none reported adverse effects in her breastfed infant.
Data
Limited published cases estimate the infant daily dose of metoprolol received from breast milk range from 0.05 mg to less than 1 mg.
In 2 women who were taking unspecified amount of metoprolol, milk samples were taken after one dose of metoprolol. The estimated amount of metoprolol and alpha-hydroxymetoprolol in breast milk is reported to be less than 2% of the mother's weight-adjusted dosage.
In a small study, breast milk was collected every 2 to 3 hours over one dosage interval, in three mothers (at least 3 months postpartum) who took metoprolol of unspecified amount. The average amount of metoprolol present in breast milk was 71.5 mcg/day (range 17.0 to 158.7). The average relative infant dosage was 0.5% of the mother's weight-adjusted dosage.
8.3 Females and Males of Reproductive Potential
Risk Summary
Based on the published literature, beta-blockers (including metoprolol) may cause erectile dysfunction and inhibit sperm motility. In animal fertility studies, metoprolol has been associated with reversible adverse effects on spermatogenesis starting at oral dose level of 3.5 mg/kg in rats, which would correspond to a dose of 34 mg/day in humans in mg/m2 equivalent, although other studies have shown no effect of metoprolol on reproductive performance in male rats.
No evidence of impaired fertility due to metoprolol was observed in rats [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of LOPRESSOR have not been established in pediatric patients.
8.5 Geriatric Use
Clinical studies of LOPRESSOR in hypertension did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience in hypertensive patients has not identified differences in responses between elderly and younger patients.
In worldwide clinical trials of Lopressor in myocardial infarction, where approximately 478 patients were over 65years of age (0 over 75 years of age), no age-related differences in safety and effectiveness were found. Other reported clinical experience in myocardial infarction has not identified differences in response between the elderly and younger patients.
In general, use a low initial starting dose in elderly patients given their greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Hepatic Impairment
No studies have been performed with LOPRESSOR in patients with hepatic impairment. Because LOPRESSOR is metabolized by the liver, metoprolol blood levels are likely to increase substantially with poor hepatic function. Therefore, initiate therapy at doses lower than those recommended for a given indication; and increase doses gradually in patients with impaired hepatic function.
8.7 Renal Impairment
The systemic availability and half-life of metoprolol in patients with renal failure do not differ to a clinically significant degree from those in normal subjects. No reduction in dosage is needed in patients with chronic renal failure [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Signs and Symptoms - Overdosage of LOPRESSOR may lead to severe bradycardia, hypotension, and cardiogenic shock. Clinical presentation can also include: atrioventricular block, heart failure, bronchospasm, hypoxia, impairment of consciousness/coma, nausea and vomiting.
Treatment – Consider treating the patient with intensive care. Patients with myocardial infarction or heart failure may be prone to significant hemodynamic instability. Beta-blocker overdose may result in significant resistance to resuscitation with adrenergic agents, including beta-agonists. On the basis of the pharmacologic actions of metoprolol, employ the following measures.
Hemodialysis is unlikely to make a useful contribution to metoprolol elimination [see Clinical Pharmacology (12.3)].
Bradycardia: Evaluate the need for atropine, adrenergic-stimulating drugs or pacemaker to treat bradycardia and conduction disorders.
Hypotension: Treat underlying bradycardia. Consider intravenous vasopressor infusion, such as dopamine or norepinephrine.
Heart failure and shock: May be treated when appropriate with suitable volume expansion, injection of glucagon (if necessary, followed by an intravenous infusion of glucagon), intravenous administration of adrenergic drugs such as dobutamine, with α1 receptor agonistic drugs added in presence of vasodilation.
Bronchospasm: Can usually be reversed by bronchodilators.
-
11 DESCRIPTION
Lopressor tablets contain metoprolol tartrate, a selective beta1-adrenoreceptor blocking agent. Metoprolol tartrate is (±)-1-(Isopropylamino)-3-[p-(2-methoxyethyl) phenoxy]-2-propanol L-(+)-tartrate (2:1) salt, and its structural formula is
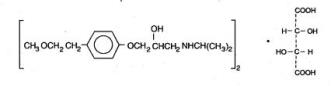
Metoprolol tartrate USP is a white, practically odorless, crystalline powder with a molecular weight of 684.82. It is very soluble in water; freely soluble in methylene chloride, in chloroform, and in alcohol; slightly soluble in acetone; and insoluble in ether.
Lopressor is available as 50 mg and 100 mg tablets for oral administration containing 50 mg and 100 mg metoprolol tartrate, respectively.
Inactive Ingredients: Tablets contain microcrystalline cellulose, colloidal silicon dioxide, lactose monohydrate, magnesium stearate, povidone, sodium starch glycolate. Film coating contains Opadry YS-1-1419 Pink (50 mg tablets) or Opadry YS-1-4281 Blue (100 mg tablets).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Metoprolol is a beta1-selective (cardioselective) adrenergic receptor blocking agent. This preferential effect is not absolute, however, and at higher plasma concentrations, metoprolol also inhibits beta2-adrenoreceptors, chiefly located in the bronchial and vascular musculature.
Metoprolol has no intrinsic sympathomimetic activity, and membrane-stabilizing activity is detectable only at plasma concentrations much greater than required for beta-blockade. Animal and human experiments indicate that metoprolol slows the sinus rate and decreases AV nodal conduction.
The relative beta1-selectivity of metoprolol has been confirmed by the following: (1) In normal subjects, metoprolol is unable to reverse the beta2-mediated vasodilating effects of epinephrine. This contrasts with the effect of nonselective beta-blockers, which completely reverse the vasodilating effects of epinephrine. (2) In asthmatic patients, metoprolol reduces FEV1 and FVC significantly less than a nonselective beta-blocker, propranolol, at equivalent beta1-receptor blocking doses.
Hypertension: The mechanism of the antihypertensive effects of beta-blocking agents has not been elucidated. However, several possible mechanisms have been proposed: (1) competitive antagonism of catecholamines at peripheral (especially cardiac) adrenergic neuron sites, leading to decreased cardiac output; (2) a central effect leading to reduced sympathetic outflow to the periphery; and (3) suppression of renin activity.
Angina Pectoris: By blocking catecholamine-induced increases in heart rate, in velocity and extent of myocardial contraction, and in blood pressure, metoprolol reduces the oxygen requirements of the heart at any given level of effort, thus making it useful in the long-term management of angina pectoris.
Heart Failure: The precise mechanism for the beneficial effects of beta-blockers in heart failure has not been elucidated.
12.2 Pharmacodynamics
Clinical pharmacology studies have confirmed the beta-blocking activity of metoprolol in man, as shown by (1) reduction in heart rate and cardiac output at rest and upon exercise, (2) reduction of systolic blood pressure upon exercise, (3) inhibition of isoproterenol-induced tachycardia, and (4) reduction of reflex orthostatic tachycardia.
The relationship between plasma metoprolol levels and reduction in exercise heart rate is independent of the pharmaceutical formulation. Beta1-blocking effects in the range of 30-80% of the maximal effect (approximately 8 to 23% reduction in exercise heart rate) correspond to metoprolol plasma concentrations from 30 to 540 nmol/L. The relative beta1-selectivity of metoprolol diminishes and blockade of beta2-adrenoceptors increases at plasma concentration above 300 nmol/L.
Significant beta-blocking effect (as measured by reduction of exercise heart rate) occurs within 1 hour after oral administration, and its duration is dose-related. For example, a 50% reduction of the maximum effect after single oral doses of 20, 50, and 100 mg occurred at 3.3, 5.0, and 6.4 hours, respectively, in normal subjects. After repeated oral dosages of 100 mg twice daily, a significant reduction in exercise systolic blood pressure was evident at 12 hours. When the drug was infused over a 10-minute period, in normal volunteers, maximum beta-blockade was achieved at approximately 20 minutes. Equivalent maximal beta-blocking effect is achieved with oral and intravenous doses in the ratio of approximately 2.5:1.
There is a linear relationship between the log of plasma levels and reduction of exercise heart rate. However, antihypertensive activity does not appear to be related to plasma levels. Because of variable plasma levels attained with a given dose and lack of a consistent relationship of antihypertensive activity to dose, selection of proper dosage requires individual titration.
In several studies of patients with acute myocardial infarction, intravenous followed by oral administration of Lopressor caused a reduction in heart rate, systolic blood pressure and cardiac output. Stroke volume, diastolic blood pressure and pulmonary artery end diastolic pressure remained unchanged.
12.3 Pharmacokinetics
Absorption: The estimated oral bioavailability of immediate release metoprolol is about 50% because of pre-systemic metabolism which is saturable leading to non-proportionate increase in the exposure with increased dose.
Distribution: Metoprolol is extensively distributed with a reported volume of distribution of 3.2 to 5.6 L/kg. About 10% of metoprolol in plasma is bound to serum albumin. Metoprolol is known to cross the placenta and is found in breast milk. Metoprolol is also known to cross the blood brain barrier following oral administration and CSF concentrations close to that observed in plasma have been reported. Metoprolol is not a significant P-glycoprotein substrate.
Elimination: Elimination of Lopressor is mainly by biotransformation in the liver. The mean elimination half-life of metoprolol is 3 to 4 hours; in poor CYP2D6 metabolizers the half-life may be 7 to 9 hours.
Metabolism: Lopressor is primarily metabolized by CYP2D6. Metoprolol is a racemic mixture of R- and S- enantiomers, and when administered orally, it exhibits stereoselective metabolism that is dependent on oxidation phenotype. CYP2D6 is absent (poor metabolizers) in about 8% of Caucasians and about 2% of most other populations. Poor CYP2D6 metabolizers exhibit several-fold higher plasma concentrations of Lopressor than extensive metabolizers with normal CYP2D6 activity thereby decreasing Lopressor’s cardioselectivity.
Excretion
Approximately 95% of the dose can be recovered in urine. In most subjects (extensive metabolizers), less than 5% of an oral dose and less than 10% of an intravenous dose are excreted as unchanged drug in the urine. In poor metabolizers, up to 30% or 40% of oral or intravenous doses, respectively, may be excreted unchanged; the rest is excreted by the kidneys as metabolites that appear to have no beta-blocking activity. The renal clearance of the stereo-isomers does not exhibit stereo-selectivity in renal excretion.
Specific populations
Geriatric patients: The geriatric population may show slightly higher plasma concentrations of metoprolol as a combined result of a decreased metabolism of the drug in elderly population and a decreased hepatic blood flow. However, this increase is not clinically significant or therapeutically relevant.
Renal impairment: The systemic availability and half-life of Lopressor in patients with renal failure do not differ to a clinically significant degree from those in normal subjects.
Hepatic Impairment: Since the drug is primarily eliminated by hepatic metabolism, hepatic impairment may impact the pharmacokinetics of metoprolol.
Drug Interactions
Metoprolol is metabolized predominantly by CYP2D6. In healthy subjects with CYP2D6 extensive metabolizer phenotype, coadministration of quinidine 100 mg, a potent CYP2D6 inhibitor, and immediate-release metoprolol 200 mg tripled the concentration of S-metoprolol and doubled the metoprolol elimination half-life. In four patients with cardiovascular disease, coadministration of propafenone 150 mg t.i.d. with immediate-release metoprolol 50 mg t.i.d. increased the steady-state metoprolol concentration 2- to 5-fold compared to metoprolol alone. Extensive metabolizers who concomitantly use CYP2D6 inhibiting drugs will have increased (several-fold) metoprolol blood levels, decreasing metoprolol's cardioselectivity [see Drug Interactions (7.2)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals have been conducted to evaluate the carcinogenic potential of metoprolol tartrate. In 2-year studies in rats at three oral dosage levels of up to 800 mg/kg/day (19 times, on a mg/m2 basis, the daily dose of 400 mg for a 60-kg patient), there was no increase in the development of spontaneously occurring benign or malignant neoplasms of any type. The only histologic changes that appeared to be drug related were an increased incidence of generally mild focal accumulation of foamy macrophages in pulmonary alveoli and a slight increase in biliary hyperplasia. In a 21-month study in Swiss albino mice at three oral dosage levels of up to 750 mg/kg/day (9 times, on a mg/m2 basis, the daily dose of 400 mg for a 60-kg patient), benign lung tumors (small adenomas) occurred more frequently in female mice receiving the highest dose than in untreated control animals. There was no increase in malignant or total (benign plus malignant) lung tumors, nor in the overall incidence of tumors or malignant tumors. This 21-month study was repeated in CD-1 mice, and no statistically or biologically significant differences were observed between treated and control mice of either sex for any type of tumor.
All genotoxicity tests performed on metoprolol tartrate (a dominant lethal study in mice, chromosome studies in somatic cells, a Salmonella/mammalian-microsome mutagenicity test, and a nucleus anomaly test in somatic interphase nuclei) and metoprolol succinate (a Salmonella/mammalian-microsome mutagenicity test) were negative.
No evidence of impaired fertility due to metoprolol tartrate was observed in a study performed in rats at doses up to 55.5 times, on a mg/m2 basis, the daily dose of 450 mg in a 60-kg patient.
-
14 CLINICAL STUDIES
14.1 Hypertension
In controlled clinical studies, Lopressor has been shown to be an effective antihypertensive agent when used alone or as concomitant therapy with thiazide-type diuretics, at dosages of 100 mg to 450 mg daily. In controlled, comparative, clinical studies, Lopressor has been shown to be as effective an antihypertensive agent as propranolol, methyldopa, and thiazide-type diuretics, to be equally effective in supine and standing positions.
14.2 Angina Pectoris
In controlled clinical trials, Lopressor, administered two or four times daily, has been shown to be an effective antianginal agent, reducing the number of angina attacks and increasing exercise tolerance. The dosage used in these studies ranged from 100 mg to 400 mg daily. A controlled, comparative, clinical trial showed that Lopressor was indistinguishable from propranolol in the treatment of angina pectoris.
14.3 Myocardial Infarction
In a large (1395 patients randomized), double-blind, placebo-controlled clinical study, Lopressor was shown to reduce 3-month mortality by 36% in patients with suspected or definite myocardial infarction.
Patients were randomized and treated as soon as possible after their arrival in the hospital, once their clinical condition had stabilized and their hemodynamic status had been carefully evaluated. Subjects were ineligible if they had hypotension, bradycardia, peripheral signs of shock, and/or more than minimal basal rales as signs of congestive heart failure. Initial treatment consisted of intravenous followed by oral administration of Lopressor or placebo, given in a coronary care or comparable unit. Oral maintenance therapy with Lopressor or placebo was then continued for 3 months. After this double-blind period, all patients were given Lopressor and followed up to 1 year.
The median delay from the onset of symptoms to the initiation of therapy was 8 hours in both the Lopressor- and placebo-treatment groups. Among patients treated with Lopressor, there were comparable reductions in 3-month mortality for those treated early (≤ 8 hours) and those in whom treatment was started later. Significant reductions in the incidence of ventricular fibrillation and in chest pain following initial intravenous therapy were also observed with Lopressor and were independent of the interval between onset of symptoms and initiation of therapy.
In this study, patients treated with metoprolol received the drug both very early (intra-venously) and during a subsequent 3-month period, while placebo patients received no beta-blocker treatment for this period. The study thus was able to show a benefit from the overall metoprolol regimen but cannot separate the benefit of very early intravenous treatment from the benefit of later beta-blocker therapy. Nonetheless, because the overall regimen showed a clear beneficial effect on survival without evidence of an early adverse effect on survival, one acceptable dosage regimen is the precise regimen used in the trial. Because the specific benefit of very early treatment remains to be defined however, it is also reasonable to administer the drug orally to patients at a later time as is recommended for certain other beta-blockers.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Lopressor (metoprolol tartrate) tablets
Tablets 50 mg – capsule-shaped, biconvex, pink, scored (imprinted LOPRESSOR on one side and 458 twice on the scored side)
Bottles of 100………………………………………….………NDC 30698-458-01
Tablets 100 mg – capsule-shaped, biconvex, light blue, scored (imprinted LOPRESSOR on one side and 459 twice on the scored side)
Bottles of 100…………………………………………………. NDC 30698-459-01
Storage:
Store at 77°F (25°C); excursions permitted to 59° to 86°F (15° to 30°C) [See USP Controlled Room Temperature]. Protect from moisture and heat.Dispense in a tight, light-resistant container (USP).
-
17 PATIENT COUNSELING INFORMATION
Advise patients to take LOPRESSOR regularly and continuously, as directed, preferably with or immediately following meals. If a dose is missed, the patient should take only the next scheduled dose (without doubling it). Patients should not interrupt or discontinue LOPRESSOR without consulting the physician.
Advise patients (1) to avoid operating automobiles and machinery or engaging in other tasks requiring alertness until the patient’s response to therapy with LOPRESSOR has been determined; (2) to contact the physician if any difficulty in breathing occurs; (3) to inform the physician or dentist before any type of surgery that he or she is taking LOPRESSOR.
Inform patients or caregivers that there is a risk of hypoglycemia when LOPRESSOR is given to patients who are fasting or who are vomiting. Monitor for symptoms of hypoglycemia.
Manufactured for and Distributed by:
Validus Pharmaceuticals LLC
Parsippany, NJ 07054
info@validuspharma.com
www.validuspharma.com
1-866-982-5438Product of Spain
© 2023 Validus Pharmaceuticals LLC
60025-06
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LOPRESSOR
metoprolol tartrate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:30698-458 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METOPROLOL TARTRATE (UNII: W5S57Y3A5L) (METOPROLOL - UNII:GEB06NHM23) METOPROLOL TARTRATE 50 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) D&C RED NO. 30 (UNII: 2S42T2808B) LACTOSE, UNSPECIFIED FORM (UNII: J2B2A4N98G) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) POVIDONE (UNII: FZ989GH94E) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color pink (pink) Score 2 pieces Shape CAPSULE (CAPSULE) Size 13mm Flavor Imprint Code LOPRESSOR;458;458 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:30698-458-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 05/05/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA017963 05/05/2014 LOPRESSOR
metoprolol tartrate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:30698-459 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METOPROLOL TARTRATE (UNII: W5S57Y3A5L) (METOPROLOL - UNII:GEB06NHM23) METOPROLOL TARTRATE 100 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) LACTOSE, UNSPECIFIED FORM (UNII: J2B2A4N98G) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) POVIDONE (UNII: FZ989GH94E) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color blue (light blue) Score 2 pieces Shape CAPSULE (CAPSULE) Size 14mm Flavor Imprint Code LOPRESSOR;459;459 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:30698-459-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 05/05/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA017963 05/05/2014 Labeler - Validus Pharmaceuticals LLC (801194619)

