Label: ORLISTAT capsule

- NDC Code(s): 61269-565-90
- Packager: H2-Pharma LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application Authorized Generic
Drug Label Information
Updated July 8, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ORLISTAT safely and effectively.
See full prescribing information for ORLISTAT.
ORLISTAT Capsules for oral use
Initial U.S. Approval: 1999
RECENT MAJOR CHANGES
- Warnings and Precautions, Oxalate Nephrolithiasis and Oxalate Nephropathy with Renal Failure ( 5.3) 11/2022
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- One 120-mg capsule three times a day with each main meal containing fat (during or up to 1 hour after the meal). ( 2)
- Advise patients to take a nutritionally balanced, reduced-calorie diet that contains approximately 30% of calories from fat. ( 2)
- Distribute the daily intake of fat, carbohydrate, and protein over three main meals. ( 2)
- Advise patients to take a multivitamin containing fat-soluble vitamins to ensure adequate nutrition. ( 2)
- Take the vitamin supplement at least 2 hours before or after the administration of ORLISTAT, such as at bedtime. ( 2)
- For patients receiving both ORLISTAT and cyclosporine therapy, administer cyclosporine 3 hours after ORLISTAT. ( 2)
- For patients receiving both ORLISTAT and levothyroxine therapy, administer levothyroxine and ORLISTAT at least 4 hours apart. ( 2)
DOSAGE FORMS AND STRENGTHS
- Capsules: 120 mg. ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- ORLISTAT has drug interactions and can decrease vitamin absorption. ( 5.1, 7)
- Take a multivitamin supplement that contains fat-soluble vitamins to ensure adequate nutrition. ( 5.1)
- Rare cases of severe liver injury with hepatocellular necrosis or acute hepatic failure have been reported. ( 5.2)
- Patients may develop oxalate nephrolithiasis and oxalate nephropathy following treatment with ORLISTAT. Monitor renal function in patients at risk for renal insufficiency. Discontinue ORLISTAT if oxalate nephropathy develops. ( 5.3)
- Substantial weight loss can increase the risk of cholelithiasis. ( 5.4)
- Exclude organic causes of obesity (eg, hypothyroidism) before prescribing ORLISTAT. ( 5.5)
- Gastrointestinal events may increase when ORLISTAT is taken with a diet high in fat (>30% total daily calories from fat). ( 5.5)
ADVERSE REACTIONS
Most common treatment emergent adverse reactions (≥5% and at least twice that of placebo) include oily spotting, flatus with discharge, fecal urgency, fatty/oily stool, oily evacuation, increased defecation and fecal incontinence. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact the Safety Call Center at 1-877-778-8969 or FDA at 1‑800‑FDA‑1088 (1-800-332-1088) or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Cyclosporine: Reduction in cyclosporine plasma levels was observed when ORLISTAT was coadministered with cyclosporine. ( 7.1)
- Fat-soluble Vitamin Supplements and Analogues: All patients should take a daily multivitamin that contains vitamins A, D, E, K, and beta-carotene. ( 7.2)
- Levothyroxine: Patients treated concomitantly with ORLISTAT and levothyroxine should be monitored for changes in thyroid function. ( 7.3)
- Warfarin: Patients on chronic stable doses of warfarin who are prescribed ORLISTAT should be monitored closely for changes in coagulation parameters. ( 7.4)
- Amiodarone: A reduction in exposure to amiodarone was observed when ORLISTAT was co-administered. ( 7.5)
- Antiepileptic Drugs: Convulsions have been reported in patients taking ORLISTAT with antiepileptic drugs. Patients should be monitored for possible changes in frequency or severity of convulsions. ( 7.6)
- Antiretroviral Drugs: Loss of virological control has been reported in HIV-infected patients. Patients should be monitored frequently for changes in HIV RNA levels. ( 7.7)
USE IN SPECIFIC POPULATIONS
- Nursing Mothers: Caution should be exercised when administered to a nursing woman. ( 8.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 7/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Drug Interactions and Decreased Vitamin Absorption
5.2 Liver Injury
5.3 Oxalate Nephrolithiasis and Oxalate Nephropathy with Renal Failure
5.4 Cholelithiasis
5.5 Miscellaneous
6 ADVERSE REACTIONS
6.1 Clinical Trials
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Cyclosporine
7.2 Fat-soluble Vitamin Supplements and Analogues
7.3 Levothyroxine
7.4 Anticoagulants including Warfarin
7.5 Amiodarone
7.6 Antiepileptic Drugs
7.7 Antiretroviral Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 One-year Results: Weight Loss, Weight Maintenance, and Risk Factors
14.2 Effect on Weight Regain
14.3 Two-year Results: Long-term Weight Control and Risk Factors
14.4 Four-year Results: Long-term Weight Control and Risk Factors
14.5 Study of Patients With Type 2 Diabetes
14.6 Glucose Tolerance in Obese Patients
14.7 Pediatric Clinical Studies
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
ORLISTAT is indicated for obesity management including weight loss and weight maintenance when used in conjunction with a reduced-calorie diet. ORLISTAT is also indicated to reduce the risk for weight regain after prior weight loss. ORLISTAT is indicated for obese patients with an initial body mass index (BMI) ≥30 kg/m 2 or ≥27 kg/m 2 in the presence of other risk factors (e.g., hypertension, diabetes, dyslipidemia).
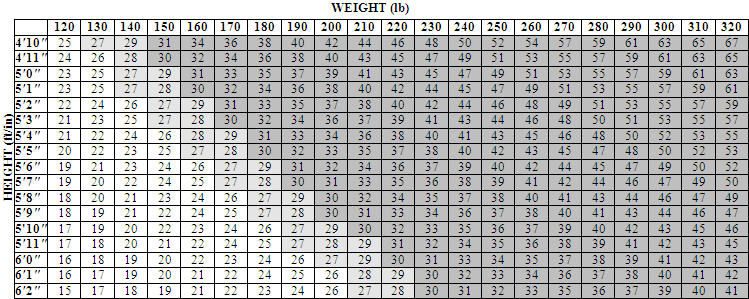
Table 1 illustrates body mass index (BMI) according to a variety of weights and heights. The BMI is calculated by dividing weight in kilograms by height in meters squared. For example, a person who weighs 180 lbs and is 5 '5 " would have a BMI of 30.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dose of ORLISTAT is one 120-mg capsule three times a day with each main meal containing fat (during or up to 1 hour after the meal).
The patient should be on a nutritionally balanced, reduced-calorie diet that contains approximately 30% of calories from fat. The daily intake of fat, carbohydrate, and protein should be distributed over three main meals. If a meal is occasionally missed or contains no fat, the dose of ORLISTAT can be omitted.
Because ORLISTAT has been shown to reduce the absorption of some fat-soluble vitamins and beta-carotene, patients should be counseled to take a multivitamin containing fat-soluble vitamins to ensure adequate nutrition [see Warnings and Precautions (5.1)] . The vitamin supplement should be taken at least 2 hours before or after the administration of ORLISTAT, such as at bedtime.
For patients receiving both ORLISTAT and cyclosporine therapy, administer cyclosporine 3 hours after ORLISTAT.
For patients receiving both ORLISTAT and levothyroxine therapy, administer levothyroxine and ORLISTAT at least 4 hours apart. Patients treated concomitantly with ORLISTAT and levothyroxine should be monitored for changes in thyroid function.
Doses above 120 mg three times a day have not been shown to provide additional benefit.
Based on fecal fat measurements, the effect of ORLISTAT is seen as soon as 24 to 48 hours after dosing. Upon discontinuation of therapy, fecal fat content usually returns to pretreatment levels within 48 to 72 hours.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
ORLISTAT is contraindicated in:
- Pregnancy [see Use in Specific Populations (8.1)]
- Patients with chronic malabsorption syndrome
- Patients with cholestasis
- Patients with known hypersensitivity to ORLISTAT or to any component of this product
-
5 WARNINGS AND PRECAUTIONS
5.1 Drug Interactions and Decreased Vitamin Absorption
ORLISTAT may interact with concomitant drugs including cyclosporine, levothyroxine, warfarin, amiodarone, antiepileptic drugs, and antiretroviral drugs [see Drug Interactions (7)].
Data from a ORLISTAT and cyclosporine drug interaction study indicate a reduction in cyclosporine plasma levels when ORLISTAT was coadministered with cyclosporine. Therefore, ORLISTAT and cyclosporine should not be simultaneously coadministered. To reduce the chance of a drug-drug interaction, cyclosporine should be taken at least 3 hours before or after ORLISTAT in patients taking both drugs. In addition, in those patients whose cyclosporine levels are being measured, more frequent monitoring should be considered.
Patients should be strongly encouraged to take a multivitamin supplement that contains fat-soluble vitamins to ensure adequate nutrition because ORLISTAT has been shown to reduce the absorption of some fat-soluble vitamins and beta-carotene [see Dosage and Administration (2), and Adverse Reactions (6.1)] . In addition, the levels of vitamin D and beta-carotene may be low in obese patients compared with non-obese subjects. The supplement should be taken once a day at least 2 hours before or after the administration of ORLISTAT, such as at bedtime.
Weight-loss may affect glycemic control in patients with diabetes mellitus. A reduction in dose of oral hypoglycemic medication (e.g., sulfonylureas) or insulin may be required in some patients [see Clinical Studies (14)] .
5.2 Liver Injury
There have been rare postmarketing reports of severe liver injury with hepatocellular necrosis or acute hepatic failure in patients treated with ORLISTAT, with some of these cases resulting in liver transplant or death. Patients should be instructed to report any symptoms of hepatic dysfunction (anorexia, pruritus, jaundice, dark urine, light-colored stools, or right upper quadrant pain) while taking ORLISTAT. When these symptoms occur, ORLISTAT and other suspect medications should be discontinued immediately and liver function tests and ALT and AST levels obtained.
5.3 Oxalate Nephrolithiasis and Oxalate Nephropathy with Renal Failure
Some patients may develop increased levels of urinary oxalate following treatment with ORLISTAT. Cases of oxalate nephrolithiasis and oxalate nephropathy with renal failure have been reported. Monitor renal function when prescribing ORLISTAT to patients at increased risk for oxalate nephropathy, including patients with renal impairment and in those with a history of hyperoxaluria or calcium oxalate nephrolithiasis. Discontinue ORLISTAT in patients who develop oxalate nephropathy.
5.4 Cholelithiasis
Substantial weight loss can increase the risk of cholelithiasis. In a clinical trial of ORLISTAT for the prevention of type 2 diabetes, the rates of cholelithiasis as an adverse event were 2.9% (47/1649) for patients randomized to ORLISTAT and 1.8% (30/1655) for patients randomized to placebo.
5.5 Miscellaneous
Organic causes of obesity (e.g., hypothyroidism) should be excluded before prescribing ORLISTAT.
Patients should be advised to adhere to dietary guidelines [see Dosage and Administration (2)] . Gastrointestinal events [see Adverse Reactions (6.1)] may increase when ORLISTAT is taken with a diet high in fat (>30% total daily calories from fat). The daily intake of fat should be distributed over three main meals. If ORLISTAT is taken with any one meal very high in fat, the possibility of gastrointestinal effects increases.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in patients.
Commonly Observed (based on first year and second year data)
Gastrointestinal (GI) symptoms were the most commonly observed treatment-emergent adverse events associated with the use of ORLISTAT in the seven double-blind, placebo-controlled clinical trials and are primarily a manifestation of the mechanism of action. (Commonly observed is defined as an incidence of ≥5% and an incidence in the ORLISTAT 120 mg group that is at least twice that of placebo.)
Table 2 Commonly Observed Adverse Events Adverse Event Year 1 Year 2 Orlistat * % Patients
(N=1913)Placebo * % Patients
(N=1466)Orlistat * % Patients
(N=613)Placebo * % Patients
(N=524)Oily Spotting † 26.6 1.3 4.4 0.2 Flatus with Discharge 23.9 1.4 2.1 0.2 Fecal Urgency 22.1 6.7 2.8 1.7 Fatty/Oily Stool † 20.0 2.9 5.5 0.6 Oily Evacuation † 11.9 0.8 2.3 0.2 Increased Defecation 10.8 4.1 2.6 0.8 Fecal Incontinence 7.7 0.9 1.8 0.2 In general, the first occurrence of these events was within 3 months of starting therapy. Overall, approximately 50% of all episodes of GI adverse events associated with ORLISTAT treatment lasted for less than 1 week, and a majority lasted for no more than 4 weeks. However, GI adverse events may occur in some individuals over a period of 6 months or longer.
Discontinuation of Treatment
In controlled clinical trials, 8.8% of patients treated with ORLISTAT discontinued treatment due to adverse events, compared with 5.0% of placebo-treated patients. For ORLISTAT, the most common adverse events resulting in discontinuation of treatment were gastrointestinal.
Other Adverse Clinical Events
The following table lists other treatment-emergent adverse events from seven multicenter, double-blind, placebo-controlled clinical trials that occurred at a frequency of ≥2% among patients treated with ORLISTAT 120 mg three times a day and with an incidence that was greater than placebo during year 1 and year 2, regardless of relationship to study medication.
Table 3 Other Treatment-Emergent Adverse Events From Seven Placebo-Controlled Clinical Trials Body System/Adverse Event Year 1 Year 2 Orlistat * % Patients
(N=1913)Placebo * % Patients
(N=1466)Orlistat * % Patients
(N=613)Placebo * % Patients
(N=524)- *
- Treatment designates ORLISTAT 120 mg three times a day plus diet or placebo plus diet
– None reported at a frequency ≥2% and greater than placebo Gastrointestinal System Abdominal Pain/Discomfort 25.5 21.4 – – Nausea 8.1 7.3 3.6 2.7 Infectious Diarrhea 5.3 4.4 – – Rectal Pain/Discomfort 5.2 4.0 3.3 1.9 Tooth Disorder 4.3 3.1 2.9 2.3 Gingival Disorder 4.1 2.9 2.0 1.5 Vomiting 3.8 3.5 – – Respiratory System Influenza 39.7 36.2 – – Upper Respiratory Infection 38.1 32.8 26.1 25.8 Lower Respiratory Infection 7.8 6.6 – – Ear, Nose & Throat Symptoms 2.0 1.6 – – Musculoskeletal System Back Pain 13.9 12.1 – – Pain Lower Extremities – – 10.8 10.3 Arthritis 5.4 4.8 – – Myalgia 4.2 3.3 – – Joint Disorder 2.3 2.2 – – Tendonitis – – 2.0 1.9 Central Nervous System Headache 30.6 27.6 – – Dizziness 5.2 5.0 – – Body as a Whole Fatigue 7.2 6.4 3.1 1.7 Sleep Disorder 3.9 3.3 – – Skin & Appendages Rash 4.3 4.0 – – Dry Skin 2.1 1.4 – – Reproductive, Female Menstrual Irregularity 9.8 7.5 – – Vaginitis 3.8 3.6 2.6 1.9 Urinary System Urinary Tract Infection 7.5 7.3 5.9 4.8 Psychiatric Disorder Psychiatric Anxiety 4.7 2.9 2.8 2.1 Depression – – 3.4 2.5 Hearing & Vestibular Disorders Otitis 4.3 3.4 2.9 2.5 Cardiovascular Disorders Pedal Edema – – 2.8 1.9 Table 4 illustrates the percentage of adult patients on ORLISTAT and placebo who developed a low vitamin level on two or more consecutive visits during 1 and 2 years of therapy in studies in which patients were not previously receiving vitamin supplementation.
Table 4 Incidence of Low Vitamin Values on Two or More Consecutive Visits (Nonsupplemented Adult Patients With Normal Baseline Values - First and Second Year) Placebo * Orlistat * - *
- Treatment designates placebo plus diet or ORLISTAT plus diet
Vitamin A 1.0% 2.2% Vitamin D 6.6% 12.0% Vitamin E 1.0% 5.8% Beta-carotene 1.7% 6.1% Table 5 illustrates the percentage of adolescent patients on ORLISTAT and placebo who developed a low vitamin level on two or more consecutive visits during the 1-year study.
Table 5 Incidence of Low Vitamin Values on Two or More Consecutive Visits (Pediatric Patients With Normal Baseline Values *) Placebo † Orlistat † Vitamin A 0.0% 0.0% Vitamin D 0.7% 1.4% Vitamin E 0.0% 0.0% Beta-carotene 0.8% 1.5% In the 4-year XENDOS study, the general pattern of adverse events was similar to that reported for the 1- and 2-year studies with the total incidence of gastrointestinal-related adverse events occurring in year 1 decreasing each year over the 4-year period.
In clinical trials in obese diabetic patients, hypoglycemia and abdominal distension were also observed.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ORLISTAT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to ORLISTAT exposure.
- Rare cases of increase in transaminases and in alkaline phosphatase and hepatitis that may be serious have been reported. There have been reports of hepatic failure observed with the use of ORLISTAT in postmarketing surveillance, with some of these cases resulting in liver transplant or death [see Warnings and Precautions (5.2)] .
- Rare cases of hypersensitivity have been reported with the use of ORLISTAT. Signs and symptoms have included pruritus, rash, urticaria, angioedema, bronchospasm and anaphylaxis. Very rare cases of bullous eruption have been reported.
- Rare cases of leukocytoclastic vasculitis have been reported. Clinical signs include palpable purpura, maculopapular lesions, or bullous eruption.
- Acute oxalate nephropathy after treatment with ORLISTAT has been reported in patients with or at risk for renal disease [see Warnings and Precautions (5.3)] .
- Pancreatitis has been reported with the use of ORLISTAT in postmarketing surveillance. No causal relationship or physiopathological mechanism between pancreatitis and obesity therapy has been definitively established.
- Lower gastrointestinal bleeding has been reported in patients treated with ORLISTAT. Most reports are nonserious; severe or persistent cases should be investigated further.
-
7 DRUG INTERACTIONS
7.1 Cyclosporine
Data from a ORLISTAT and cyclosporine drug interaction study indicate a reduction in cyclosporine plasma levels when ORLISTAT was coadministered with cyclosporine. ORLISTAT and cyclosporine should not be simultaneously coadministered. Cyclosporine should be administered 3 hours after the administration of ORLISTAT [see Dosage and Administration (2), and Warnings and Precautions (5.1)] .
7.2 Fat-soluble Vitamin Supplements and Analogues
Data from a pharmacokinetic interaction study showed that the absorption of beta-carotene supplement is reduced when concomitantly administered with ORLISTAT. ORLISTAT inhibited absorption of a vitamin E acetate supplement. The effect of ORLISTAT on the absorption of supplemental vitamin D, vitamin A, and nutritionally-derived vitamin K is not known at this time [see Clinical Pharmacology (12.3), and Warnings and Precautions (5.1)] .
7.3 Levothyroxine
Hypothyroidism has been reported in patients treated concomitantly with ORLISTAT and levothyroxine postmarketing. Patients treated concomitantly with ORLISTAT and levothyroxine should be monitored for changes in thyroid function. Administer levothyroxine and ORLISTAT at least 4 hours apart [see Dosage and Administration (2)] .
7.4 Anticoagulants including Warfarin
Vitamin K absorption may be decreased with ORLISTAT. Reports of decreased prothrombin, increased INR and unbalanced anticoagulant treatment resulting in change of hemostatic parameters have been reported in patients treated concomitantly with ORLISTAT and anticoagulants. Patients on chronic stable doses of warfarin or other anticoagulants who are prescribed ORLISTAT should be monitored closely for changes in coagulation parameters [see Clinical Pharmacology (12.3)] .
7.5 Amiodarone
A pharmacokinetic study, where amiodarone was orally administered during orlistat treatment, demonstrated a reduction in exposure to amiodarone and its metabolite, desethylamiodarone [see Clinical Pharmocology (12.3)] . A reduced therapeutic effect of amiodarone is possible. The effect of commencing orlistat treatment in patients on stable amiodarone therapy has not been studied.
7.6 Antiepileptic Drugs
Convulsions have been reported in patients treated concomitantly with orlistat and antiepileptic drugs. Patients should be monitored for possible changes in the frequency and/or severity of convulsions.
7.7 Antiretroviral Drugs
Loss of virological control has been reported in HIV-infected patients taking orlistat concomitantly with antiretroviral drugs such as atazanavir, ritonavir, tenofovir disoproxil fumarate, emtricitabine, and with the combinations lopinavir/ritonavir and emtricitabine/efavirenz/tenofovir disoproxil fumarate. The exact mechanism for this is unclear, but may include a drug-drug interaction that inhibits systemic absorption of the antiretroviral drug. HIV RNA levels should be frequently monitored in patients who take ORLISTAT while being treated for HIV infection. If there is a confirmed increase in HIV viral load, ORLISTAT should be discontinued.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category X
ORLISTAT is contraindicated during pregnancy, because weight loss offers no potential benefit to a pregnant woman and may result in fetal harm. A minimum weight gain, and no weight loss, is currently recommended for all pregnant women, including those who are already overweight or obese, due to the obligatory weight gain that occurs in maternal tissues during pregnancy. No embryotoxicity or teratogenicity was seen in animals that received orlistat at doses much higher than the recommended human dose. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard of maternal weight loss to the fetus.
8.3 Nursing Mothers
It is not known if ORLISTAT is present in human milk. Caution should be exercised when ORLISTAT is administered to a nursing woman.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients below the age of 12 have not been established.
The safety and efficacy of ORLISTAT have been evaluated in obese adolescent patients aged 12 to 16 years. Use of ORLISTAT in this age group is supported by evidence from adequate and well-controlled studies of ORLISTAT in adults with additional data from a 54-week efficacy and safety study and a 21-day mineral balance study in obese adolescent patients aged 12 to 16 years. Patients treated with ORLISTAT in the 54-week efficacy and safety study (64.8% female, 75% Caucasians, 18.8% Blacks, and 6.3% Other) had a mean reduction in BMI of 0.55 kg/m 2 compared with an average increase of 0.31 kg/m 2 in placebo-treated patients (p=0.001). In both adolescent studies, adverse effects were generally similar to those described in adults and included fatty/oily stool, oily spotting, and oily evacuation. In a subgroup of 152 ORLISTAT and 77 placebo patients from the 54-week study, changes in body composition measured by DEXA were similar in both treatment groups with the exception of fat mass, which was significantly reduced in patients treated with ORLISTAT compared to patients treated with placebo (-2.5 kg vs -0.6 kg, p=0.033). Because ORLISTAT can interfere with the absorption of fat-soluble vitamins, all patients should take a daily multivitamin that contains vitamins A, D, E, K, and beta-carotene. The vitamin supplement should be taken at least 2 hours before or after ORLISTAT [see Dosage and Administration (2), Warnings and Precautions (5.1), and Clinical Pharmacology (12.3)] .
Plasma concentrations of orlistat and its metabolites M1 and M3 were similar to those found in adults at the same dose level. Daily fecal fat excretions were 27% and 7% of dietary intake in ORLISTAT and placebo treatment groups, respectively.
8.5 Geriatric Use
Clinical studies of ORLISTAT did not include sufficient numbers of patients aged 65 years and older to determine whether they respond differently from younger patients [see Clinical Studies (14)] .
-
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
As with any weight-loss agent, the potential exists for abuse of ORLISTAT in inappropriate patient populations (e.g., patients with anorexia nervosa or bulimia). See Indications and Usage (1) for recommended prescribing guidelines.
-
10 OVERDOSAGE
Single doses of 800 mg ORLISTAT and multiple doses of up to 400 mg three times a day for 15 days have been studied in normal weight and obese subjects without significant adverse findings.
Should a significant overdose of ORLISTAT occur, it is recommended that the patient be observed for 24 hours. Based on human and animal studies, systemic effects attributable to the lipase-inhibiting properties of ORLISTAT should be rapidly reversible.
-
11 DESCRIPTION
ORLISTAT is a gastrointestinal lipase inhibitor for obesity management that acts by inhibiting the absorption of dietary fats.
Orlistat is (S)-2-formylamino-4-methyl-pentanoic acid (S)-1-[[(2S, 3S)-3-hexyl-4-oxo-2-oxetanyl] methyl]-dodecyl ester. Its empirical formula is C 29H 53NO 5, and its molecular weight is 495.7. It is a single diastereomeric molecule that contains four chiral centers, with a negative optical rotation in ethanol at 529 nm. The structure is:
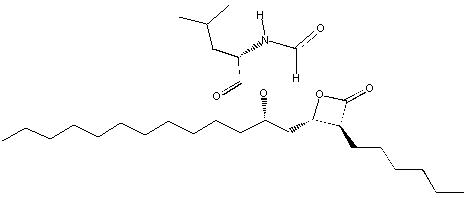
Orlistat is a white to off-white crystalline powder. Orlistat is practically insoluble in water, freely soluble in chloroform, and very soluble in methanol and ethanol. Orlistat has no p Ka within the physiological pH range.
ORLISTAT is available for oral administration as a turquoise hard-gelatin capsule. The capsule is imprinted with black. Each capsule contains a pellet formulation consisting of 120 mg of the active ingredient, orlistat, as well as the inactive ingredients microcrystalline cellulose, sodium starch glycolate, sodium lauryl sulfate, povidone, and talc. The capsule shell contains gelatin, titanium dioxide, and FD&C Blue No. 2 with black printing ink containing pharmaceutical grade shellac, propylene glycol, strong ammonium solution, potassium hydroxide and black iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Orlistat is a reversible inhibitor of gastrointestinal lipases. It exerts its therapeutic activity in the lumen of the stomach and small intestine by forming a covalent bond with the active serine residue site of gastric and pancreatic lipases. The inactivated enzymes are thus unavailable to hydrolyze dietary fat in the form of triglycerides into absorbable free fatty acids and monoglycerides. As undigested triglycerides are not absorbed, the resulting caloric deficit may have a positive effect on weight control.
12.2 Pharmacodynamics
Dose-response Relationship
The dose-response relationship for orlistat in human volunteers is shown in Figure 1. The effect is the percentage of ingested fat excreted, referred to as fecal fat excretion percentage. Both individual data (open circles) and the curve predicted for the population with the maximum-effect model (continuous line) are shown in Figure 1.
Figure 1 Dose-Response Relationship for Orlistat in Human Volunteers
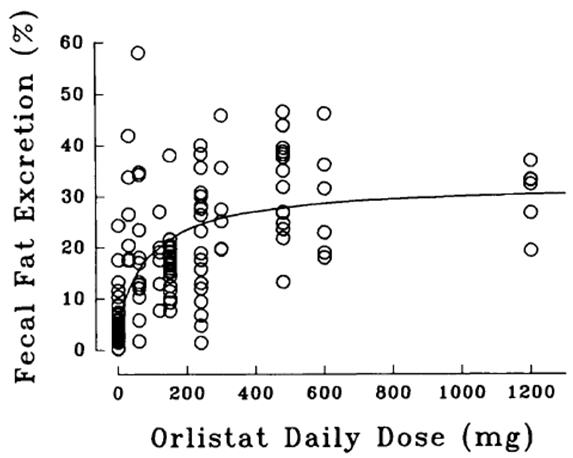
At the recommended therapeutic dose of 120 mg three times a day, orlistat inhibits dietary fat absorption by approximately 30%.
Ethanol does not affect orlistat's effect on preventing the absorption of fat.
Other Short-term Studies
Adults
In several studies of up to 6-weeks duration, the effects of therapeutic doses of ORLISTAT on gastrointestinal and systemic physiological processes were assessed in normal weight and obese subjects. Postprandial cholecystokinin plasma concentrations were lowered after multiple doses of ORLISTAT in two studies but not significantly different from placebo in two other experiments. There were no clinically significant changes observed in gallbladder motility, bile composition or lithogenicity, or colonic cell proliferation rate, and no clinically significant reduction of gastric emptying time or gastric acidity. In addition, no effects on plasma triglyceride levels or systemic lipases were observed with the administration of ORLISTAT in these studies. In a 3-week study of 28 healthy male volunteers, ORLISTAT (120 mg three times a day) did not significantly affect the balance of calcium, magnesium, phosphorus, zinc, copper, and iron.
Pediatrics
In a 3-week study of 32 obese adolescents aged 12 to 16 years, ORLISTAT (120 mg three times a day) did not significantly affect the balance of calcium, magnesium, phosphorus, zinc, or copper. The iron balance was decreased by 64.7 µmole/24 hours and 40.4 µmole/24 hours in ORLISTAT and placebo treatment groups, respectively.
12.3 Pharmacokinetics
Absorption
Systemic exposure to orlistat is minimal. Following oral dosing with 360 mg 14C-orlistat, plasma radioactivity peaked at approximately 8 hours; plasma concentrations of intact orlistat were near the limits of detection (<5 ng/mL). In therapeutic studies involving monitoring of plasma samples, detection of intact orlistat in plasma was sporadic and concentrations were low (<10 ng/mL or 0.02 µM), without evidence of accumulation, and consistent with minimal absorption.
Distribution
In vitro orlistat was >99% bound to plasma proteins (lipoproteins and albumin were major binding proteins). Orlistat minimally partitioned into erythrocytes.
Metabolism
Based on an oral 14C-orlistat mass balance study in obese patients, two metabolites, M1 ((the hydrolyzed β-lactone ring product of orlistat) and M3 (sequential metabolite after M1's cleavage of the N-formyl leucine side-chain), accounted for approximately 42% of total radioactivity in plasma. M1 and M3 have an open β-lactone ring and extremely weak lipase inhibitory activity (1000- and 2500-fold less than orlistat, respectively). In view of this low inhibitory activity and the low plasma levels at the therapeutic dose (average of 26 ng/mL and 108 ng/mL for M1 and M3, respectively, 2 to 4 hours after a dose), these metabolites are considered pharmacologically inconsequential. The primary metabolite M1 had a short half-life (approximately 3 hours) whereas the secondary metabolite M3 eliminated at a slower rate (half-life approximately 13.5 hours).
Elimination
Following a single oral dose of 360 mg 14C-orlistat in both normal weight and obese subjects, fecal excretion of the unabsorbed drug was found to be the major route of elimination. Orlistat and its M1 and M3 metabolites were also subject to biliary excretion. Approximately 97% of the administered radioactivity was excreted in feces; 83% of that was found to be unchanged orlistat. The cumulative renal excretion of total radioactivity was <2% of the given dose of 360 mg 14C-orlistat. The time to reach complete excretion (fecal plus urinary) was 3 to 5 days. The disposition of orlistat appeared to be similar between normal weight and obese subjects. Based on limited data, the half-life of the absorbed orlistat is in the range of 1 to 2 hours.
Specific Populations
No pharmacokinetic study was conducted for specific populations such as geriatric, different races, and patients with renal and hepatic impairment.
Drug Interactions
Alcohol
In a multiple-dose study in 30 normal-weight subjects, coadministration of ORLISTAT and 40 grams of alcohol (e.g., approximately 3 glasses of wine) did not result in alteration of alcohol pharmacokinetics, orlistat pharmacodynamics (fecal fat excretion), or systemic exposure to orlistat.
Amiodarone
In a pharmacokinetic study conducted in healthy volunteers who received 120 mg orlistat three times daily for 13 days and a single dose of 120 mg orlistat on the morning of Day 14 co-administered with a single dose of 1200 mg amiodarone on Day 4, a 23 – 27% reduction in the systemic exposure to amiodarone and desethylamiodarone was observed [see Drug Interactions (7.5)] . The effect of commencing orlistat treatment in patients on stable amiodarone therapy has not been studied.
Cyclosporine
In a multiple-dose study, coadministration of 50 mg cyclosporine twice daily with 120 mg ORLISTAT three times daily decreased cyclosporine AUC and C max by 31% and 25%, respectively. In the same study, administration of 50 mg cyclosporine twice daily three hours after the administration of 120 mg ORLISTAT three times daily decreased cyclosporine AUC and C max by 17% and 4%, respectively.
Digoxin
In 12 normal-weight subjects receiving ORLISTAT 120 mg three times a day for 6 days, ORLISTAT did not alter the pharmacokinetics of a single dose of digoxin.
Fat-soluble Vitamin Supplements and Analogues
A pharmacokinetic interaction study showed a 30% reduction in beta-carotene supplement absorption when concomitantly administered with ORLISTAT. ORLISTAT inhibited absorption of a vitamin E acetate supplement by approximately 60%. The effect of ORLISTAT on the absorption of supplemental vitamin D, vitamin A, and nutritionally-derived vitamin K is not known at this time.
Glyburide
In 12 normal-weight subjects receiving orlistat 80 mg three times a day for 5 days, orlistat did not alter the pharmacokinetics or pharmacodynamics (blood glucose-lowering) of glyburide.
Nifedipine (extended-release tablets)
In 17 normal-weight subjects receiving ORLISTAT 120 mg three times a day for 6 days, ORLISTAT did not alter the bioavailability of nifedipine (extended-release tablets).
Oral Contraceptives
In 20 normal-weight female subjects, the treatment of ORLISTAT 120 mg three times a day for 23 days resulted in no changes in the ovulation-suppressing action of oral contraceptives.
Phenytoin
In 12 normal-weight subjects receiving ORLISTAT 120 mg three times a day for 7 days, ORLISTAT did not alter the pharmacokinetics of a single 300-mg dose of phenytoin.
Pravastatin
In a 2-way crossover study of 24 normal-weight, mildly hypercholesterolemic patients receiving ORLISTAT 120 mg three times a day for 6 days, ORLISTAT did not affect the pharmacokinetics of pravastatin.
Warfarin
In 12 normal-weight subjects, administration of ORLISTAT 120 mg three times a day for 16 days did not result in any change in either warfarin pharmacokinetics (both R- and S-enantiomers) or pharmacodynamics (prothrombin time and serum Factor VII). Although undercarboxylated osteocalcin, a marker of vitamin K nutritional status, was unaltered with ORLISTAT administration, vitamin K levels tended to decline in subjects taking ORLISTAT. Therefore, as vitamin K absorption may be decreased with ORLISTAT, patients on chronic stable doses of warfarin who are prescribed ORLISTAT should be monitored closely for changes in coagulation parameters.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies in rats and mice did not show a carcinogenic potential for orlistat at doses up to 1000 mg/kg/day and 1500 mg/kg/day, respectively. For mice and rats, these doses are 38 and 46 times the daily human dose calculated on an area under concentration vs time curve basis of total drug-related material.
Orlistat had no detectable mutagenic or genotoxic activity as determined by the Ames test, a mammalian forward mutation assay (V79/HPRT), an in vitro clastogenesis assay in peripheral human lymphocytes, an unscheduled DNA synthesis assay (UDS) in rat hepatocytes in culture, and an in vivo mouse micronucleus test.
When given to rats at a dose of 400 mg/kg/day in a fertility and reproduction study, orlistat had no observable adverse effects. This dose is 12 times the daily human dose calculated on a body surface area (mg/m 2) basis.
-
14 CLINICAL STUDIES
The long-term effects of ORLISTAT on morbidity and mortality associated with obesity have not been established.
The effects of ORLISTAT on weight loss, weight maintenance, and weight regain and on a number of comorbidities (e.g., type 2 diabetes, lipids, blood pressure) were assessed in the 4-year XENDOS study and in seven long-term (1- to 2-years duration) multicenter, double-blind, placebo-controlled clinical trials. During the first year of therapy, the studies of 2-year duration assessed weight loss and weight maintenance. During the second year of therapy, some studies assessed continued weight loss and weight maintenance and others assessed the effect of ORLISTAT on weight regain. These studies included over 2800 patients treated with ORLISTAT and 1400 patients treated with placebo (age range 17-78 years, 80.2% women, 91.0% Caucasians, 5.7% Blacks, 2.3% Hispanics, 0.9% Other). The majority of these patients had obesity-related risk factors and comorbidities. In the XENDOS study, which included 3304 patients (age range 30-58 years, 55% women, 99% Caucasians, 1% other), the time to onset of type 2 diabetes was assessed in addition to weight management. In all these studies, treatment with ORLISTAT and placebo designates treatment with ORLISTAT plus diet and placebo plus diet, respectively.
During the weight loss and weight maintenance period, a well-balanced, reduced-calorie diet that was intended to result in an approximate 20% decrease in caloric intake and provide 30% of calories from fat was recommended to all patients. In addition, all patients were offered nutritional counseling.
14.1 One-year Results: Weight Loss, Weight Maintenance, and Risk Factors
Pooled data from five clinical trials indicated that the overall mean weight loss from randomization to the end of 1 year of treatment in the intent-to-treat population was 13.4 lbs in the patients treated with ORLISTAT and 5.8 lbs in the placebo-treated patients. After 1 year of treatment, the mean percent weight loss difference between ORLISTAT-treated patients and placebo-treated patients was 3%. One thousand seventy two (69%) patients treated with ORLISTAT and 701 (63%) patients treated with placebo completed 1 year of treatment. Of the patients who completed 1 year of treatment, 57% of the patients treated with ORLISTAT (120 mg three times a day) and 31% of the placebo-treated patients lost at least 5% of their baseline body weight.
The percentages of patients achieving ≥5% and ≥10% weight loss after 1 year in five large multicenter studies for the intent-to-treat populations are presented in Table 6.
Table 6 Percentage of Patients Losing ≥5% and ≥10% of Body Weight From Randomization After 1-Year Treatment * Study No. Intent-to-Treat Population † ≥5% Weight Loss ≥10% Weight Loss Orlistat n Placebo n p-value Orlistat n Placebo n p-value - *
- Treatment designates ORLISTAT 120 mg three times a day plus diet or placebo plus diet
- †
- Last observation carried forward
- ‡
- All studies, with the exception of 14161, were conducted at centers specialized in treating obesity and complications of obesity. Study 14161 was conducted with primary care physicians.
The diet utilized during year 1 was a reduced-calorie diet. 14119B 35.5% 110 21.3% 108 0.021 16.4% 110 6.5% 108 0.022 14119C 54.8% 343 27.4% 340 <0.001 24.8% 343 8.2% 340 <0.001 14149 50.6% 241 26.3% 236 <0.001 22.8% 241 11.9% 236 0.02 14161 ‡ 37.1% 210 16.0% 212 <0.001 19.5% 210 3.8% 212 <0.001 14185 42.6% 657 22.4% 223 <0.001 17.7% 657 9.9% 223 0.006 The relative changes in risk factors associated with obesity following 1 year of therapy with ORLISTAT and placebo are presented for the population as a whole and for the population with abnormal values at randomization.
Population as a Whole
The changes in metabolic, cardiovascular and anthropometric risk factors associated with obesity based on pooled data for five clinical studies, regardless of the patient's risk factor status at randomization, are presented in Table 7. One year of therapy with ORLISTAT resulted in relative improvement in several risk factors.
Table 7 Mean Change in Risk Factors From Randomization Following 1-Year Treatment * Population as a Whole Risk Factor Orlistat 120 mg † Placebo † Metabolic: Total Cholesterol -2.0% +5.0% LDL-Cholesterol -4.0% +5.0% HDL-Cholesterol +9.3% +12.8% LDL/HDL -0.37 -0.20 Triglycerides +1.34% +2.9% Fasting Glucose, mmol/L -0.04 +0.0 Fasting Insulin, pmol/L -6.7 +5.2 Cardiovascular: Systolic Blood Pressure, mm Hg -1.01 +0.58 Diastolic Blood Pressure, mm Hg -1.19 +0.46 Anthropometric: Waist Circumference, cm -6.45 -4.04 Hip Circumference, cm -5.31 -2.96 Population With Abnormal Risk Factors at Randomization
The changes from randomization following 1-year treatment in the population with abnormal lipid levels (LDL ≥130 mg/dL, LDL/HDL ≥3.5, HDL <35 mg/dL) were greater for ORLISTAT compared to placebo with respect to LDL-cholesterol (-7.83% vs +1.14%) and the LDL/HDL ratio (-0.64 vs -0.46). HDL increased in the placebo group by 20.1% and in the ORLISTAT group by 18.8%. In the population with abnormal blood pressure at baseline (systolic BP ≥140 mm Hg), the change in SBP from randomization to 1 year was greater for ORLISTAT (-10.89 mm Hg) than placebo (-5.07 mm Hg). For patients with a diastolic blood pressure ≥90 mm Hg, ORLISTAT patients decreased by -7.9 mm Hg while the placebo patients decreased by -5.5 mm Hg. Fasting insulin decreased more for ORLISTAT than placebo (-39 vs -16 pmol/L) from randomization to 1 year in the population with abnormal baseline values (≥120 pmol/L). A greater reduction in waist circumference for ORLISTAT vs placebo (-7.29 vs -4.53 cm) was observed in the population with abnormal baseline values (≥100 cm).
14.2 Effect on Weight Regain
Three studies were designed to evaluate the effects of ORLISTAT compared to placebo in reducing weight regain after a previous weight loss achieved following either diet alone (one study, 14302) or prior treatment with ORLISTAT (two studies, 14119C and 14185). The diet utilized during the 1-year weight regain portion of the studies was a weight-maintenance diet, rather than a weight-loss diet, and patients received less nutritional counseling than patients in weight-loss studies. For studies 14119C and 14185, patients' previous weight loss was due to 1 year of treatment with ORLISTAT in conjunction with a mildly hypocaloric diet. Study 14302 was conducted to evaluate the effects of 1 year of treatment with ORLISTAT on weight regain in patients who had lost 8% or more of their body weight in the previous 6 months on diet alone.
In study 14119C, patients treated with placebo regained 52% of the weight they had previously lost while the patients treated with ORLISTAT regained 26% of the weight they had previously lost (p<0.001). In study 14185, patients treated with placebo regained 63% of the weight they had previously lost while the patients treated with ORLISTAT regained 35% of the weight they had lost (p<0.001). In study 14302, patients treated with placebo regained 53% of the weight they had previously lost while the patients treated with ORLISTAT regained 32% of the weight that they had lost (p<0.001).
14.3 Two-year Results: Long-term Weight Control and Risk Factors
The treatment effects of ORLISTAT were examined for 2 years in four of the five 1-year weight management clinical studies previously discussed (see Table 6). At the end of year 1, the patients' diets were reviewed and changed where necessary. The diet prescribed in the second year was designed to maintain patient's current weight. ORLISTAT was shown to be more effective than placebo in long-term weight control in four large, multicenter, 2-year double-blind, placebo-controlled studies.
Pooled data from four clinical studies indicate that 74% of all patients treated with 120 mg three times a day of ORLISTAT and 76% of patients treated with placebo completed 2 years of the same therapy. Pooled data from four clinical studies indicate that the mean weight loss difference between ORLISTAT 120 mg three times a day and placebo treatment groups at year 2 in those patients who completed 1 year of treatment (ITT LOCF) was 3%. In the same studies cited in the One-year Results (see Table 6), the percentages of patients achieving a ≥5% and ≥10% weight loss after 2 years are shown in Table 8.
Table 8 Percentage of Patients Losing ≥5% and ≥10% of Body Weight From Randomization After 2-Year Treatment * Study No. Intent-to-Treat Population † ≥5% Weight Loss ≥10% Weight Loss Orlistat n Placebo n p-value Orlistat n Placebo n p-value - *
- Treatment designates ORLISTAT 120 mg three times a day plus diet or placebo plus diet
- †
- Last observation carried forward
- ‡
- All studies, with the exception of 14161, were conducted at centers specializing in treating obesity or complications of obesity. Study 14161 was conducted with primary care physicians.
The diet utilized during year 2 was designed for weight maintenance and not weight loss. 14119C 45.1% 133 23.6% 123 <0.001 24.8% 133 6.5% 123 <0.001 14149 43.3% 178 27.2% 158 0.002 18.0% 178 9.5% 158 0.025 14161 ‡ 25.0% 148 15.0% 113 0.049 16.9% 148 3.5% 113 0.001 14185 34.0% 147 27.9% 122 0.279 17.7% 147 11.5% 122 0.154 The relative changes in risk factors associated with obesity following 2 years of therapy were also assessed in the population as a whole and the population with abnormal risk factors at randomization.
Population as a Whole
The relative differences in risk factors between treatment with ORLISTAT and placebo were similar to the results following 1 year of therapy for total cholesterol, LDL-cholesterol, LDL/HDL ratio, triglycerides, fasting glucose, fasting insulin, diastolic blood pressure, waist circumference, and hip circumference. The relative differences between treatment groups for HDL cholesterol and systolic blood pressure were less than that observed in the year one results.
Population With Abnormal Risk Factors at Randomization
The relative differences in risk factors between treatment with ORLISTAT and placebo were similar to the results following 1 year of therapy for LDL- and HDL-cholesterol, triglycerides, fasting insulin, diastolic blood pressure, and waist circumference. The relative differences between treatment groups for LDL/HDL ratio and isolated systolic blood pressure were less than that observed in the year one results.
14.4 Four-year Results: Long-term Weight Control and Risk Factors
In the 4-year double-blind, placebo-controlled XENDOS study, the effects of ORLISTAT in delaying the onset of type 2 diabetes and on body weight were compared to placebo in 3304 obese patients who had either normal or impaired glucose tolerance at baseline. Thirty-four percent of the 1655 patients who were randomized to the placebo group and 52% of the 1649 patients who were randomized to the ORLISTAT group completed the 4-year study.
At the end of the study, the mean percent weight loss in the placebo group was -2.75% compared with -5.17% in the ORLISTAT group (p<0.001) (see Figure 2). Forty-five percent of the placebo patients and 73% of the ORLISTAT patients lost ≥5% of their baseline body weight, and 21% of the placebo patients and 41% of the ORLISTAT patients lost ≥10% of their baseline body weight following the first year of treatment. Following 4 years of treatment, 28% of the placebo patients and 45% of the ORLISTAT patients lost ≥5% of their baseline body weight and 10% of the placebo patients and 21% of the ORLISTAT patients lost ≥10% of their baseline body weight. After 4 years of treatment, the mean % difference in weight loss between ORLISTAT treated patients and placebo was 2.5%.
Figure 2 Mean Change from Baseline Body Weight (Kgs) Over Time*
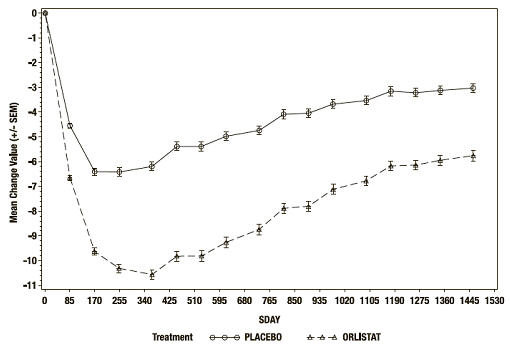
*ITT LOCF study population
The relative changes from baseline in risk factors associated with obesity following 4 years of therapy were assessed in the XENDOS study population (see Table 9).
Table 9 Mean Change in Risk Factors From Randomization Following 4-Years Treatment * Risk Factor Orlistat 120 mg † Placebo † Metabolic: Total Cholesterol -7.02% -2.03% LDL-Cholesterol -11.66% -3.85% HDL-Cholesterol +5.92% +7.01% LDL/HDL -0.53 -0.33 Triglycerides +3.64% +1.30 Fasting Glucose, mmol/L +0.12 +0.23 Fasting Insulin, pmol/L -24.93 -15.71 Cardiovascular: Systolic Blood Pressure, mm Hg -4.12 -2.60 Diastolic Blood Pressure, mm Hg -1.93 -0.87 Anthropometric: Waist Circumference, cm -5.78 -3.99 Onset of Type 2 Diabetes in Obese Patients
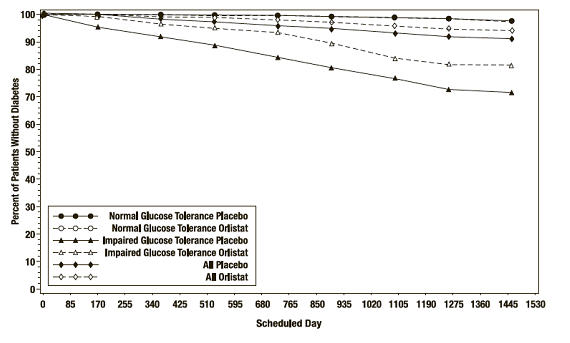
In the XENDOS trial, in the overall population, ORLISTAT delayed the onset of type 2 diabetes such that at the end of four years of treatment the cumulative incidence rate of diabetes was 8.3% for the placebo group compared to 5.5% for the ORLISTAT group, p=0.01 (see Table 10). This finding was driven by a statistically-significant reduction in the incidence of developing type 2 diabetes in those patients who had impaired glucose tolerance at baseline ( Table 10 and Figure 3). ORLISTAT did not reduce the risk for the development of diabetes in patients with normal glucose tolerance at baseline.
The effect of ORLISTAT to delay the onset of type 2 diabetes in obese patients with IGT is presumably due to weight loss, and not to any independent effects of the drug on glucose or insulin metabolism. The effect of ORLISTAT on weight loss is adjunctive to diet and exercise.
Table 10 Incidence Rate of Diabetes at Year 4 by OGTT Status at Baseline * OGTT at Baseline Normal Impaired All Treatment Placebo Orlistat Placebo Orlistat Placebo Orlistat Number of patients * 1148 1235 324 337 1472 1572 # pts developing diabetes 16 21 62 48 78 69 Life table rate † 2.1% 1.7% 27.2% 18.7% 8.3% 5.5% Observed percent 1.4% 1.7% 19.1% 14.2% 5.3% 4.4% Absolute risk reduction Life table 0.4% 8.5% 2.8% Observed -0.3% 4.9% 0.9% Relative risk reduction ‡ 8% 42% 34% p-value 0.79 <0.01 0.01 Figure 3 Percentage of Patients Without Diabetes Over Time
14.5 Study of Patients With Type 2 Diabetes
A 1-year double-blind, placebo-controlled study in type 2 diabetics (N=321) stabilized on sulfonylureas was conducted. Thirty percent of patients treated with ORLISTAT achieved at least a 5% or greater reduction in body weight from randomization compared to 13% of the placebo-treated patients (p<0.001). Table 11 describes the changes over 1 year of treatment with ORLISTAT compared to placebo, in sulfonylurea usage and dose reduction as well as in hemoglobin HbA1c, fasting glucose, and insulin.
Table 11 Mean Changes in Body Weight and Glycemic Control From Randomization Following 1-Year Treatment in Patients With Type 2 Diabetes Orlistat 120 mg *
(n=162)Placebo *
(n=159)Statistical Significance Statistical significance based on intent-to-treat population, last observation carried forward. % patients who discontinued dose of oral sulfonylurea 11.7% 7.5% † % patients who decreased dose of oral sulfonylurea 31.5% 21.4% Average reduction in sulfonylurea medication dose -22.8% -9.1% † Body weight change (lbs) -8.9 -4.2 † HbA1c -0.18% +0.28% † Fasting glucose, mmol/L -0.02 +0.54 † Fasting insulin, pmol/L -19.68 -18.02 ns In addition, ORLISTAT (n=162) compared to placebo (n=159) was associated with significant lowering for total cholesterol (-1.0% vs +9.0%, p≤0.05), LDL-cholesterol (-3.0% vs +10.0%, p≤0.05), LDL/HDL ratio (-0.26 vs -0.02, p≤0.05) and triglycerides (+2.54% vs +16.2%, p≤0.05), respectively. For HDL cholesterol, there was a +6.49% increase on ORLISTAT and +8.6% increase on placebo, p>0.05. Systolic blood pressure increased by +0.61 mm Hg on ORLISTAT and increased by +4.33 mm Hg on placebo, p>0.05. Diastolic blood pressure decreased by -0.47 mm Hg for ORLISTAT and by -0.5 mm Hg for placebo, p>0.05.
14.6 Glucose Tolerance in Obese Patients
Two-year studies that included oral glucose tolerance tests were conducted in obese patients not previously diagnosed or treated for type 2 diabetes and whose baseline oral glucose tolerance test (OGTT) status at randomization was either normal, impaired, or diabetic.
The progression from a normal OGTT at randomization to a diabetic or impaired OGTT following 2 years of treatment with ORLISTAT (n=251) or placebo (n=207) were compared. Following treatment with ORLISTAT, 0.0% and 7.2% of the patients progressed from normal to diabetic and normal to impaired, respectively, compared to 1.9% and 12.6% of the placebo treatment group, respectively.
In patients found to have an impaired OGTT at randomization, the percent of patients improving to normal or deteriorating to diabetic status following 1 and 2 years of treatment with ORLISTAT compared to placebo are presented. After 1 year of treatment, 45.8% of the placebo patients and 73% of the ORLISTAT patients had a normal oral glucose tolerance test while 10.4% of the placebo patients and 2.6% of the ORLISTAT patients became diabetic. After 2 years of treatment, 50% of the placebo patients and 71.7% of the ORLISTAT patients had a normal oral glucose tolerance test while 7.5% of placebo patients were found to be diabetic and 1.7% of ORLISTAT patients were found to be diabetic after treatment.
14.7 Pediatric Clinical Studies
The effects of ORLISTAT on body mass index (BMI) and weight loss were assessed in a 54-week multicenter, double-blind, placebo-controlled study in 539 obese adolescents (357 receiving ORLISTAT 120 mg three times a day, 182 receiving placebo), aged 12 to 16 years. All study participants had a baseline BMI that was 2 units greater than the US weighted mean for the 95 th percentile based on age and gender. Body mass index was the primary efficacy parameter because it takes into account changes in height and body weight, which occur in growing children.
During the study, all patients were instructed to take a multivitamin containing fat-soluble vitamins at least 2 hours before or after ingestion of ORLISTAT. Patients were also maintained on a well-balanced, reduced-calorie diet that was intended to provide 30% of calories from fat. In addition, all patients were placed on a behavior modification program and offered exercise counseling.
Approximately 65% of patients in each treatment group completed the study.
Following one year of treatment, BMI decreased by an average of 0.55 kg/m 2 in the ORLISTAT-treated patients and increased by an average of 0.31 kg/m 2 in the placebo-treated patients (p=0.001).
The percentages of patients achieving ≥5% and ≥10% reduction in BMI and body weight after 52 weeks of treatment for the intent-to-treat population are presented in Table 12.
Table 12 Percentages of Patients with ≥5% and ≥10% Decrease in Body Mass Index and Body Weight After 1-Year Treatment * (Protocol NM16189) Intent-to-Treat Population † ≥5% Decrease ≥10% Decrease Orlistat n Placebo n Orlistat n Placebo n BMI 26.5% 347 15.7% 178 13.3% 347 4.5% 178 Body Weight 19.0% 348 11.7% 180 9.5% 348 3.3% 180 - 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information).
Information for Patients
Patients should not take ORLISTAT if they are pregnant, have chronic malabsorption syndrome, cholestasis or hypersensitivity to ORLISTAT or to any component of this product [see Contraindications (4)] .
Concomitant Medications
Patients should be asked if they are taking cyclosporine, beta carotene or vitamin E supplements, levothyroxine, warfarin, antiepileptic drugs, amiodarone, or antiretroviral drugs due to potential interactions [see Drug Interactions (7)] .
Commonly Observed Adverse Events
Patients should be informed of the commonly-observed adverse events associated with the use of ORLISTAT which include oily spotting, flatus with discharge, fecal urgency, fatty/oily stool, oily evacuation, increased defecation, fecal incontinence [see Adverse Reactions (6.1)] .
Potential Risks and Benefits
Patients should be informed of potential risks which include lowered absorption of fat-soluble vitamins and potential liver injury, increases in urinary oxalate, and cholelithiasis [see Warnings and Precautions (5)] . Treatment with ORLISTAT may result in weight loss and improvement in obesity-related risk factors due to weight loss [see Clinical Studies (14)] .
Dosing Instructions
Patients should be counseled to take ORLISTAT as directed with meals or up to one hour after a meal. Patients should also be advised to take multivitamin supplementation at least two hours before or after the administration of ORLISTAT, or at bedtime [see Dosage and Administration (2)] .
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
Patient Information
ORLISTAT (or-lis-tat)
CapsulesRead this Patient Information before you start taking ORLISTAT and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or your treatment.
What is ORLISTAT?
ORLISTAT is a prescription medicine used with a low calorie diet to increase weight loss in people with obesity. ORLISTAT may help obese people lose weight and keep the weight off.
It is not known if ORLISTAT is safe and effective in children under 12 years old.
Who should not take ORLISTAT?
Do not take ORLISTAT if you:
- are pregnant. A minimum weight gain, and no weight loss, is currently recommended for all pregnant women, including those who are already overweight or obese.
- always have problems absorbing food (chronic malabsorption).
- have gallbladder problems (cholestasis).
- are allergic to orlistat or any of the ingredients in ORLISTAT. See the end of this leaflet for a complete list of ingredients in ORLISTAT.
What should I tell my doctor before taking ORLISTAT?
Before you take ORLISTAT, tell your doctor about all of your medical conditions, including if you:
- have liver problems.
- have kidney problems.
- have problems with your thyroid.
- have eating problems such as anorexia or bulimia.
- have diabetes.
- have a seizure disorder (epilepsy).
- have an abnormal heart rhythm (arrhythmia).
- have the human immunodeficiency virus (HIV).
- are breastfeeding or plan to breastfeed. It is not known if ORLISTAT passes into your breast milk. Talk to your doctor before you breastfeed and take ORLISTAT.
- are pregnant or plan to become pregnant. Do not take ORLISTAT if you are pregnant or plan to become pregnant.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
ORLISTAT and other medicines may affect each other causing side effects. ORLISTAT may affect the way other medicines work, and other medicines may affect the way ORLISTAT works.
Especially tell your doctor if you are taking:
- cyclosporine (Gengraf, Neoral, Sandimmune, Restasis, Sangcya).
- beta-carotene or vitamin E supplements.
- levothyroxine (Levo-T, Levolet, Levothyroid, Levothyroxine Sodium, Levoxyl, Novothyrox, Synthroid, Tirosint, Unithroid).
- warfarin (Athrombin, Athrombin-K, Coumadin, Jantoven, Panwarfin, Warfarin Sodium).
- amiodarone (Cordarone, Pacerone).
- medicines used to treat seizures. They may not work as well while you take ORLISTAT. Talk to your doctor right away if your seizures happen more often or get worse while you take ORLISTAT.
- antiretroviral medicines used to treat HIV. They may not work as well while you take ORLISTAT.
Know the medicines you take. Keep a list of your medicines and show it to your doctor and pharmacist when you get a new medicine.
How should I take ORLISTAT?
- Take ORLISTAT exactly as your doctor tells you to take it.
- Your doctor will tell you how much ORLISTAT to take and when to take it.
- Take ORLISTAT with your meals or up to one hour after your meal. If you miss a meal or have a meal without fat, you can skip your dose of ORLISTAT. If you take a cyclosporine medicine, take ORLISTAT and cyclosporine at least 3 hours apart. See " What should I tell my doctor before taking ORLISTAT?" for a complete list of cyclosporine medicines.
- If you take a multivitamin, take it at least 2 hours before or after you take ORLISTAT. Bedtime is a good time to take your multivitamin.
- If you take a levothyroxine medicine, take ORLISTAT and levothyroxine at least 4 hours apart. See " What should I tell my doctor before taking ORLISTAT?" for a complete list of levothyroxine medicines.
- Take ORLISTAT with a nutritionally balanced, low calorie diet that has no more than about 30% of calories from fat. Taking ORLISTAT with any meal high in fat (more than 30% fat) may make the common side effects worse. See Table 1.
Table 1 IF YOUR DAILY CALORIE LEVEL IS: THE RECOMMENDED DAILY GRAMS OF FAT (in a 30% fat diet) ARE: 1500 50 1600 53 1800 60 2000 67 - If you take too much ORLISTAT call your doctor or go to the nearest hospital emergency room right away.
What are the possible risks of ORLISTAT?
ORLISTAT may cause serious side effects, including:
- Lowered absorption of certain vitamins in your body. Take a multivitamin containing vitamins A, D, E, K, and beta-carotene one time each day. Take a multivitamin at least 2 hours before or after you take ORLISTAT, such as at bedtime.
-
Severe liver problems.Stop taking ORLISTAT and call your doctor right away if you have the following symptoms of liver problems:
- loss of appetite
- itchy skin
- yellowing of your skin or the white part of your eyes
- amber-colored urine
- light-colored bowel movements (stools)
- pain in the upper right portion of your stomach
-
Kidney problems.Your doctor may do certain tests to check your kidney function during treatment with ORLISTAT. Call your doctor right away if you have the following symptoms of kidney problems:
- swelling, especially of the legs and feet
- little or no urine output
- frequent or painful urination
- blood in the urine
- loss of appetite, nausea and vomiting
- severe pain in the back, belly or groin
-
Gallbladder problems (gallstones).Call your doctor right away if you have the following symptoms of gallstones:
- pain in the upper right portion of your stomach
- nausea
- vomiting
The most common side effects of ORLISTAT include:
- oily 1 rectal discharge
- passing gas with oily discharge 1
- urgent need to have a bowel movement
- oily 1 or fatty stools
- increased number of bowel movements
- being unable to control your bowel movements
These are not all the possible side effects of ORLISTAT.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ORLISTAT?
- Store ORLISTAT at 59°F to 86°F (15°C to 30°C).
- Keep ORLISTAT in a tightly closed container.
- Do not use ORLISTAT after the expiration date on the bottle.
- Safely throw away medicine that is out of date or no longer needed.
Keep ORLISTAT and all medicines out of the reach of children.
General information about the safe and effective use of ORLISTAT.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ORLISTAT for a condition for which it was not prescribed. Do not give ORLISTAT to other people, even if they have the same symptoms that you have. It may harm them.
This Patient Information leaflet summarizes the most important information about ORLISTAT. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about ORLISTAT that is written for health professionals.
For more information, call the Safety Call Center at 1-877-778-8969.
What are the ingredients in ORLISTAT?
Active Ingredient: orlistat
Inactive Ingredients: microcrystalline cellulose, sodium starch glycolate, sodium lauryl sulfate, povidone, talc, gelatin and titanium dioxide.
Turquoise capsule shell: FD&C Blue No. 2, with black printing ink containing pharmaceutical grade shellac, propylene glycol, strong ammonium solution, potassium hydroxide and black iron oxide.
Other Information: Body Mass Index
The chart below illustrates body mass index (BMI) according to a variety of weights and heights. ORLISTAT is intended for patients with a BMI of greater than or equal to 30 kg/m 2 or a BMI of greater than or equal to 27 kg/m 2 in the presence of other risk factors such as hypertension, diabetes, or high cholesterol. The BMI is calculated by dividing your weight in kilograms by your height in meters squared. To use this chart:
- Find the height closest to your height in the left-hand column.
- Then move across the top row to find the weight closest to your weight.
- The number where these two meet is your BMI. (For example, a person who weighs 180 lbs and is 5'5" has a BMI of 30.)
This Patient Information has been approved by the U.S. Food and Drug Administration.
Licensed by:
CHEPLAPHARM Arzneimittel GmbHZiegelhof 24, 17489 Greifswald, Germany
Distributed by:
H2-Pharma, LLC611 Industrial Park Blvd, Montgomery, AL 36117, USA
PPI Revised: 11/2022
- 1
- Oily discharge may be clear or have an orange or brown color.
-
SPL UNCLASSIFIED SECTION
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ORLISTAT
orlistat capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:61269-565 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ORLISTAT (UNII: 95M8R751W8) (ORLISTAT - UNII:95M8R751W8) ORLISTAT 120 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) SODIUM LAURYL SULFATE (UNII: 368GB5141J) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) TALC (UNII: 7SEV7J4R1U) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) SHELLAC (UNII: 46N107B71O) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) AMMONIA (UNII: 5138Q19F1X) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color turquoise Score no score Shape CAPSULE Size 19mm Flavor Imprint Code XENICAL;120 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:61269-565-90 90 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 06/01/2022 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA020766 06/01/2022 Labeler - H2-Pharma LLC (028473634) Registrant - Cheplapharm Arzneimittel GmbH (329834878)
