Label: CILOSTAZOL tablet

- NDC Code(s): 63629-8796-1
- Packager: Bryant Ranch Prepack
- This is a repackaged label.
- Source NDC Code(s): 0093-2065
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated October 30, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CILOSTAZOL TABLETS safely and effectively. See full prescribing information for CILOSTAZOL TABLETS.
CILOSTAZOL tablets, for oral use
Initial U.S. Approval: 1999WARNING: CONTRAINDICATED IN HEART FAILURE PATIENTS
See full prescribing information for complete boxed warning.
- Cilostazol is contraindicated in patients with heart failure of any severity. Cilostazol and several of its metabolites are inhibitors of phosphodiesterase III. Several drugs with the pharmacologic effect have caused decreased survival compared to placebo patients with class III-IV heart failure. (4)
INDICATIONS AND USAGE
Cilostazol tablets are a phosphodiesterase III inhibitor (PDE III inhibitor) indicated for the reduction of symptoms of intermittent claudication, as demonstrated by an increased walking distance (1)
DOSAGE AND ADMINISTRATION
- The recommended dosage of cilostazol tablets is 100 mg twice daily taken at least half an hour before or two hours after breakfast and dinner (2.1)
- Reduce the dose to 50 mg twice daily when coadministered with CYP3A4 inhibitors such as ketoconazole, itraconazole, erythromycin, and diltiazem, or CYP2C19 inhibitors such as ticlopidine, fluconazole, and omeprazole (2.2)
DOSAGE FORMS AND STRENGTHS
- Tablets: 50 mg and 100 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Risks of tachycardia, palpitation, tachyarrhythmia or hypotension. Risks of exacerbations of angina pectoris or myocardial infarction in patients with a history of ischemic heart disease (5.2)
- Left ventricular outflow tract obstruction has been reported in patients with sigmoid shaped interventricular septum (5.1)
- Risks of thrombocytopenia or leukopenia progressing to agranulocytosis-monitor platelets and white blood cell counts (5.3)
- Avoid use in patients with hemostatic disorders or active pathologic bleeding (5.4)
ADVERSE REACTIONS
Most common adverse reactions greater than or equal to 2% and at least twice that for placebo in patients on 100 mg twice daily are headache, diarrhea, abnormal stools, and palpitation (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Teva at 1-888-838-2872 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CONTRAINDICATED IN HEART FAILURE PATIENTS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dose Reduction with CYP3A4 and CYP2C19 Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Tachycardia
5.2 Left Ventricular Outflow Tract Obstruction
5.3 Hematologic Adverse Reactions
5.4 Hemostatic Disorders or Active Pathologic Bleeding
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Inhibitors of CYP3A4 or CYP2C19
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CONTRAINDICATED IN HEART FAILURE PATIENTS
Cilostazol is contraindicated in patients with heart failure of any severity. Cilostazol and several of its
metabolites are inhibitors of phosphodiesterase III. Several drugs with this pharmacologic effect have caused
decreased survival compared to placebo in patients with class III-IV heart failure [see Contraindications (4)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of cilostazol tablets is 100 mg twice daily taken at least half an hour before or two hours after breakfast and dinner.
Patients may respond as early as 2 to 4 weeks after the initiation of therapy, but treatment for up to 12 weeks may beneeded before a beneficial effect is experienced. If symptoms are unimproved after 3 months, discontinue cilostazol tablets.
2.2 Dose Reduction with CYP3A4 and CYP2C19 Inhibitors
Reduce dose to 50 mg twice daily when coadministered with strong or moderate inhibitors of CYP3A4 (e.g.,
ketoconazole, itraconazole, erythromycin, and diltiazem) or inhibitors of CYP2C19 (e.g., ticlopidine, fluconazole, and
omeprazole) [see Drug Interactions (7.1)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Cilostazol tablets are contraindicated in patients with:
- Heart failure of any severity: Cilostazol and several of its metabolites are inhibitors of phosphodiesterase III. Several drugs with this pharmacologic effect have caused decreased survival compared to placebo in patients with class III-IV heart failure.
- Hypersensitivity to cilostazol or any components of cilostazol tablets (e.g., anaphylaxis, angioedema)
-
5 WARNINGS AND PRECAUTIONS
5.1 Tachycardia
Cilostazol may induce tachycardia, palpitation, tachyarrhythmia or hypotension. The increase in heart rate associated with
cilostazol is approximately 5 to 7 bpm. Patients with a history of ischemic heart disease may be at risk for exacerbations
of angina pectoris or myocardial infarction.
5.2 Left Ventricular Outflow Tract Obstruction
Left ventricular outflow tract obstruction has been reported in patients with sigmoid shaped interventricular septum. Monitor patients for the development of a new systolic murmur or cardiac symptoms after starting cilostazol.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Patients with Heart Failure [see Boxed Warning]
- Tachycardia [see Warnings and Precautions (5.1)]
- Left Ventricular Outflow Tract Obstruction [see Warnings and Precautions (5.2)]
- Hematologic Adverse Reactions [see Warnings and Precautions (5.3)]
- Hemostatic Disorders or Active Pathologic Bleeding [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials
of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed
in practice.
Adverse reactions were assessed in eight placebo-controlled clinical trials involving patients exposed to either 50 or 100 mg twice daily cilostazol (n = 1301) or placebo (n = 973), with a median treatment duration of 127 days for patients on cilostazol and 134 days for patients on placebo.
The most frequent adverse reaction resulting in discontinuation of therapy in more than 3% of patients treated with
cilostazol was headache [50 mg twice daily (1.3%), 100 mg twice daily (3.5%) and placebo (0.3%)]. Other frequent causes of discontinuation included palpitation and diarrhea, both 1.1% for cilostazol (all doses) versus 0.1% for placebo.
The most common adverse reactions, occurring in at least 2% of patients treated with cilostazol 50 or 100 mg twice daily,
are shown in Table 1.
Table 1: Most Common Adverse Reactions in Patients on Cilostazol 50 or 100 mg Twice Daily (Incidence at least 2% and Occurring More Frequently (≥ 2%) in the 100 mg Twice Daily Group than on Placebo) Adverse Reactions
Placebo
(N = 973)
Cilostazol 50 mg twice daily
(N = 303)
Cilostazol 100 mg twice daily
(N = 998)
Headache
14%
27%
34%
Diarrhea
7%
12%
19%
Abnormal stools
4%
12%
15%
Palpitation
1%
5%
10%
Dizziness
6%
9%
10%
Pharyngitis
7%
7%
10%
Infection
8%
14%
10%
Peripheral edema
4%
9%
7%
Rhinitis
5%
12%
7%
Dyspepsia
4%
6%
6%
Abdominal pain
3%
4%
5%
Tachycardia
1%
4%
4%
Less frequent clinical significant adverse reactions (less than 2%) that were experienced by patients treated with cilostazol50 mg twice daily or 100 mg twice daily in the eight controlled clinical trials and that occurred at a frequency in the
100 mg twice daily group greater than in the placebo group are listed below.
Body as a whole: fever, generalized edema, malaise
Cardiovascular: atrial fibrillation, heart failure, myocardial infarction, nodal arrhythmia, supraventricular tachycardia, ventricular extrasystoles, ventricular tachycardia
Digestive: anorexia, melena
Hematologic and Lymphatic: anemia
Metabolic and Nutritional: increased creatinine, hyperuricemia
Nervous: insomnia
Respiratory: epistaxis
Skin and Appendages: urticaria
Special Senses: conjunctivitis, retinal hemorrhage, tinnitus
Urogenital: urinary frequency
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of cilostazol. Because these reactions are
reported voluntarily from a population of an unknown size, it is not always possible to reliably estimate their frequency or
establish a causal relationship to drug exposure.
Blood and lymphatic system disorders:
Aplastic anemia, granulocytopenia, pancytopenia, bleeding tendency
Cardiac disorders:
Torsade de pointes and QTc prolongation in patients with cardiac disorders (e.g., complete atrioventricular block, heart failure; and bradyarrhythmia), angina pectoris.
Gastrointestinal disorders:
Gastrointestinal hemorrhage, vomiting, flatulence, nausea
General disorders and administration site conditions:
Pain, chest pain, hot flushes
Hepatobiliary disorders:
Hepatic dysfunction/abnormal liver function tests, jaundice
Immune system disorders:
Anaphylaxis, angioedema, and hypersensitivity
Investigations:
Blood glucose increased, blood uric acid increased, increase in BUN (blood urea increased), blood pressure increase
Nervous system disorders:
Intracranial hemorrhage, cerebral hemorrhage, cerebrovascular accident, extradural hematoma and subdural hematoma
Renal and urinary disorders:
Hematuria
Respiratory, thoracic and mediastinal disorders:
Pulmonary hemorrhage, interstitial pneumonia
Skin and subcutaneous tissue disorders:
Hemorrhage subcutaneous, pruritus, skin eruptions including Stevens-Johnson syndrome, skin drug eruption (dermatitis medicamentosa), rash
Vascular disorders:
Subacute stent thrombosis, hypertension.
-
7 DRUG INTERACTIONS
7.1 Inhibitors of CYP3A4 or CYP2C19
Inhibitors of CYP3A4
Coadministration of strong (e.g., ketoconazole) and moderate (e.g., erythromycin, diltiazem and grapefruit juice) CYP3A4
inhibitors can increase exposure to cilostazol. Reduce cilostazol dose to 50 mg twice daily when coadministered with
strong or moderate inhibitors of CYP3A4 [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
Inhibitors of CYP2C19
Coadministration with CYP2C19 inhibitors (e.g., omeprazole) increases systemic exposure of cilostazol active
metabolites. Reduce cilostazol dose to 50 mg twice daily when coadministered with strong or moderate inhibitors of
CYP2C19 [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects
Cilostazol has been shown to be teratogenic in rats at doses that are greater than 5-times the human MRHD on a body
surface area basis. There are no adequate and well-controlled studies in pregnant women.
In a rat developmental toxicity study, oral administration of 1000 mg cilostazol/kg/day was associated with decreased
fetal weights, and increased incidences of cardiovascular, renal, and skeletal anomalies (ventricular septal, aortic arch and
subclavian artery abnormalities, renal pelvic dilation, 14th rib, and retarded ossification). At this dose, systemic exposure
to unbound cilostazol in nonpregnant rats was about 5 times the exposure in humans given the MRHD. Increased
incidences of ventricular septal defect and retarded ossification were also noted at 150 mg/kg/day (5 times the MRHD on
a systemic exposure basis). In a rabbit developmental toxicity study, an increased incidence of retardation of ossification
of the sternum was seen at doses as low as 150 mg/kg/day. In nonpregnant rabbits given 150 mg/kg/day, exposure to
unbound cilostazol was considerably lower than that seen in humans given the MRHD, and exposure to 3,4-dehydrocilostazol was barely detectable.
When cilostazol was administered to rats during late pregnancy and lactation, an increased incidence of stillborn and
decreased birth weights of offspring was seen at doses of 150 mg/kg/day (5 times the MRHD on a systemic exposure
basis).
8.3 Nursing Mothers
Transfer of cilostazol into milk has been reported in rats. Because many drugs are excreted in human milk and because of
the potential for serious adverse reactions in nursing infants from cilostazol, discontinue nursing or discontinue cilostazol.
8.4 Pediatric Use
Safety and effectiveness of cilostazol in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of subjects (n = 2,274) in clinical studies of cilostazol, 56 percent were 65 years old and over, while
16 percent were 75 years old and over. No overall differences in safety or effectiveness were observed between these
subjects and younger subjects, and other reported clinical experience has not identified differences in responses between
the elderly and younger patients, but greater sensitivity of some older individuals cannot be excluded. Pharmacokinetic
studies have not disclosed any age-related effects on the absorption, distribution, metabolism, and elimination of
cilostazol and its metabolites.
8.6 Hepatic Impairment
No dose adjustment is required in patients with mild hepatic impairment. Patients with moderate or severe hepatic
impairment have not been studied in clinical trials and dosing recommendations cannot be provided [see Clinical
Pharmacology (12.3)].
8.7 Renal Impairment
No dose adjustment is required in patients with renal impairment. Patients on dialysis have not been studied, but, it is
unlikely that cilostazol can be removed efficiently by dialysis because of its high protein binding (95 to 98%) [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Information on acute overdosage with cilostazol in humans is limited. The signs and symptoms of an acute overdose can
be anticipated to be those of excessive pharmacologic effect: severe headache, diarrhea, hypotension, tachycardia, and
possibly cardiac arrhythmias. The patient should be carefully observed and given supportive treatment. Since cilostazol is
highly protein-bound, it is unlikely that it can be efficiently removed by hemodialysis or peritoneal dialysis. The oral LD50
of cilostazol is greater than 5 g per kg in mice and rats and greater than 2 g per kg in dogs.
-
11 DESCRIPTION
Cilostazol, USP is a quinolinone derivative that inhibits cellular phosphodiesterase (more specific for phosphodiesterase III). Cilostazol, USP is 6-[4-(1-cyclohexyl-1H-tetrazol-5-yl)butoxy]-3,4-dihydro-2(1H)-quinolinone, CAS-73963-72-1.
The structural formula is:
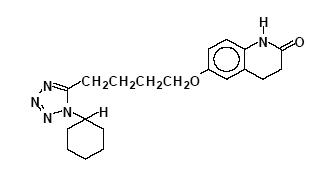
C20H27N5O2 M.W. 369.46
Cilostazol, USP occurs as white to off-white crystals or as a crystalline powder that is slightly soluble in methanol and ethanol, and is practically insoluble in water, 0.1 N HCl, and 0.1 N NaOH.
Cilostazol tablets, USP for oral administration are available in 50 mg pillow-shaped and 100 mg round, white to off-white debossed tablets. Each tablet, in addition to the active ingredient, contains the following inactive ingredients: colloidal silicon dioxide, corn starch, crospovidone, magnesium stearate, microcrystalline cellulose, and povidone.
Meets USP Dissolution Test 3.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Cilostazol and several of its metabolites inhibit phosphodiesterase III activity and suppress cAMP degradation with a
resultant increase in cAMP in platelets and blood vessels, leading to inhibition of platelet aggregation and vasodilation,
respectively.
Cilostazol reversibly inhibits platelet aggregation induced by a variety of stimuli, including thrombin, ADP, collagen,
arachidonic acid, epinephrine, and shear stress.
Cardiovascular effects:
Cilostazol affects both vascular beds and cardiovascular function. It produces heterogeneous dilation of vascular beds,
with greater dilation in femoral beds than in vertebral, carotid or superior mesenteric arteries. Renal arteries were not
responsive to the effects of cilostazol.
In dogs or cynomolgus monkeys, cilostazol increased heart rate, myocardial contractile force, and coronary blood flow as
well as ventricular automaticity, as would be expected for a PDE III inhibitor. Left ventricular contractility was increased
at doses required to inhibit platelet aggregation. A-V conduction was accelerated. In humans, heart rate increased in a
dose-proportional manner by a mean of 5.1 and 7.4 beats per minute in patients treated with 50 and 100 mg twice daily,
respectively.
12.2 Pharmacodynamics
Cilostazol's effects on platelet aggregation were evaluated in both healthy subjects and in patients with stable symptoms of cerebral thrombosis, cerebral embolism, transient ischemic attack, or cerebral arteriosclerosis over a range of doses from 50 mg every day to 100 mg three times a day. Cilostazol significantly inhibited platelet aggregation in a dose-dependent manner. The effects were observed as early as 3 hours post-dose and lasted up to 12 hours following a single dose.
Following chronic administration and withdrawal of cilostazol, the effects on platelet aggregation began to subside 48 hours after withdrawal and returned to baseline by 96 hours with no rebound effect. A cilostazol dosage of 100 mg twice daily consistently inhibited platelet aggregation induced with arachidonic acid, collagen and adenosine diphosphate (ADP). Bleeding time was not affected by cilostazol administration.
Effects on circulating plasma lipids have been examined in patients taking cilostazol. After 12 weeks, as compared to placebo, cilostazol 100 mg twice daily produced a reduction in triglycerides of 29.3 mg/dL (15%) and an increase in HDL-cholesterol of 4.0 mg/dL (≅ 10%).
Drug Interactions
Aspirin
Short-term (less than or equal to 4 days) coadministration of aspirin with cilostazol increased the inhibition of ADP- induced ex vivo platelet aggregation by 22% to 37% when compared to either aspirin or cilostazol alone. Short-term (less than or equal to 4 days) coadministration of aspirin with cilostazol increased the inhibition of arachidonic acid-induced ex vivo platelet aggregation by 20% compared to cilostazol alone and by 48% compared to aspirin alone. However, short- term coadministration of aspirin with cilostazol had no clinically significant impact on PT, aPTT, or bleeding time compared to aspirin alone. Effects of long-term coadministration in the general population are unknown.
In eight randomized, placebo-controlled, double-blind clinical trials, aspirin was coadministered with cilostazol to 201 patients. The most frequent doses and mean durations of aspirin therapy were 75 to 81 mg daily for 137 days (107 patients) and 325 mg daily for 54 days (85 patients). There was no apparent increase in frequency of hemorrhagic adverse effects in patients taking cilostazol and aspirin compared to patients taking placebo and equivalent doses of aspirin.
Warfarin
Cilostazol did not inhibit the pharmacologic effects (PT, aPTT, bleeding time, or platelet aggregation) of R- and S-warfarin after a single 25-mg dose of warfarin. The effect of concomitant multiple dosing of warfarin and cilostazol on the pharmacodynamics of both drugs is unknown.
12.3 Pharmacokinetics
Cilostazol is absorbed after oral administration. A high fat meal increases absorption, with an approximately 90% increase in Cmax and a 25% increase in AUC. Absolute bioavailability is not known. Cilostazol is extensively metabolized by hepatic cytochrome P-450 enzymes, mainly 3A4, and, to a lesser extent, 2C19, with metabolites largely excreted in urine.
Two metabolites are active, with one metabolite appearing to account for at least 50% of the pharmacologic (PDE III inhibition) activity after administration of cilostazol.
Pharmacokinetics are approximately dose proportional. Cilostazol and its active metabolites have apparent elimination half-lives of about 11 to 13 hours. Cilostazol and its active metabolites accumulate about 2-fold with chronic administration and reach steady state blood levels within a few days. The pharmacokinetics of cilostazol and its two major active metabolites were similar in healthy subjects and patients with intermittent claudication due to peripheral arterial disease (PAD). Figure 1 shows the mean plasma concentration-time profile at steady state after multiple dosing of cilostazol 100 mg twice daily.
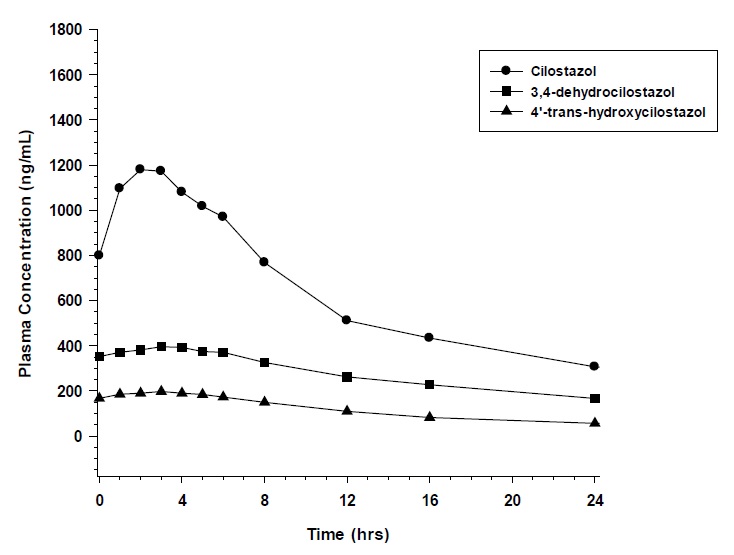
Figure 1: Mean Plasma Concentration-time Profile at Steady State after Multiple Dosing of Cilostazol 100 mg Twice Daily
Distribution
Cilostazol is 95 to 98% protein bound, predominantly to albumin. The binding for 3,4-dehydrocilostazol is 97.4% and for 4´-trans-hydroxy-cilostazol is 66%. Mild hepatic impairment did not affect protein binding. The free fraction of cilostazol
was 27% higher in subjects with renal impairment than in healthy volunteers. The displacement of cilostazol from plasma proteins by erythromycin, quinidine, warfarin, and omeprazole was not clinically significant.
Metabolism
Cilostazol is eliminated predominantly by metabolism and subsequent urinary excretion of metabolites. Based on in vitro studies, the primary isoenzymes involved in cilostazol’s metabolism are CYP3A4 and, to a lesser extent, CYP2C19. The enzyme responsible for metabolism of 3,4-dehydrocilostazol, the most active of the metabolites, is unknown.
Following oral administration of 100 mg radiolabeled cilostazol, 56% of the total analytes in plasma was cilostazol, 15% was 3,4-dehydrocilostazol (4 to 7 times as active as cilostazol), and 4% was 4´-trans-hydroxy-cilostazol (20% as active as cilostazol).
Elimination
The primary route of elimination was via the urine (74%), with the remainder excreted in feces (20%). No measurable amount of unchanged cilostazol was excreted in the urine, and less than 2% of the dose was excreted as 3,4-dehydrocilostazol. About 30% of the dose was excreted in urine as 4´-trans-hydroxy-cilostazol. The remainder was excreted as other metabolites, none of which exceeded 5%. There was no evidence of induction of hepatic microenzymes.
Special Populations
Age and Gender
The total and unbound oral clearances, adjusted for body weight, of cilostazol and its metabolites were not significantly different with respect to age (50 to 80 years) or gender.
Smokers
Population pharmacokinetic analysis suggests that smoking decreased cilostazol exposure by about 20%.
Hepatic Impairment
The pharmacokinetics of cilostazol and its metabolites were similar in subjects with mild hepatic disease as compared to healthy subjects.
Patients with moderate or severe hepatic impairment have not been studied.
Renal Impairment
The total pharmacologic activity of cilostazol and its metabolites was similar in subjects with mild to moderate renal impairment and in healthy subjects. Severe renal impairment increases metabolite levels and alters protein binding of the parent. The expected pharmacologic activity, however, based on plasma concentrations and relative PDE III inhibiting potency of parent drug and metabolites, appeared little changed. Patients on dialysis have not been studied, but, it is unlikely that cilostazol can be removed efficiently by dialysis because of its high protein binding (95 to 98%).
Drug Interactions
Cilostazol does not appear to inhibit CYP3A4.
Warfarin
Cilostazol did not inhibit the metabolism of R- and S-warfarin after a single 25-mg dose of warfarin.
Clopidogrel
Multiple doses of clopidogrel do not significantly increase steady state plasma concentrations of cilostazol.
Strong Inhibitors of CYP3A4
A priming dose of ketoconazole 400 mg (a strong inhibitor of CYP3A4), was given one day prior to coadministration of single doses of ketoconazole 400 mg and cilostazol 100 mg. This regimen increased cilostazol Cmax by 94% and AUC by 117%. Other strong inhibitors of CYP3A4, such as itraconazole, voriconazole, clarithromycin, ritonavir, saquinavir, and nefazodone would be expected to have a similar effect [see Dosage and Administration (2.2), Drug Interactions (7.1)].
Moderate Inhibitors of CYP3A4
Erythromycin and other macrolide antibiotics: Erythromycin is a moderately strong inhibitor of CYP3A4.
Coadministration of erythromycin 500 mg every 8h with a single dose of cilostazol 100 mg increased cilostazol Cmax by 47% and AUC by 73%. Inhibition of cilostazol metabolism by erythromycin increased the AUC of 4´-trans-hydroxy-cilostazol by 141% [see Dosage and Administration (2.2)].
Diltiazem:
Diltiazem 180 mg decreased the clearance of cilostazol by ~30%. Cilostazol Cmax increased ~30% and AUC increased ~40% [see Dosage and Administration (2.2)].
Grapefruit Juice:
Grapefruit juice increased the Cmax of cilostazol by ~50%, but had no effect on AUC.
Inhibitors of CYP2C19
Omeprazole: Coadministration of omeprazole did not significantly affect the metabolism of cilostazol, but the systemic exposure to 3,4-dehydrocilostazol was increased by 69%, probably the result of omeprazole’s potent inhibition of CYP2C19 [see Dosage and Administration (2.2)].
Quinidine
Concomitant administration of quinidine with a single dose of cilostazol 100 mg did not alter cilostazol pharmacokinetics.
Lovastatin
The concomitant administration of lovastatin with cilostazol decreases cilostazol Css, max and AUCτ by 15%. There is also a decrease, although nonsignificant, in cilostazol metabolite concentrations. Coadministration of cilostazol with lovastatin increases lovastatin and β-hydroxylovastatin AUC approximately 70% and is not expected to be clinically significant.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Dietary administration of cilostazol to male and female rats and mice for up to 104 weeks, at doses up to 500 mg/kg/day
in rats and 1000 mg/kg/day in mice, revealed no evidence of carcinogenic potential. The maximum doses administered in
both rat and mouse studies were, on a systemic exposure basis, less than the human exposure at the MRHD of the drug.
Cilostazol tested negative in bacterial gene mutation, bacterial DNA repair, mammalian cell gene mutation, and mouse in
vivo bone marrow chromosomal aberration assays. It was, however, associated with a significant increase in chromosomal
aberrations in the in vitro Chinese Hamster Ovary Cell assay.
In female mice, cilostazol caused a reversible contraceptive effect at a dose (300 mg/kg) that was approximately 7.4-fold greater than the Maximum Recommended Human Dose (MRHD) on a body surface area basis. These findings have not been demonstrated in other animal species.
Cilostazol did not affect fertility or mating performance of male and female rats at doses as high as 1000 mg/kg/day. At
this dose, systemic exposures (AUCs) to unbound cilostazol were less than 1.5 times in males, and about 5 times in
females, the exposure in humans at the MRHD.
13.2 Animal Toxicology and/or Pharmacology
Repeated oral administration of cilostazol to dogs (30 or more mg/kg/day for 52 weeks, 150 or more mg/kg/day for 13
weeks, and 450 mg/kg/day for 2 weeks), produced cardiovascular lesions that included endocardial hemorrhage,
hemosiderin deposition and fibrosis in the left ventricle, hemorrhage in the right atrial wall, hemorrhage and necrosis of
the smooth muscle in the wall of the coronary artery, intimal thickening of the coronary artery, and coronary arteritis and
periarteritis. At the lowest dose associated with cardiovascular lesions in the 52-week study, systemic exposure (AUC) to
unbound cilostazol was less than that seen in humans at the maximum recommended human dose (MRHD) of 100 mg
twice daily. Similar lesions have been reported in dogs following the administration of other positive inotropic agents
(including PDE III inhibitors) and/or vasodilating agents. No cardiovascular lesions were seen in rats following 5 or 13
weeks of administration of cilostazol at doses up to 1500 mg/kg/day. At this dose, systemic exposures (AUCs) to unbound
cilostazol were only about 1.5 and 5 times (male and female rats, respectively) the exposure seen in humans at the
MRHD. Cardiovascular lesions were also not seen in rats following 52 weeks of administration of cilostazol at doses up to
150 mg/kg/day. At this dose, systemic exposures (AUCs) to unbound cilostazol were about 0.5 and 5 times (male and
female rats, respectively) the exposure in humans at the MRHD. In female rats, cilostazol AUCs were similar at 150 and
1500 mg/kg/day. Cardiovascular lesions were also not observed in monkeys after oral administration of cilostazol for 13
weeks at doses up to 1800 mg/kg/day. While this dose of cilostazol produced pharmacologic effects in monkeys, plasma
cilostazol levels were less than those seen in humans given the MRHD, and those seen in dogs given doses associated
with cardiovascular lesions.
-
14 CLINICAL STUDIES
The ability of cilostazol to improve walking distance in patients with stable intermittent claudication was studied in eight, randomized, placebo-controlled, double-blind trials of 12 to 24 weeks’ duration involving 2,274 patients using dosages of 50 mg twice daily (n = 303), 100 mg twice daily (n = 998), and placebo (n = 973). Efficacy was determined primarily by the change in maximal walking distance from baseline (compared to change on placebo) on one of several standardized exercise treadmill tests.
Compared to patients treated with placebo, patients treated with cilostazol 50 or 100 mg twice daily experienced statistically significant improvements in walking distances both for the distance before the onset of claudication pain and the distance before exercise-limiting symptoms supervened (maximal walking distance). The effect of cilostazol on walking distance was seen as early as the first on-therapy observation point of two or four weeks.
Figure 2 depicts the percent mean improvement in maximal walking distance, at study end for each of the eight studies.
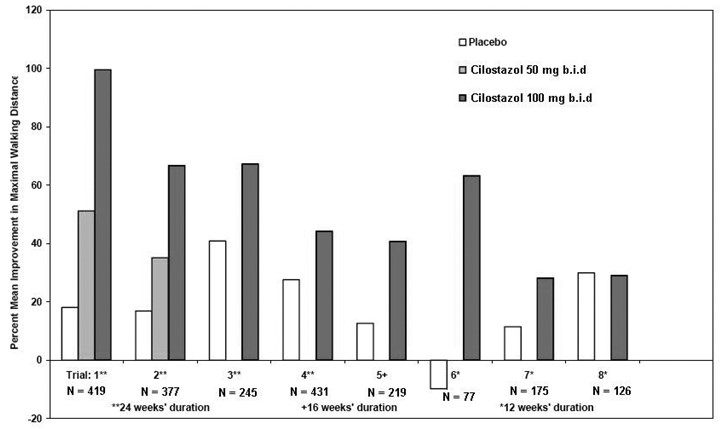
Figure 2: Percent Mean Improvement in Maximal Walking Distance at Study End for the Eight Randomized, Double-Blind, Placebo-Controlled Clinical Trials
Across the eight clinical trials, the range of improvement in maximal walking distance in patients treated with cilostazol 100 mg twice daily, expressed as the change from baseline, was 28% to 100%.
The corresponding changes in the placebo group were –10% to 41%.
The Walking Impairment Questionnaire, which was administered in six of the eight clinical trials, assesses the impact of a therapeutic intervention on walking ability. In a pooled analysis of the six trials, patients treated with either cilostazol 100 mg twice daily or 50 mg twice daily reported improvements in their walking speed and walking distance as compared to placebo. Improvements in walking performance were seen in the various subpopulations evaluated, including those defined by gender, smoking status, diabetes mellitus, duration of peripheral artery disease, age, and concomitant use of beta blockers or calcium channel blockers. Cilostazol has not been studied in patients with rapidly progressing claudication or in patients with leg pain at rest, ischemic leg ulcers, or gangrene. Its long-term effects on limb preservation and hospitalization have not been evaluated.
A randomized, double-blind, placebo-controlled Phase IV study was conducted to assess the long-term effects of cilostazol, with respect to mortality and safety, in 1,439 patients with intermittent claudication and no heart failure. The trial stopped early due to enrollment difficulties and a lower than expected overall death rate. With respect to mortality, the observed 36-month Kaplan-Meier event rate for deaths on study drug with a median time on study drug of 18 months was 5.6% (95% CI of 2.8 to 8.4 %) on cilostazol and 6.8% (95% CI of 1.9 to 11.5 %) on placebo. These data appear to be sufficient to exclude a 75% increase in the risk of mortality on cilostazol, which was the a priori study hypothesis.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Cilostazol Tablets 50 mg - white to off-white, pillow-shaped tablets, debossed with “TEVA” on one side and “7230” on the other, available in bottles of 60 (NDC 63629-8796-01).
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
KEEP THIS AND ALL MEDICATIONS OUT OF THE REACH OF CHILDREN.
Repackaged/Relabeled by:
Bryant Ranch Prepack, Inc.
Burbank, CA 91504
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information)
Advise the patient:
- to take cilostazol at least one-half hour before or two hours after food.
- to discuss with their doctor before taking any CYP3A4 or CYP2C19 inhibitors (e.g., omeprazole).
- that the beneficial effects of cilostazol on the symptoms of intermittent claudication may not be immediate. Although the patient may experience benefit in 2 to 4 weeks after initiation of therapy, treatment for up to 12 weeks may be required before a beneficial effect is experienced. Discontinue cilostazol if symptoms do not improve after 3 months.
Manufactured In India By:
Piramal Pharma Limited
Pithampur 454775,
Dist. Dhar, Madhya Pradesh, IndiaManufactured For:
Teva Pharmaceuticals
Parsippany, NJ 07054Rev. P 7/2022
-
PATIENT INFORMATION
Cilostazol (sye loe' sta zol) Tablets
for oral useRead this Patient Information leaflet before you start taking cilostazol tablets and each time you get a refill. There may
be new information. This information does not take the place of talking to your doctor about your medical condition or your treatment.
What is the most important information I should know about cilostazol tablets?
Cilostazol tablets can cause serious side effects:
- Cilostazol stops a protein called phosphodiesterase III from working. Other similar drugs which affect this protein may cause death if you already have heart problems, called class 3 to 4 (III-IV) heart failure. Do not take cilostazol tablets if you have heart failure of any kind.
What are cilostazol tablets?
Cilostazol tablets are a prescription medicine used to reduce the symptoms of intermittent claudication and can increase
your ability to walk further distances.
It is not known if cilostazol tablets are safe and effective for use in children.
How do cilostazol tablets work?
Improvement in symptoms may occur as soon as 2 weeks, but could take up to 12 weeks.
Who should not take cilostazol tablets?
Do not take cilostazol tablets if you:
- have heart problems (heart failure)
- are allergic to cilostazol or any of the ingredients in cilostazol tablets. See the end of this leaflet for a complete list of ingredients in cilostazol tablets.
Tell your doctor before taking this medicine if you have any of these conditions.
What should I tell my doctor before taking cilostazol tablets?
Before you take cilostazol tablets, tell your doctor if you:
- drink grapefruit juice. Taking cilostazol tablets and drinking grapefruit juice can increase the amount of cilostazol causing side effects.
- have any other medical conditions
- are pregnant or planning to become pregnant. It is not known if cilostazol tablets will harm your unborn baby.
- are breastfeeding or planning to breastfeed. It is not known if cilostazol passes into your breast milk. You and your doctor should decide if you will take cilostazol tablets or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines,
vitamins and herbal supplements.
Ask your doctor for a list of these medicines if you are not sure. You can ask your pharmacist for a list of
medicines that interact with cilostazol tablets. Know the medicines you take. Keep a list of them to show to your doctor
and pharmacist when you get a new medicine.
How should I take cilostazol tablets?
- Take cilostazol tablets exactly as your doctor tells you to take them.
- Your doctor will tell you how many cilostazol tablets to take and when to take them.
- Your doctor may change your dose if needed.
- Take cilostazol tablets 30 minutes before you eat or 2 hours after you eat.
What are the possible side effects of cilostazol tablets?
Cilostazol tablets may cause serious side effects, including:
- heart problems. Taking cilostazol tablets may cause you to have heart problems, including a fast heart beat, palpitations, irregular heartbeat, and low blood pressure.
- See “What is the most important information I should know about cilostazol tablets?”
-
severe allergic reactions (anaphylaxis, angioedema). Call your doctor or go to the nearest emergency room right away if you have any of the following signs or symptoms of a severe allergic reaction:
- hives
- swelling of your face, lips, mouth, or tongue
- trouble breathing or wheezing
- dizziness
- changes in your blood cell counts (thrombocytopenia or leukopenia). Your doctor should do blood tests to check your blood cell counts while you take cilostazol tablets.
The most common side effects of cilostazol tablets include:
- headache
- abnormal stools
- diarrhea
Tell your doctor if you have any side effect that bothers you or does not go away. These are not all the possible
side effects of cilostazol tablets. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store cilostazol tablets?
Store cilostazol tablets at 68° to 77°F (20° to 25°C).
Keep cilostazol tablets and all medicines out of the reach of children.
General information about the safe and effective use of cilostazol tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use cilostazol tablets for a condition for which they were not prescribed. Do not give cilostazol tablets to other people, even if they have the same symptoms that you have. They may harm them.
This Patient Information summarizes the most important information about cilostazol tablets. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about cilostazol tablets that is written for health professionals.
For more information, call 1-888-838-2872.
What are the ingredients in cilostazol tablets?
Active ingredient: cilostazol
Inactive ingredients: colloidal silicon dioxide, corn starch, crospovidone, magnesium stearate, microcrystalline cellulose, and povidone.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Brands listed are the trademarks of their respective owners.
Manufactured In India By:
Piramal Pharma Limited
Pithampur 454775,
Dist. Dhar, Madhya Pradesh, IndiaManufactured For:
Teva Pharmaceuticals
Parsippany, NJ 07054Rev. H 7/2022
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
CILOSTAZOL
cilostazol tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63629-8796(NDC:0093-2065) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CILOSTAZOL (UNII: N7Z035406B) (CILOSTAZOL - UNII:N7Z035406B) CILOSTAZOL 50 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, CORN (UNII: O8232NY3SJ) CROSPOVIDONE (120 .MU.M) (UNII: 68401960MK) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POVIDONE K30 (UNII: U725QWY32X) Product Characteristics Color white (white to off-white) Score no score Shape SQUARE (pillow-shaped) Size 6mm Flavor Imprint Code TEVA;7230 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63629-8796-1 60 in 1 BOTTLE; Type 0: Not a Combination Product 08/30/2022 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077027 04/24/2012 Labeler - Bryant Ranch Prepack (171714327) Registrant - Bryant Ranch Prepack (171714327) Establishment Name Address ID/FEI Business Operations Bryant Ranch Prepack 171714327 REPACK(63629-8796) , RELABEL(63629-8796)
