Label: ISTURISA- osilodrostat tablet, coated



-
NDC Code(s):
55292-320-20,
55292-320-60,
55292-321-20,
55292-321-60, view more55292-322-20, 55292-322-60
- Packager: Recordati Rare Diseases, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated November 3, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use
ISTURISA ® safely and effectively. See full prescribing information for
ISTURISA.
ISTURISA (osilodrostat) tablets, for oral use
Initial U.S. Approval: 2020INDICATIONS AND USAGE
ISTURISA is a cortisol synthesis inhibitor indicated for the treatment of adult patients with Cushing's disease for whom pituitary surgery is not an option or has not been curative (1)
DOSAGE AND ADMINISTRATION
- Correct hypokalemia and hypomagnesemia, and obtain baseline electrocardiogram prior to starting ISTURISA (2.1, 5.2, 5.3)
- Initiate dosage at 2 mg orally twice daily, with or without food (2.2)
- Titrate dosage by 1 to 2 mg twice daily, no more frequently than every 2 weeks based on rate of cortisol changes, individual tolerability and improvement in signs and symptoms (2.2)
- Maximum recommended dosage is 30 mg twice daily (2.2)
- See Full Prescribing Information for complete titration, laboratory, and dosage modification recommendations (2.1,2.2, 2.3)
- Patients with Hepatic Impairment:
DOSAGE FORMS AND STRENGTHS
Tablets: 1 mg, 5 mg, and 10 mg (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Hypocortisolism: Monitor patients closely for hypocortisolism and potentially life-threatening adrenal insufficiency. Dosage reduction or interruption may be necessary (5.1)
- QTc Prolongation: Perform electrocardiogram in all patients Use with caution in patients with risk factors for QTc prolongation (5.2)
- Elevations in Adrenal Hormone Precursors and Androgens: Monitor for hypokalemia, worsening of hypertension, edema, and hirsutism (5.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence > 20%) are adrenal insufficiency, fatigue, nausea, headache, edema (6)
To report SUSPECTED ADVERSE REACTIONS, contact Recordati Rare Diseases Inc. at 1-888-575-8344 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- CYP3A4 Inhibitor: Reduce the dose of ISTURISA by half with concomitant use of a strong CYP3A4 inhibitor (7.1)
- CYP3A4 and CYP2B6 Inducers: An increase of ISTURISA dosage may be needed if ISTURISA is used concomitantly with strong CYP3A4 and CYP2B6 inducers. A reduction in ISTURISA dosage may be needed if strong CYP3A4 and CYP2B6 inducers are discontinued while using ISTURISA (7.1)
USE IN SPECIFIC POPULATIONS
- Lactation: Breastfeeding is not recommended during treatment with ISTURISA and for at least one week after treatment (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Laboratory Testing Prior to ISTURISA Initiation
2.2 Recommended Dosage, Titration, and Monitoring
2.3 Dosage Interruptions and Modifications
2.4 Recommended Dosage and Monitoring in Patients with Renal Impairment
2.5 Recommended Dosage and Monitoring in Patients with Hepatic Impairment
2.6 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypocortisolism
5.2 QTc Prolongation
5.3 Elevations in Adrenal Hormone Precursors and Androgens
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ISTURISA
7.2 Effect of ISTURISA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.2 Recommended Dosage, Titration, and Monitoring
- Initiate dosing at 2 mg orally twice daily, with or without food.
- Initially, titrate the dosage by 1 to 2 mg twice daily, no more frequently than every 2 weeks based on the rate of cortisol changes, individual tolerability and improvement in signs and symptoms of Cushing's disease. If a patient tolerates ISTURISA dosage of 10 mg twice daily and continues to have elevated 24-hour urine free cortisol (UFC) levels above upper normal limit, the dosage can be titrated further by 5 mg twice daily every 2 weeks. Monitor cortisol levels from at least two 24-hour urine free cortisol collections every 1-2 weeks until adequate clinical response is maintained.
- The maintenance dosage of ISTURISA is individualized and determined by titration based on cortisol levels and patient's signs and symptoms.
- The maintenance dosage varied between 2 mg and 7 mg twice daily in clinical trials. The maximum recommended maintenance dosage of ISTURISA is 30 mg twice daily.
- Once the maintenance dosage is achieved, monitor cortisol levels at least every 1-2 months or as indicated.
2.3 Dosage Interruptions and Modifications
- Decrease or temporarily discontinue ISTURISA if urine free cortisol levels fall below the target range, there is a rapid decrease in cortisol levels, and/or patients report symptoms of hypocortisolism. If necessary, glucocorticoid replacement therapy should be initiated.
- Stop ISTURISA and administer exogenous glucocorticoid replacement therapy if serum or plasma cortisol levels are below target range and patients have symptoms of adrenal insufficiency [see Warnings and Precautions (5.1)].
- If treatment is interrupted, re-initiate ISTURISA at a lower dose when cortisol levels are within target ranges and patient symptoms have been resolved.
2.4 Recommended Dosage and Monitoring in Patients with Renal Impairment
- No dose adjustment is required for patients with renal impairment. Use caution in interpreting urine free cortisol levels in patients with moderate to severe renal impairment, due to reduced urine free cortisol excretion [see Clinical Pharmacology (12.3)].
2.5 Recommended Dosage and Monitoring in Patients with Hepatic Impairment
- For patients with moderate hepatic impairment (Child-Pugh B), the recommended starting dose is 1 mg twice daily. For patients with severe hepatic impairment (Child-Pugh C), the recommended starting dose is 1 mg once daily in the evening.
- No dose adjustment is required for patients with mild hepatic impairment (Child-Pugh A).
- More frequent monitoring of adrenal function may be required during dose titration in all patients with hepatic impairment [see Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
ISTURISA is available as:
- 1 mg tablets: Pale yellow, unscored, round, biconvex with beveled edge tablet, debossed "1" on one side.
- 5 mg tablets: Yellow, unscored, round, biconvex with beveled edge tablet, debossed "5" on one side.
- 10 mg tablets: Pale orange brown, unscored, round, biconvex with beveled edge tablet, debossed "10" on one side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypocortisolism
ISTURISA lowers cortisol levels and can lead to hypocortisolism and sometimes life-threatening adrenal insufficiency. Lowering of cortisol can cause nausea, vomiting, fatigue, abdominal pain, loss of appetite, dizziness. Significant lowering of serum cortisol may result in hypotension, abnormal electrolyte levels, and hypoglycemia [see Adverse Reactions (6)].
Hypocortisolism can occur at any time during ISTURISA treatment. Evaluate patients for precipitating causes of hypocortisolism (infection, physical stress, etc.). Monitor 24-hour urine free cortisol, serum or plasma cortisol, and patient's signs and symptoms periodically during ISTURISA treatment.
Decrease or temporarily discontinue ISTURISA if urine free cortisol levels fall below the target range, there is a rapid decrease in cortisol levels, and/or patients report symptoms of hypocortisolism. Stop ISTURISA and administer exogenous glucocorticoid replacement therapy if serum or plasma cortisol levels are below target range and patients have symptoms of adrenal insufficiency. Re-initiate ISTURISA at a lower dose when urine free cortisol, serum or plasma cortisol levels are within target range, and/or patient symptoms have resolved. After ISTURISA discontinuation, cortisol suppression may persist beyond the 4 hour half-life of ISTURISA.
Educate patients on the symptoms associated with hypocortisolism and advise them to contact a healthcare provider if they occur.
5.2 QTc Prolongation
ISTURISA is associated with a dose-dependent QT interval prolongation (maximum mean estimated QTcF increase of up to 5.3 ms at 30 mg), which may cause cardiac arrhythmias [see Adverse Reactions (6), Clinical Pharmacology (12.2)].
Perform an ECG to obtain a baseline QTc interval measurement prior to initiating therapy with ISTURISA and monitor for an effect on the QTc interval thereafter. Correct hypokalemia and/or hypomagnesemia prior to ISTURISA initiation and monitor periodically during treatment with ISTURISA. Correct electrolyte abnormalities if indicated. Consider temporary discontinuation of ISTURISA in the case of an increase in QTc interval > 480 ms.
Use caution in patients with risk factors for QT prolongation, (such as congenital long QT syndrome, congestive heart failure, bradyarrythmias, uncorrected electrolyte abnormalities, and concomitant medications known to prolong the QT interval) and consider more frequent ECG monitoring.
5.3 Elevations in Adrenal Hormone Precursors and Androgens
ISTURISA blocks cortisol synthesis and may increase circulating levels of cortisol and aldosterone precursors (11-deoxy cortisol and 11-deoxycorticosterone) and androgens.
Elevated 11-deoxycorticosterone levels may activate mineralocorticoid receptors and cause hypokalemia, edema and hypertension [see Adverse Reactions (6)]. Hypokalemia should be corrected prior to initiating ISTURISA. Monitor patients treated with ISTURISA for hypokalemia, worsening of hypertension and edema. ISTURISA-induced hypokalemia should be treated with intravenous or oral potassium supplementation based on event severity. If hypokalemia persists despite potassium supplementation, consider adding mineralocorticoid antagonists. ISTURISA dose reduction or discontinuation may be necessary.
Accumulation of androgens may lead to hirsutism, hypertrichosis and acne (in females). Inform patients of the symptoms associated with hyperandrogenism and advise them to contact a healthcare provider if they occur.
-
6 ADVERSE REACTIONS
Clinically significant adverse reactions that appear in other sections of the labeling include:
- Hypocortisolism [see Warnings and Precautions (5.1)]
- QT Prolongation [see Warnings and Precautions (5.2)]
- Elevations in Adrenal Hormone Precursors and Androgens [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
A total of 137 Cushing's disease patients were exposed to ISTURISA in the study [see Clinical Studies (14)]. The adverse reactions that occurred with frequency higher than 10% during the core 48-week period are shown in Table 1.
Table 1: Adverse Reactions With a Frequency of More Than 10% in 48-week Clinical Study in Cushing's Disease Patients a Adrenal insufficiency includes glucocorticoid deficiency, adrenocortical insufficiency acute, steroid withdrawal syndrome, cortisol free urine decreased, cortisol decreased. One-third of the subjects with this event had low cortisol levels indicative of Adrenal Insufficiency. The majority of subjects had normal cortisol levels suggesting a cortisol withdrawal syndrome.
b Fatigue includes lethargy, asthenia.
c Headache includes head discomfort.
d Edema includes edema peripheral, generalized edema, localized edema.
e Rash includes rash erythematous, rash generalized, rash maculopapular, rash papular.
f Dizziness includes dizziness postural.
g Abdominal pain includes abdominal pain upper, abdominal discomfort
h Hypokalaemia includes blood potassium decreased.
i Hypotension includes orthostatic hypotension, blood pressure decreased, blood pressure diastolic decreased, blood pressure systolic decreased.Adverse Reaction Type
(N = 137)
%Adrenal insufficiencya
43.1
Fatigueb
38.7
Nausea
37.2
Headachec
30.7
Edemad
21.2
Nasopharyngitis
19.7
Vomiting
19
Arthralgia
17.5
Back pain
15.3
Rashe
15.3
Diarrhea
14.6
Blood corticotrophin increased
13.9
Dizzinessf
13.9
Abdominal paing
13.1
Hypokalaemiah
12.4
Myalgia
12.4
Decreased appetite
11.7
Hormone level abnormal
11.7
Hypotensioni
11.7
Urinary tract infection
11.7
Blood testosterone increased
10.9
Pyrexia
10.9
Anemia
10.2
Cough
10.2
Hypertension
10.2
Influenza
10.2
Other notable adverse reactions which occurred with a frequency less than 10% were: hirsutism (9.5%), acne (8.8%), dyspepsia (8%), insomnia (8%), anxiety (7.3%), depression (7.3%), gastroenteritis (7.3%), malaise (6.6%), tachycardia (6.6%), alopecia (5.8%), transaminases increased (4.4%), electrocardiogram QT prolongation (3.6%), and syncope (1.5%).
Description of Selected Adverse Reactions
Gastrointestinal Disorders
Gastrointestinal disorders, predominantly nausea, vomiting, diarrhea and abdominal pain were reported in 69% of patients. In many cases, the episodes were of short duration (1-2 days) and the severity was mild to moderate.
Hypocortisolism
Hypocortisolism was reported at a rate of 31% up to 12 weeks, and 18% from Weeks 12 to 26. The majority of cases were manageable by reducing the dose of ISTURISA and/or adding low-dose, short-term glucocorticoid therapy.
Changes in Pituitary Tumor Volume
An increase in the pituitary corticotroph tumor volume by greater than 20% from baseline was observed in 21/137 (15%) patients, while a decrease in tumor volume by greater than 20% from baseline was observed in 24/137 (18%) patients at Week 48. Eight patients discontinued because of an increase in tumor volume. There was no correlation between tumor volume increase and increase in adrenocorticotrophic hormone (ACTH). There was no specific pattern of timing of the tumor volume increase and no relationship with the total and the last dose of ISTURISA used in the study.
QTc Interval Prolongation
Adverse reactions of QT prolongation and clinically relevant ECG findings were reported. Five (4%) patients had an event of QT prolongation, 3 (2%) patients had a QTcF increase of > 60 ms from baseline, and 18 (13%) had a new QTcF value of > 450 ms [see Clinical Pharmacology (12.2)].
Accumulation of Adrenal Hormone Precursors
CYP11B1 inhibition by ISTURISA is associated with adrenal steroid precursor accumulation and testosterone increases [see Warnings and Precautions (5.3)]. The incidence of adverse reactions potentially related to accumulation of adrenal hormone precursors was 42%. Hypertension and hypokalemia were the most common adrenal hormone precursor-related adverse reactions and occurred in 14% of patients and 17% of patients, respectively; edema was reported in 7% of patients, elevated blood pressure in 15% of patients. All cases of hypokalemia responded to treatment with potassium supplementation and/or mineralocorticoid antagonist therapy (e.g., spironolactone). One patient discontinued the study because of hypokalemia. In male patients testosterone levels generally increased but remained within normal limits; all patients were asymtomatic with no values above upper limit of normal (ULN) at last available value. In female patients, mean testosterone levels increased above the normal range from baseline and reversed when treatment was interrupted. The testosterone increase was associated with mild to moderate cases of hirsutism (12%) or acne (11%) in a subset of female patients.
Other Abnormal Laboratory Findings
Decreased Absolute Neutrophil Count
Of the 137 patients from the 48-week study, 18 patients had at least one measured absolute neutrophil count below the normal limit, 2 patients had an adverse reaction of neutropenia. No concomitant infections and/or fever were reported in patients with decreased absolute neutrophil count.
Elevated Liver Function Tests
Liver enzyme elevations in patients treated with ISTURISA were infrequent, typically mild and reversed spontaneously or following dose adjustment. Most liver abnormal parameters occurred during the dose-titration period and no patients discontinued ISTURISA drug due to abnormal liver chemistry parameters. Five (4%) patients had ALT or AST > 3 x ULN during the 48-week clinical study.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ISTURISA
The effect of other drugs on ISTURISA can be found in Table 2.
Table 2: Effect of Other Drugs on ISTURISA CYP3A4 Inhibitors
Clinical Impact:
Concomitant use of ISTURISA with a strong CYP3A4 inhibitor (e.g., itraconazole, clarithromycin) may cause an increase in osilodrostat concentration and may increase the risk of ISTURISA-related adverse reactions [see Clinical Pharmacology (12.3)].
Intervention:
Reduce the dose of ISTURISA by half with concomitant use of a strong CYP3A4
inhibitor.
CYP3A4 and CYP2B6 Inducers
Clinical Impact:
Concomitant use of ISTURISA with strong CYP3A4 and/or CYP2B6 inducers (e.g., carbamazepine, rifampin, phenobarbital) may cause a decrease in osilodrostat concentration and may reduce the efficacy of ISTURISA [see Clinical Pharmacology (12.3)].
Discontinuation of strong CYP3A4 and/or CYP2B6 inducers while using ISTURISA may cause an increase in osilodrostat concentration and may increase the risk of ISTURISA-related adverse reactions [see Clinical Pharmacology (12.3)].
Intervention:
During concomitant use of ISTURISA with strong CYP3A4 and CYP2B6 inducers, monitor cortisol concentration and patient's signs and symptoms. An increase in ISTURISA dosage may be needed.
Upon discontinuation of strong CYP3A4 and CYP2B6 inducers during ISTURISA treatment, monitor cortisol concentration and patient's signs and symptoms. A reduction in ISTURISA dosage may be needed.
7.2 Effect of ISTURISA on Other Drugs
ISTURISA should be used with caution when coadministered with CYP1A2 and CYP2C19 substrates with a narrow therapeutic index, such as theophylline, tizanidine, and S-mephenytoin [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on osilodrostat use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with active Cushing's Syndrome during pregnancy (see Clinical Considerations).
No adverse developmental outcomes were observed in reproduction studies in pregnant rats and rabbits when exposed to osilodrostat during organogenesis at doses that produced maternal exposures of 7 and 0.5-times the 30 mg twice daily maximum clinical dose, by AUC. In rabbits, exposures associated with maternal toxicity at 7-times the maximum clinical dose resulted in decreased fetal viability. No adverse developmental outcomes were observed in a pre- and postnatal development study with administration of osilodrostat to pregnant rats from organogenesis through lactation at 8-times the 30 mg twice daily maximum clinical dose (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Active Cushing Syndrome during pregnancy has been associated with an increased risk of maternal and fetal morbidity and mortality (including gestational diabetes, gestational hypertension, pre-eclampsia, maternal death, miscarriage, fetal loss, and preterm birth).
Data
Animal Data
Osilodrostat administered to pregnant Wistar Han rats from gestation day 6-17 at doses of 0.5, 5, 50 mg/kg did not adversely affect embryo-fetal development up to 5 mg/kg (8-times the 30 mg twice daily maximum clinical dose, by AUC). Maternal toxicity, increased embryonic and fetal deaths, decreased fetal weights, and malformations occurred at 50 mg/kg (118-times the maximum clinical dose, by AUC).
Osilodrostat administered to pregnant New Zealand rabbits from gestation day 7-20 at doses of 3, 10, and 30 mg/kg did not adversely affect embryo-fetal development at 3 mg/kg (0.5-times the 30 mg twice daily maximum clinical dose, by AUC). Maternal toxicity, increased embryo resorption and decreased fetal viability was observed at ≥ 10mg/kg (7-times the maximum clinical dose, by AUC).
Osilodrostat administered to Wistar Han rats from gestation day 6 through lactation day 20 at doses of 1, 5, and 20 mg/kg did not adversely impact behavioral, developmental, or reproductive parameters up to 5 mg/kg (~ 8 times the 30 mg twice daily maximum clinical dose, by AUC). Delayed parturition and dystocia in maternal rats and decreased pup survival were observed at 20 mg/kg (43-times the maximum clinical dose, by AUC).
8.2 Lactation
Risk Summary
There are no available data on the presence of osilodrostat in human or animal milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions (such as adrenal insufficiency) in the breastfed infant, advise patients that breastfeeding is not recommended during treatment with ISTURISA and for one week after the final dose.
8.4 Pediatric Use
The safety and effectiveness of ISTURISA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 167 patients in clinical trials with ISTURISA, 10 (6%) were 65 years and older. There were no patients above 75 years of age. Based on the available data on the use of ISTURISA in patients older than 65 years, no dosage adjustment is required [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dosage adjustment of ISTURISA in patients with impaired renal function is required [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)]. In patients with moderate to severe renal impairment, UFC levels should be interpreted with caution due to reduced UFC excretion.
8.7 Hepatic Impairment
Dosage adjustment is not required in patients with mild hepatic impairment (Child-Pugh A) but is required for patients with moderately impaired hepatic function (Child-Pugh B) and for patients with severe hepatic impairment (Child-Pugh C) [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)]. More frequent monitoring of adrenal function may be required during dose titration in all patients with hepatic impairment.
-
10 OVERDOSAGE
Overdosage may result in severe hypocortisolism. Signs and symptoms suggestive of hypocortisolism may include nausea, vomiting, fatigue, low blood pressure, abdominal pain, loss of appetite, dizziness, and syncope.
In case of suspected overdosage, ISTURISA should be temporarily discontinued, cortisol levels should be checked, and if necessary, corticosteroid supplementation should be initiated. Close surveillance may be necessary, including monitoring of the QT interval, blood pressure, glucose, fluid, and electrolyte until the patient's condition is stable.
-
11 DESCRIPTION
ISTURISA (osilodrostat) is a cortisol synthesis inhibitor.
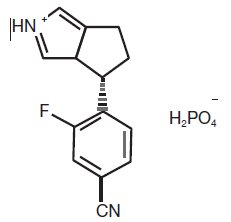
The chemical name of osilodrostat is 4-[(5R)-6,7-Dihydro-5H-pyrrolo[1,2-c]imidazol-5-yl]-3-fluorobenzonitrile dihydrogen phosphate.
Molecular formula of osilodrostat salt (phosphate) form on anhydrous basis is: (C13H11FN3) (H2PO4). Relative molecular mass of osilodrostat phosphate salt form is 325.24 g/mol.
ISTURISA tablets for oral administration contains 1 mg, 5 mg, or 10 mg of osilodrostat equivalent to 1.4 mg, 7.2 mg, and 14.3 mg of osilodrostat phosphate respectively, and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, mannitol, microcrystalline cellulose, and magnesium stearate. The film coat is composed of hypromellose, titanium dioxide, ferric oxide (yellow), ferric oxide (red) (1 mg and 10 mg only), ferrosoferric oxide (10 mg only), polyethylene glycol 4000, and talc.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Osilodrostat is a cortisol synthesis inhibitor. It inhibits 11beta-hydroxylase (CYP11B1), the enzyme responsible for the final step of cortisol biosynthesis in the adrenal gland. In a Chinese hamster lung cell line V79-4 that overexpresses human CYP11B1, adrenodoxin and adrenodoxin reductase, osilodrostat inhibited the activity of human CYP11B1 dose-dependently with IC50 values of 2.5 ± 0.1 nM (n = 4).
12.2 Pharmacodynamics
A dose dependent increase was observed in 11-deoxycortisol, the cortisol precursor, and ACTH levels in patients with Cushing's disease.
Cardiac Electrophysiology
A thorough QT study in 86 male and female healthy volunteers showed a maximum mean placebo-corrected QTcF interval increase of 1.73 ms [90% confidence interval (CI): 0.15, 3.31] at a 10 mg dose, and 25.38 ms (90% CI: 23.53, 27.22) at a 150 mg dose (up to 2.5 times the maximum recommended dosage) [see Warnings and Precautions (5.2)].
The predicted mean placebo-corrected QTcF change from baseline at the highest recommended dose in clinical practice (30 mg twice daily) was estimated as 5.3 ms (90% CI: 4.2, 6.5), based on an interpolation of the data from the thorough QT Study and population PK analysis [see Warnings and Precautions (5.2)].
12.3 Pharmacokinetics
Absorption
Osilodrostat is absorbed with a time of maximum observed concentration (Tmax) of approximately 1 hour. Exposure (AUCinf and Cmax) slightly increases over dose-proportionally within the therapeutic dose range of 1 mg to 30 mg.
Effect of Food
In a healthy volunteer study (N = 20), subjects administered with a single, 30 mg oral dose of ISTURISA film-coated tablets with a high-fat meal resulted in reduction of AUC by 11% and Cmax by 21%, respectively. The median Tmax was delayed from 1 to 2.5 hours. These changes are not considered to be clinically significant, therefore ISTURISA can be administered with or without food.
Distribution
The median apparent volume of distribution of osilodrostat is approximately 100 L. Protein binding is low (36.4%). The osilodrostat blood-to-plasma concentration ratio is 0.85.
Elimination
The elimination half-life of osilodrostat is approximately 4 hours.
In an absorption, distribution, metabolism, and excretion study, the majority of the radioactivity dose of osilodrostat is eliminated in the urine (mean: 90.6% of administered dose) with only a minor amount eliminated in the feces (1.58% of dose). The low percentage of the dose eliminated in the urine as unchanged osilodrostat (5.2%) indicates that metabolism is the major clearance pathway in humans.
Metabolism
Multiple CYP enzymes (i.e., CYP3A4, CYP2B6, and CYP2D6) and UDP-glucuronosyltransferases contribute to osilodrostat metabolism and no single enzyme contributes greater than 25% to the total clearance. The metabolites are not expected to contribute to the pharmacological effect of osilodrostat.
Specific Populations
Age and gender have no significant impact on osilodrostat exposure in adults.
Race/Ethnicity
The relative bioavailability in Asian patients is approximately 20% higher compared to that of non-Asian, along with higher Tmax and Cmax, compared to other ethnicities. However, the difference is not clinically significant.
Patients with Renal Impairment
Osilodrostat exposure was similar in the three renal function groups [normal, severe, and end stage renal disease (ESRD) groups] and thus a study was not conducted in mild and moderate renal impairment groups. The results showed that the PK of osilodrostat was not influenced by varying degrees of renal impairment to any clinically significant extent [see Dosage and Administration (2.4), Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
There was a trend of increasing AUCinf to osilodrostat in moderate and severe hepatic impaired subjects (geo-mean ratios are 1.44 and 2.66, respectively) as compared to normal subjects. Exposures (Cmax and AUC) of osilodrostat in the mild hepatic impairment group were similar to those in the normal group [see Dosage and Administration (2.5), Use in Specific Populations (8.7)].
Drug Interaction
In a healthy volunteer study (N = 20) using a single dose of osilodrostat (50 mg) and a probe drugs cocktail, osilodrostat showed inhibition potential on CYP1A2, CYP2C19, CYP2D6, and CYP3A4/5 isozymes with 2.5-, 1.9-, 1.5- and 1.5-fold increase in caffeine, omeprazole, dextromethorphan, and midazolam exposure, respectively [see Drug Interactions (7.1), (7.2)].
There was no significant impact of osilodrostat (30 mg twice daily for 12 days) on the exposure of oral contraceptives containing 0.03 mg estradiol and 0.15 mg levonorgestrel in healthy female subjects.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis
Carcinogenicity studies were conducted in Wistar Han rats and CD1 mice. Hepatocellular adenomas and carcinomas occurred in male rats at ≥ 10 mg/kg and in females at 30 mg/kg (18- and 65-times the 30 mg twice daily maximum clinical dose, by AUC, respectively). Thyroid follicular adenoma/carcinoma was also observed in male rats at 30 mg/kg. Hepatocellular adenomas and carcinomas occurred in male mice at ≥ 10 mg kg (6 times the maximum clinical dose, by AUC) but not in female mice at any dose ≤ 30 mg/kg (31 times the maximum clinical dose, by AUC). These findings are likely rodent specific and considered not relevant to humans. Genetic profiling studies support activation of hepatic constitutive androstane receptors as the likely tumorigenic mechanism in rodents and is not a significant concern for human risk at clinical exposure to osilodrostat.
Genotoxicity
Genotoxicity assays conducted in vitro in bacterial systems and in vitro and in vivo in mammalian systems with and without metabolic activation indicate that there is no genotoxic risk in humans with osilodrostat.
Impairment of Fertility
In a fertility and early embryonic-development study in Wistar Han rats, doses of 50 mg/kg (118 times the 30 mg twice daily maximum clinical dose, by AUC) resulted in changes to estrous cyclicity and impaired female fertility and embryo viability. No effect on reproductive performance in females was observed at 5 mg/kg (8 times the maximum clinical dose). Fertility and reproductive performance were not affected in male rats up to 50 mg/kg (77 times the maximum clinical dose, by AUC).
-
14 CLINICAL STUDIES
The safety and efficacy of ISTURISA was assessed in a 48-week, multicenter study (called the Core Period) that consisted of four study periods as follows:
- Period 1: 12-week, open-label, dose titration period
- Period 2: 12-week, open-label, maintenance treatment period
- Period 3: 8-week, double-blind, placebo-controlled, randomized withdrawal treatment period which provided the data for the primary efficacy endpoint
- Period 4: open-label treatment period of 14 to 24 weeks duration
The trial enrolled Cushing's disease patients with persistent or recurrent disease despite pituitary surgery or de novo patients for whom surgery was not indicated or who had refused surgery. The mean age at enrollment was 41 years; 77% of patients were female. There were 65% Caucasian, 28% Asian, 3% black, and 4% other race. Overall, 96% patients had received previous treatments for Cushing's disease prior to entering the study, of which 88% had undergone surgery. Persistence or recurrence of Cushing's disease was evidenced by the mean of three 24-hour UFC (mUFC) > 1.5x upper limit of normal (ULN). The mUFC (SD) at baseline was 1006 nmol/24 hr (1589), which corresponds to approximately 7 x ULN. The median mUFC at baseline was 476 nmol/24 hr, which corresponds to approximately 3.5 x ULN.
Period 1 (Week 1 to 12)
137 patients received a starting dose of 2 mg ISTURISA orally twice daily that could be titrated up to a maximum of 30 mg twice daily at no greater than 2-week intervals to achieve a mUFC within the normal range. Individual dose adjustments were based on mUFC. The dose was increased if mUFC was above ULN and was reduced if mUFC was below the lower limit of normal (LLN), or if the patient had symptoms consistent with hypocortisolism and mUFC was in the lower part of the normal range.
Period 2 (Week 13 to 24)
130 patients entered Period 2. The daily dose for patients that achieved a mUFC within the normal range in Period 1 was maintained during Period 2. Patients who did not require further dose increase, tolerated the drug, and had a mUFC ≤ ULN at Week 24 (end of Period 2) were to be considered responders and eligible to enter the Randomization Withdrawal phase (Period 3). Patients whose mUFC became elevated during Period 2 could have their dose increased further, if tolerated, up to 30 mg twice daily. These patients were considered non-responders and did not enter Period 3 but continued open-label treatment together with the patients who did not achieve normal mUFC at Week 12 and were followed for long-term safety and response to treatment.
Period 3 (Week 26 to 34)
At Week 26, 71 patients were considered responders and were randomized 1:1 to continue receiving ISTURISA (n = 36) or to switch to placebo (n = 35) for 8 weeks. Patients were stratified at randomization according to dose received at Week 24 (≤ 5 mg twice daily vs 5 mg twice daily) and history of pituitary irradiation (yes/no).
Patients were to remain on their assigned treatment and dose throughout Period 3 if mUFC were within the normal range. Blinded dose reduction or temporary discontinuation for safety or tolerability reasons were permitted. Dose increases were not permitted during Period 3. Patients with mUFC increase > 1.5 x ULN or who required a dose increase were considered non-responders and discontinued from Period 3 but allowed to receive open-label treatment during Period 4.
Period 4 (Week 26 or 34 to 48)
This period included patients who were not eligible for randomization (n = 47) at Week 26, patients who were considered non-responders during Period 3 (n = 29), and patients who were considered responders during Period 3 (n = 41). Open-label treatment with ISTURISA continued in these patients until Week 48 when patients who maintained clinical benefit on ISTURISA, as judged by the Investigator, had an option to enter an extension period.
Efficacy Assessment
The primary efficacy endpoint of the study was to compare the percentage of complete responders at the end of the 8-week randomized withdrawal period (Period 3) between patients randomized to continue ISTURISA versus the patients switched to placebo. A complete responder for the primary endpoint was defined as a patient who had mUFC ≤ ULN based on central laboratory result at the end of Period 3 (Week 34), and who neither discontinued randomized treatment or the study nor had any dose increase above their Week 26 dose.
The key secondary endpoint was to assess the complete responder rate at the end of Period 2 (Week 24). A complete responder for the key secondary endpoint was defined as a patient with mUFC ≤ ULN at Week 24 who did not require an increase in dose above the level established at the end of Period 1 (Week 12). Patients who were missing mUFC assessment at Week 24 were counted as non-responders for the key secondary endpoint.
Results
At the end of Period 3, the percentage of complete responders for the primary endpoint was 86% and 29% in the ISTURISA and placebo groups, respectively (Table 3). The difference in percentage of complete responders between ISTURISA and placebo groups was 57%, with 95% two-sided CI of (38, 76). The 95% CI were not presented by individual strata due to the small sample sizes of some of these strata.
Table 3: Percentage of Cushing's Disease Patients with Normal mUFC at End of Period 3 (8-week randomized withdrawal period) Primary Endpoint
ISTURISA
(N = 36)
n (%)Placebo
(N = 34)
n (%)Complete Responder Rate
Difference
(Differences in Percentages)
Complete responder rate at the end of the 8-week randomized withdrawal period (Week 34)
31 (86)
10 (29)
osilodrostat vs placebo
57 (38, 76)(95% CI)
(71, 95)
(15, 47)
2-sided p-value < 0.001
Abbreviation: CI, Confidence Interval.
The key secondary endpoint, complete responder rate after 24 weeks of treatment with ISTURISA was achieved by 72/137 patients (52.6%) with 95% two-sided CI of (43.9, 61.1). The lower bound of this 95% CI exceeded 30%, the pre-specified threshold for statistical significance and minimum threshold for clinical benefit.
At Week 48, 91/137 patients (66%) had normal mUFC levels.
Variable decreases from baseline for blood pressure, glucose parameters, weight and weight circumference were observed at Week 48. However, because the study allowed initiation of anti-hypertensive and anti-diabetic medications and dose increases in patients already receiving such medications and the absence of a control group, the individual contribution of ISTURISA or of anti-hypertensive and anti-diabetic medication adjustments cannot be clearly established.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ISTURISA (osilodrostat) tablets are supplied as follows:
Tablet Strength
Description
Package Configuration
NDC No.
1 mg
Pale yellow, unscored, round, biconvex with beveled edge tablet, debossed "1" on one side.
Each carton contains 3 blister packs. Each blister pack contains 20 tablets.
55292-320-60
5 mg
Yellow, unscored, round, biconvex with beveled edge tablet, debossed "5" on one side.
55292-321-60
10 mg
Pale orange brown, unscored, round, biconvex with beveled edge tablet, debossed "10" on one side.
55292-322-60
Storage and Handling
Store at room temperature between 68°F to 77°F (20°C to 25°C); excursions permitted 15°C to 30°C (59°F to 86°F); protect from moisture.
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Monitoring
Instruct patients on the importance of laboratory monitoring and adhering to their return visit schedule [see Dosage and Administration (2.2, 2.3)].
Hypocortisolism
Advise patients that ISTURISA is associated with hypocortisolism-related events. Advise patients to report symptoms of hypercortisolism to their healthcare provider [see Warnings and Precautions (5.1)].
QT Prolongation
Advise patients of the signs and symptoms of QT prolongation. Advise patients to contact their healthcare provider immediately for signs or symptoms of QT prolongation.
Advise patients that an ECG will be taken before treatment and periodically thereafter. Advise patients with cardiac disease and risk factors for QT prolongation that adjustments in cardiac medications may be made and electrolyte disturbances may require correction [see Warnings and Precautions (5.2)].
Adrenal Hormone Precursors/Androgens
Advise patients that elevation of adrenal hormone precursors may occur and lead to low potassium levels, worsening of hypertension, and edema. Advise patients to report the occurrence of these symptoms to their healthcare provider.
Advise patients that elevations of androgens may occur and may lead to hirsutism, hypertrichosis, and acne (in females). Advise patients to report the occurrence of these symptoms to their healthcare provider [see Warnings and Precautions (5.3)].
Lactation
Advise females not to breastfeed during treatment with ISTURISA and for at least one week after treatment [see Use in Specific Populations (8.2)].

Manufactured for:
Recordati Rare Diseases Inc. Bridgewater, NJ 08807 U.S.A.
© Recordati -
PATIENT PACKAGE INSERT
PATIENT INFORMATION
ISTURISA® (is tur ee' sah)
(osilodrostat) TabletsWhat is the most important information I should know about ISTURISA?
ISTURISA can cause serious side effects, including:
- Low cortisol levels in your blood (hypocortisolism).
Tell your healthcare provider right away if you experience more than one of the following symptoms, as these may be symptoms of very low cortisol level, known as adrenal insufficiency:
- nausea
- vomiting
- tiredness (fatigue)
- low blood pressure
- stomach (abdominal) pain
- loss of appetite
- dizziness
If you get symptoms of hypocortisolism while taking ISTURISA, your healthcare provider may change your dose or ask you to stop taking it.
Heart problem or a heart rhythm problem, such as an irregular heartbeat which could be a sign of a heart problem called QT prolongation. Call your healthcare provider right away if you have irregular heartbeats.
See "What are the possible side effects of ISTURISA?" for more information about side effects.
What is ISTURISA?
ISTURISA is a prescription medicine that is used to treat adults with Cushing's disease:
- who cannot have pituitary surgery, or
- who have had pituitary surgery, but the surgery did not cure their Cushing's disease
It is not known if ISTURISA is safe and effective in children.
Before you take ISTURISA, tell your healthcare provider about all of your medical conditions, including if you:
- have or had heart problems, such as an irregular heartbeat, including a condition called prolonged QT syndrome (QT internal prolongation). Your healthcare provider will check the electrical signal of your heart (called an electrocardiogram) before you start taking ISTURISA, 1 week after starting ISTURISA, and as needed after that.
- have a history of low levels of potassium or magnesium in your blood.
- have liver problems.
- are breastfeeding or plan to breastfeed. It is not known if ISTURISA passes into your breast milk.
You should not breastfeed if you take ISTURISA and for 1 week after stopping treatment.
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements.
Especially tell your healthcare provider if you take medicines used to treat certain heart problems. Ask your healthcare provider if you are not sure whether your medicine is used to treat heart problems.
How should I take ISTURISA?
- Take ISTURISA exactly as your healthcare provider tells you. Your healthcare provider will tell you exactly how many tablets of ISTURISA to take. Do not change your dose or stop taking ISTURISA unless your healthcare provider tells you to.
- Start ISTURISA by taking 2 mg two times a day by mouth or as directed by your healthcare provider.
- After you have started treatment, your healthcare provider may change your dose, depending on how you respond to the treatment with ISTURISA.
- Take ISTURISA with or without food.
- If you miss a dose of ISTURISA, take the next dose at your regularly scheduled time.
What are the possible side effects of ISTURISA?
ISTURISA may cause serious side effects, including:
- See "What is the most important information I should know about ISTURISA?"
-
Increase in other adrenal hormone levels. Your other adrenal hormones may increase when you take ISTURISA. Your healthcare provider may monitor you for the symptoms associated with these hormonal changes while you are taking ISTURISA:
- Low potassium (hypokalemia).
- High blood pressure (hypertension).
- Swelling (edema) in the legs, ankles or other signs of fluid retention.
- Excessive facial or body hair growth (hirsutism).
- Acne (in women).
Call your healthcare provider if you have any of these side effects.
The most common side effects of ISTURISA include:
- very low cortisol levels (adrenal insufficiency)
- tiredness (fatigue)
- nausea
- headache
- swelling of the legs, ankles or other signs of fluid retention (edema)
These are not all of the possible side effects of ISTURISA.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ISTURISA?
- Store ISTURISA at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep ISTURISA dry.
Keep ISTURISA and all medicines out of the reach of children.
General information about the safe and effective use of ISTURISA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ISTURISA for a condition for which it was not prescribed. Do not give ISTURISA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about ISTURISA that is written for healthcare professionals.
What are the ingredients in ISTURISA?
Active ingredient: osilodrostat phosphate
Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, mannitol, microcrystalline cellulose, and magnesium stearate. Tablet film-coating: hypromellose, titanium dioxide, ferric oxide (yellow), ferric oxide (red) (1 mg and 10 mg only) ferrosoferric oxide (10 mg only), polyethylene glycol 4000, and talc.
For more information, go to www.ISTURISA.com or call 1-888-575-8344.
Manufactured for: Recordati Rare Diseases Inc., Bridgewater, NJ 08807 USA
© Recordati
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: 11/2023
- PRINCIPAL DISPLAY PANEL - 1 mg Blister Pack Carton
- PRINCIPAL DISPLAY PANEL - 5 mg Blister Pack Carton
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ISTURISA
osilodrostat tablet, coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55292-320 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OSILODROSTAT PHOSPHATE (UNII: Y6581YAW9V) (OSILODROSTAT - UNII:5YL4IQ1078) OSILODROSTAT 1 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) POLYETHYLENE GLYCOL 4000 (UNII: 4R4HFI6D95) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color YELLOW (PALE YELLOW) Score no score Shape ROUND (ROUND, BICONVEX, BEVELED EDGE) Size 6mm Flavor Imprint Code Y1;NVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55292-320-60 60 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/31/2020 2 NDC:55292-320-20 20 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/31/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212801 03/31/2020 ISTURISA
osilodrostat tablet, coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55292-321 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OSILODROSTAT PHOSPHATE (UNII: Y6581YAW9V) (OSILODROSTAT - UNII:5YL4IQ1078) OSILODROSTAT 5 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) POLYETHYLENE GLYCOL 4000 (UNII: 4R4HFI6D95) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color YELLOW Score no score Shape ROUND (ROUND, BICONVEX, BEVELED EDGE) Size 7mm Flavor Imprint Code Y2;NVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55292-321-60 60 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/31/2020 2 NDC:55292-321-20 20 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/31/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212801 03/31/2020 ISTURISA
osilodrostat tablet, coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55292-322 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OSILODROSTAT PHOSPHATE (UNII: Y6581YAW9V) (OSILODROSTAT - UNII:5YL4IQ1078) OSILODROSTAT 10 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POLYETHYLENE GLYCOL 4000 (UNII: 4R4HFI6D95) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color BROWN (PALE ORANGE BROWN) Score no score Shape ROUND (ROUND, BICONVEX, BEVELED EDGE) Size 9mm Flavor Imprint Code Y3;NVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55292-322-60 60 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/31/2020 04/03/2023 2 NDC:55292-322-20 20 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/31/2020 04/03/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212801 03/31/2020 04/03/2023 Labeler - Recordati Rare Diseases, Inc. (181699406) Establishment Name Address ID/FEI Business Operations Novartis Pharma Stein AG 488152505 ANALYSIS(55292-320, 55292-321, 55292-322) , MANUFACTURE(55292-320, 55292-321, 55292-322) , API MANUFACTURE(55292-320, 55292-321, 55292-322) , PACK(55292-320, 55292-321, 55292-322) Establishment Name Address ID/FEI Business Operations PENN PHARMACEUTICAL SERVICES LIMITED 226277259 ANALYSIS(55292-320, 55292-321, 55292-322) , MANUFACTURE(55292-320, 55292-321, 55292-322) , PACK(55292-320, 55292-321, 55292-322)