Label: TACLONEX- calcipotriene and betamethasone dipropionate ointment

- NDC Code(s): 50222-227-04, 50222-227-81
- Packager: LEO Pharma Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated May 24, 2022
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TACLONEX® Ointment safely and effectively. See Full Prescribing Information for TACLONEX® Ointment.
TACLONEX® (calcipotriene and betamethasone dipropionate ointment), for topical use
Initial U.S. Approval: 2006INDICATIONS AND USAGE
Taclonex® Ointment is a vitamin D analogue and corticosteroid combination product indicated for the topical treatment of plaque psoriasis in patients 12 years of age and older. (1)
DOSAGE AND ADMINISTRATION
- Apply Taclonex® Ointment to affected area(s) once daily for up to 4 weeks. Discontinue therapy when control is achieved. (2)
- Adult patients should not use more than 100 g per week. (2)
- Patients ages 12 to 17 years should not use more than 60 g per week. (2)
- Treatment of more than 30% body surface area is not recommended. (2)
- Do not use with occlusive dressings unless directed by a physician. (2)
- Avoid use on the face, groin, or axillae, or if skin atrophy is present at the treatment site. (2)
- Not for oral, ophthalmic, or intravaginal use. (2)
DOSAGE FORMS AND STRENGTHS
Ointment, 0.005%/0.064% (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Hypercalcemia and hypercalciuria have been observed. If either occurs, discontinue treatment until parameters of calcium metabolism normalize. (5.1)
- Taclonex® Ointment can produce reversible hypothalamic-pituitary-adrenal (HPA) axis suppression with the potential for glucocorticosteroid insufficiency during and after withdrawal of treatment. Risk factors include the use of high-potency topical corticosteroid, use over a large surface area or to areas under occlusion, prolonged use, concomitant use of more than one corticosteroid-containing product, altered skin barrier, liver failure, and use in pediatric patients. Modify use should HPA axis suppression develop. (5.2, 8.4)
- Taclonex® Ointment may increase the risk of cataract and glaucoma. If visual symptoms occur, consider referral to an ophthalmologist. (5.3)
ADVERSE REACTIONS
The most common adverse reactions (≥1%) are pruritus and scaly rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact LEO Pharma Inc. at 1-877-494-4536 or FDA at 1-800-FDA-1088 or
www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypercalcemia and Hypercalciuria
5.2 Effects on Endocrine System
5.3 Ophthalmic Adverse Reactions
5.4 Allergic Contact Dermatitis with Topical Corticosteroids
5.5 Allergic Contact Dermatitis with Topical Calcipotriene
5.6 Skin Irritation
5.7 Risk of Ultraviolet Light Exposure
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
16.3 Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
Apply an adequate layer of Taclonex® Ointment to the affected area(s) once daily for up to 4 weeks. Taclonex® Ointment should be rubbed in gently and completely. Patients should wash their hands after applying Taclonex® Ointment. Therapy should be discontinued when control is achieved.
Patients 18 years and older should not use more than 100 g per week and patients 12 to 17 years should not use more than 60 g per week. Treatment of more than 30% body surface area is not recommended.
Taclonex® Ointment should not be used with occlusive dressings unless directed by a physician. Avoid use on the face, groin, or axillae, or if skin atrophy is present at the treatment site. Taclonex® Ointment is not for oral, ophthalmic, or intravaginal use.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypercalcemia and Hypercalciuria
Hypercalcemia and hypercalciuria have been observed with use of Taclonex® Ointment. If hypercalcemia or hypercalciuria develops, treatment should be discontinued until parameters of calcium metabolism have normalized. In the trials that included assessment of the effects of Taclonex® Ointment on calcium metabolism, such testing was done after 4 weeks of treatment. The effects of Taclonex® Ointment on calcium metabolism following treatment durations of longer than 4 weeks have not been evaluated [see Clinical Pharmacology (12.2)].
5.2 Effects on Endocrine System
Taclonex® Ointment can cause reversible hypothalamic-pituitary-adrenal (HPA) axis suppression with the potential for clinical glucocorticosteroid insufficiency. This may occur during treatment or upon withdrawal of treatment. Factors that predispose a patient to HPA axis suppression include the use of high-potency corticosteroids, large treatment surface areas, prolonged use, concomitant use of more than one corticosteroid-containing product, use of occlusive dressings, altered skin barrier, liver failure, and young age. Evaluation for HPA axis suppression may be done by using the cosyntropin stimulation test [see Clinical Pharmacology (12.2)].
In a trial evaluating the effects of Taclonex® Topical Suspension and Taclonex® Ointment on the HPA axis, 32 adult subjects were treated with Taclonex® Topical Suspension on the scalp and Taclonex® Ointment on the body. Adrenal suppression was identified in 5 of 32 subjects (15.6%) after 4 weeks of treatment [see Clinical Pharmacology (12.2)]. The effects of Taclonex® Ointment on the HPA axis following treatment durations of longer than 4 weeks have not been adequately studied.
If HPA axis suppression is documented, gradually withdraw the drug, reduce the frequency of application, or substitute with a less potent corticosteroid.
Cushing's syndrome and hyperglycemia may also occur due to the systemic effects of topical corticosteroids. These complications are rare and generally occur after prolonged exposure to excessively large doses, especially of high-potency topical corticosteroids.
Pediatric patients may be more susceptible to systemic toxicity due to their higher skin surface to body mass ratios [see Use in Specific Populations (8.4), Clinical Pharmacology (12.2)].
Use of more than one corticosteroid-containing product at the same time may increase the total systemic corticosteroid exposure.
5.3 Ophthalmic Adverse Reactions
Use of topical corticosteroids, including Taclonex® Ointment, may increase the risks of glaucoma and posterior subcapsular cataract. Glaucoma and cataracts have been reported in postmarketing experience with the use of topical corticosteroid products, including topical clobetasol products.
Avoid contact of Taclonex® Ointment with eyes. Advise patients to report any visual symptoms and consider referral to an ophthalmologist for evaluation.
5.4 Allergic Contact Dermatitis with Topical Corticosteroids
Allergic contact dermatitis to any component of topical corticosteroids is usually diagnosed by a failure to heal rather than a clinical exacerbation. Clinical diagnosis of allergic contact dermatitis can be confirmed by patch testing.
5.5 Allergic Contact Dermatitis with Topical Calcipotriene
Allergic contact dermatitis has been observed with use of topical calcipotriene. Clinical diagnosis of allergic contact dermatitis can be confirmed by patch testing.
5.6 Skin Irritation
If irritation develops, treatment with Taclonex® Ointment should be discontinued and appropriate therapy instituted.
5.7 Risk of Ultraviolet Light Exposure
Patients who apply Taclonex® Ointment to exposed skin should avoid excessive exposure to either natural or artificial sunlight, including tanning booths, sun lamps, etc. Physicians may wish to limit or avoid use of phototherapy in patients who use Taclonex® Ointment.
-
6 ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience
Clinical Trials Conducted in Subjects 18 years and older with Plaque Psoriasis
The data described below reflect exposure to Taclonex® Ointment in 2448 subjects with plaque psoriasis, including 1992 exposed for 4 weeks, and 289 exposed for 8 weeks.
Taclonex® Ointment was studied primarily in placebo- and active-controlled trials (N = 1176, and N = 1272, respectively). The population was 15-97 years old, 61% males and 39% females, mostly white (97%) and had a baseline disease severity ranging from mild to very severe. Most subjects received once daily application, and the median weekly dose was 24.5 g.
The percentage of subjects reporting at least one adverse event was 27.1% in the Taclonex® Ointment group, 33.0% in the calcipotriene group, 28.3% in the betamethasone group, and 33.4% in the vehicle group.
Table 1
Adverse Events Reported by ≥1% of Subjects by Preferred Term
Taclonex® Ointment Calcipotriene Betamethasone dipropionate Vehicle N = 2448 N = 3197 N = 1164 N = 470 Any Adverse Event 663 (27.1) 1055 (33.0) 329 (28.3) 157 (33.4) Preferred Term # of subjects (%) Pruritus 75 (3.1) 183 (5.7) 38 (3.3) 43 (9.1) Headache 69 (2.8) 75 (2.3) 44 (3.8) 12 (2.6) Nasopharyngitis 56 (2.3) 77 (2.4) 34 (2.9) 9 (1.9) Psoriasis 30 (1.2) 47 (1.5) 14 (1.2) 5 (1.1) Rash scaly 30 (1.2) 40 (1.3) 0 (0.0) 1 (0.2) Influenza 23 (0.9) 34 (1.1) 14 (1.2) 6 (1.3) Upper respiratory tract infection 20 (0.8) 19 (0.6) 12 (1.0) 3 (0.6) Erythema 15 (0.6) 54 (1.7) 3 (0.3) 5 (1.1) Application site pruritus 13 (0.5) 24 (0.8) 10 (0.9) 6 (1.3) Skin irritation 11 (0.4) 60 (1.9) 8 (0.7) 5 (1.1) Pain 7 (0.3) 12 (0.4) 3 (0.3) 5 (1.1) Burning sensation 6 (0.2) 30 (0.9) 3 (0.3) 6 (1.3)
A lesional/perilesional adverse event was generally defined as an adverse event located ≤ 2 cm from the lesional border.
Table 2
Lesional/Perilesional Adverse Events Reported by ≥1% of SubjectsTaclonex® Ointment Calcipotriene Betamethasone dipropionate Vehicle N = 2448 N = 3197 N = 1164 N = 470 Any Adverse Event 213 (8.7) 419 (13.1) 85 (7.3) 76 (16.2) Preferred Term # of subjects (%) Pruritus 69 (2.8) 170 (5.3) 31 (2.7) 41 (8.7) Rash scaly 29 (1.2) 38 (1.2) 0 (0.0) 0 (0.0) Application site pruritus 12 (0.5) 24 (0.8) 10 (0.9) 6 (1.3) Erythema 9 (0.4) 36 (1.1) 2 (0.2) 4 (0.9) Skin irritation 9 (0.4) 51 (1.6) 8 (0.7) 5 (1.1) Burning sensation 6 (0.2) 25 (0.8) 3 (0.3) 5 (1.1)
For subjects who reported lesional/perilesional adverse events, the median time to onset was 7 days for Taclonex® Ointment, 7 days for calcipotriene, 5 days for betamethasone dipropionate, and 3 days for vehicle.
Other less common reactions (less than 1% but more than 0.1%) were, in decreasing order of incidence, folliculitis, rash papular, rash pustular, and skin hypopigmentation. Skin atrophy, telangiectasia and skin hyperpigmentation were reported infrequently (0.1%).
In a separate trial, subjects (N = 207) with at least moderate disease severity were given Taclonex® Ointment intermittently on an "as needed" basis for up to 52 weeks. The median use was 15.4 g per week. The effects of Taclonex® Ointment on calcium metabolism were not studied and the effects on the HPA axis were not adequately studied. The following adverse reactions were reported by 1% or more of the subjects: pruritus (7.2%), psoriasis (3.4%), skin atrophy (1.9%), folliculitis (1.4%), burning sensation (1.4%), skin depigmentation (1.4%), ecchymosis (1.0%), erythema (1.0%) and hand dermatitis (1.0%). One case of serious flare-up of psoriasis was reported.6.2 Postmarketing Experience
The following adverse reactions associated with the use of Taclonex® Ointment have been identified post-approval: pustular psoriasis and rebound effect.
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Postmarketing reports for local adverse reactions to topical corticosteroids may also include: striae, dryness, acneiform eruptions, perioral dermatitis, secondary infection and miliaria.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Taclonex® Ointment contains calcipotriene and betamethasone dipropionate. The limited data with Taclonex® Ointment and calcipotriene use in pregnant women are not sufficient to evaluate a Taclonex® Ointment-associated or calcipotriene-associated risk for major birth defects, miscarriages, or adverse maternal or fetal outcomes.
Observational studies suggest an increased risk of having low birthweight infants with the maternal use of potent or very potent topical corticosteroids (see Data). Advise pregnant women that Taclonex® Ointment may increase the potential risk of having a low birth weight infant and to use Taclonex® Ointment on the smallest area of skin and for the shortest duration possible.
In animal reproduction studies, oral administration of calcipotriene to pregnant rats during the period of organogenesis resulted in an increased incidence of minor skeletal abnormalities, including enlarged fontanelles and extra ribs (see Data). Oral administration of calcipotriene to pregnant rabbits during the period of organogenesis had no apparent effects on embryo-fetal development. Subcutaneous administration of betamethasone dipropionate to pregnant rats and rabbits during the period of organogenesis resulted in fetal toxicity, including fetal deaths, reduced fetal weight, and fetal malformations (cleft palate and crooked or short tail) (see Data). The available data do not allow the calculation of relevant comparisons between the systemic exposures of calcipotriene and betamethasone dipropionate observed in animal studies to the systemic exposures that would be expected in humans after topical use of Taclonex® Ointment.
The estimated background risk of major birth defects and miscarriage of the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Human Data
Available observational studies in pregnant women did not identify a drug-associated risk of major birth defects, preterm delivery, or fetal mortality with the use of topical corticosteroids of any potency. However, when the dispensed amount of potent or very potent topical corticosteroids exceeded 300 g during the entire pregnancy, maternal use was associated with an increased risk of low birth weight in infants.
Animal Data
Embryo-fetal development studies with calcipotriene were performed by the oral route in rats and rabbits. Pregnant rats received dosages of 0, 6, 18, or 54 mcg/kg/day (0, 36, 108, and 324 mcg/m2/day, respectively) on days 6-15 of gestation (the period of organogenesis). There were no apparent effects on maternal survival, behavior, or body weight gain, no effects on litter parameters, and no effects on the incidence of major malformations in fetuses. Fetuses from dams dosed at 54 mcg/kg/day exhibited a significantly increased incidence of minor skeletal abnormalities, including enlarged fontanelles and extra ribs.
Pregnant rabbits were dosed daily with calcipotriene at exposures of 0, 4, 12, or 36 mcg/kg/day (0, 48, 144, and 432 mcg/m2/day, respectively) on days 6-18 of gestation (the period of organogenesis). Mean maternal body weight gain was reduced in animals dosed at 12 or 36 mcg/kg/day. The incidence of fetal deaths was increased in the group dosed at 36 mcg/kg/day; reduced fetal weight was also observed in this group. The incidence of major malformations among fetuses was not affected. An increase in the incidence of minor skeletal abnormalities, including incomplete ossification of sternebrae, pubic bones, and forelimb phalanges, was observed in the group dosed at 36 mcg/kg/day.
Embryo-fetal development studies with betamethasone dipropionate were performed via subcutaneous injection in mice and rabbits. Pregnant mice were administered doses of 0, 156, 625, or 2500 mcg/kg/day (0, 468, 1875, and 7500 mcg/m2/day, respectively) on days 7 through 13 of gestation (the period of organogenesis). Betamethasone dipropionate induced fetal toxicity, including fetal deaths, reduced fetal weight, malformations (increased incidence of the cleft palate and crooked or short tail), and minor skeletal abnormalities (delayed ossification of vertebra and sternebrae). Fetal toxicity was observed at the lowest exposure that was evaluated (156 mcg/kg/day).
Pregnant rabbits were injected subcutaneously at dosages of 0, 0.625, 2.5, and 10 mcg/kg/day (0, 7.5, 30, and 120 mcg/m2/day, respectively) on days 6 through 18 of gestation (the period of organogenesis). Betamethasone dipropionate induced fetal toxicity, including fetal deaths, reduced fetal weight, external malformations (including malformed ears, cleft palate, umbilical hernia, kinked tail, club foot, and club hand), and skeletal malformations (including absence of phalanges of the first digit and cranial dysplasia) at dosages of 2.5 mcg/kg/day and above.
Calcipotriene was evaluated for effects on peri- and post-natal development when orally administered to pregnant rats at dosages of 0, 6, 18 or 54 mcg/kg/day (0, 36, 108, and 324 mcg/m2/day, respectively) from gestation day 15 through day 20 postpartum. No remarkable effects were observed on any parameter, including survival, behavior, body weight, litter parameters, or the ability to nurse or rear pups.
Betamethasone dipropionate was evaluated for effects on peri- and post-natal development when orally administered to pregnant rats at dosages of 0, 100, 300, and 1000 mcg/kg/day (0, 600, 1800, and 6000 mcg/m2/day, respectively) from gestation day 6 through day 20 postpartum. Mean maternal body weight was significantly reduced on gestation day 20 in animals dosed at 300 and 1000 mcg/kg/day. The mean duration of gestation was slightly, but statistically significantly, increased at 100, 300, and 1000 mcg/kg/day. The mean percentage of pups that survived to day 4 was reduced in relation to dosage. On lactation day 5, the percentage of pups with a reflex to right themselves when placed on their back was significantly reduced at 1000 mcg/ kg/day. No effects on the ability of pups to learn were observed, and the ability of the offspring of treated rats to reproduce was not affected.
8.2 Lactation
Risk Summary
There is no information regarding the presence of topically administered calcipotriene and betamethasone dipropionate in human milk, the effects on the breastfed infant, or the effects on milk production. It is not known whether topically administered calcipotriene or corticosteroids could result in sufficient systemic absorption to produce detectable quantities in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for Taclonex® Ointment and any potential adverse effects on the breastfed child from Taclonex® Ointment or from the underlying maternal condition.
Clinical Considerations
To minimize potential exposure to the breastfed infant via breast milk, use Taclonex® Ointment on the smallest area of skin and for the shortest duration possible while breastfeeding. Advise breastfeeding women not to apply Taclonex® Ointment directly to the nipple and areola to avoid direct infant exposure [see Use in Specific Populations (8.4)].
8.4 Pediatric Use
Safety and effectiveness of the use of Taclonex® Ointment in pediatric patients under the age of 12 years have not been established.
The safety and effectiveness of Taclonex® Ointment for the treatment of plaque psoriasis have been established in the age group 12 to 17 years. In a prospective, uncontrolled trial, 33 pediatric subjects ages 12-17 years with plaque psoriasis on the body were treated with Taclonex® Ointment for 4 weeks up to a maximum of 55.8 g per week. Subjects were assessed for HPA axis suppression and effects on calcium metabolism. No adverse effects on adrenal suppression were observed. No hypercalcemia was observed but one subject had a possible treatment-related increase in urinary calcium [see Clinical Pharmacology (12.2)].
Because of a higher ratio of skin surface area to body mass, pediatric patients are at a greater risk than adults of systemic toxicity when treated with topical drugs. They are, therefore, also at greater risk of HPA axis suppression and adrenal insufficiency upon the use of topical corticosteroids [see Warnings and Precautions (5.2)].
Rare systemic toxicities such as Cushing's syndrome, linear growth retardation, delayed weight gain, and intracranial hypertension have been reported in pediatric patients, especially those with prolonged exposure to large doses of high potency topical corticosteroids.
Local adverse reactions including striae have also been reported with use of topical corticosteroids in pediatric patients.
8.5 Geriatric Use
Of the total number of subjects in the clinical studies of Taclonex® Ointment, approximately 14% were 65 years and older and approximately 3% were 75 years and over.
No overall differences in safety or effectiveness of Taclonex® Ointment were observed between these subjects and younger subjects. All other reported clinical experience has not identified any differences in response between elderly and younger patients. However, greater sensitivity of some older individuals cannot be ruled out.
- 10 OVERDOSAGE
-
11 DESCRIPTION
Taclonex® (calcipotriene and betamethasone dipropionate) Ointment, 0.005%/0.064% contains calcipotriene hydrate and betamethasone dipropionate. It is intended for topical use only.
Calcipotriene hydrate is a synthetic vitamin D3 analogue.
Chemically, calcipotriene hydrate is (5Z,7E,22E,24S)-24-cyclopropyl-9,10-secochola-5,7,10(19),22-tetraene-1(alpha),3(beta),24-triol,hydrate, with the empirical formula C27H40O3,H2O, a molecular weight of 430.6, and the following structural formula:
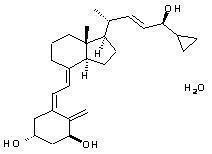
Calcipotriene hydrate is a white to almost white crystalline compound.Betamethasone dipropionate is a synthetic corticosteroid.
Betamethasone dipropionate has the chemical name 9-fluoro-11(beta), 17,21-trihydroxy-16(beta)-methylpregna-1,4-diene-3,20-dione17,21-dipropionate, with the empirical formula C28H37FO7, a molecular weight of 504.6, and the following structural formula:
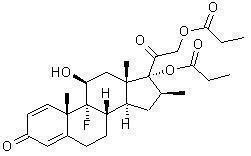
Betamethasone dipropionate is a white to almost white odorless powder.
Each gram of Taclonex® Ointment contains 52.18 mcg of calcipotriene hydrate (equivalent to 50 mcg of calcipotriene) and 0.643 mg of betamethasone dipropionate (equivalent to 0.5 mg of betamethasone) in an off-white to yellow paraffin ointment base of butylhydroxytoluene, mineral oil, polyoxypropylene stearyl ether, all-rac-alpha-tocopherol, and white petrolatum. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Taclonex® Ointment combines the pharmacological effects of calcipotriene hydrate as a synthetic vitamin D3 analogue and betamethasone dipropionate as a synthetic corticosteroid. However, while their pharmacologic and clinical effects are known, the exact mechanisms of their actions in plaque psoriasis are unknown.
12.2 Pharmacodynamics
Vasoconstriction:
In a vasoconstrictor trial in healthy subjects, the skin blanching response of Taclonex® Ointment was consistent with that of a potent corticosteroid when compared with other topical corticosteroids. However, similar blanching scores do not necessarily imply therapeutic equivalence.
Hypothalamic-Pituitary-Adrenal (HPA) Axis Suppression:HPA axis suppression was evaluated in four trials (Trial A, B, C and D) following the application of Taclonex® Ointment.
In Trial A, Taclonex® Ointment was applied once daily for 4 weeks to adult subjects (N = 12) with plaque psoriasis to study its effects on the hypothalamic-pituitary-adrenal (HPA) axis. Of eleven subjects tested, none demonstrated adrenal suppression as indicated by a 30-minute post-stimulation cortisol level ≤ 18 mcg/dL.
In Trial B, Taclonex® Ointment was evaluated in adult subjects with plaque psoriasis (N = 19). One subject demonstrated adrenal suppression.
In Trial C, HPA axis suppression was evaluated in adult subjects (N=32) with extensive plaque psoriasis involving at least 30% of the scalp and, in total, 15-30% of the body surface area. Treatment consisted of once daily application of Taclonex Scalp® Topical Suspension on the scalp in combination with Taclonex® Ointment on the body. Adrenal suppression as indicated by a 30-minutes post-stimulation cortisol level < 18 mcg/dL was observed in 5 of 32 subjects (15.6%) after 4 weeks of treatment as per the recommended duration of use [see Dosage and Administration (2)].
In Trial D, HPA axis suppression was evaluated in subjects 12 to 17 years (N=32) with plaque psoriasis of the body involving 5-30% of the body surface area. Treatment consisted of once daily application of Taclonex® Ointment to the affected areas for up to 4 weeks. Mean weekly dose was 29.6 g with a range of 8.1-55.8 g/week. Adrenal suppression as indicated by a 30-minute post-stimulation cortisol level ≤18 mcg/dL was observed in none of 32 evaluable subjects after 4 weeks of treatment [see Use in Specific Populations (8.4)].Effects on Calcium Metabolism
In Trial C described above, the effects of once daily application of Taclonex® Ointment on the body in combination with Taclonex Scalp® Topical Suspension on the scalp on calcium metabolism were also examined. Elevated urinary calcium levels outside the normal range were observed in 1 of 35 subjects (2.9%) after 4 weeks of treatment.
In Trial D described above, calcium metabolism was evaluated in a total of 33 subjects aged 12 to 17 years with plaque psoriasis involving 5-30% of the body surface area who underwent once daily application of Taclonex® Ointment for up to 4 weeks. No cases of hypercalcemia and no clinically relevant changes in urinary calcium were reported. However, one subject had a normal urinary calcium:creatinine ratio at baseline (3.75 mmol/g), which increased above the normal range at week 4 (16 mmol/g). There were no relevant changes in albumin-corrected serum calcium or other markers of calcium metabolism for this subject. The clinical significance of this finding is unknown.12.3 Pharmacokinetics
Absorption
In Trial C described above, the systemic effect of Taclonex® Ointment in extensive plaque psoriasis was investigated. In this trial, the serum levels of calcipotriene and betamethasone dipropionate and their major metabolites were measured after 4 weeks (maximum recommended duration of treatment) and also after 8 weeks of once daily application of Taclonex® Ointment on the body in combination with Taclonex Scalp® Topical Suspension on the scalp. Both calcipotriene and betamethasone dipropionate were below the lower limit of quantification in all serum samples of the 34 subjects evaluated. However, one major metabolite of calcipotriene (MC1080) was quantifiable in 10 of 34 (29.4%) subjects at week 4 and in five of 12 (41.7%) subjects at week 8. The major metabolite of betamethasone dipropionate, betamethasone 17-propionate (B17P) was also quantifiable in 19 of 34 (55.9%) subjects at week 4 and seven of 12 (58.3%) subjects at week 8. The serum concentrations for MC1080 ranged from 20-75 pg/mL. The clinical significance of this finding is unknown.
Metabolism
Calcipotriene:
Calcipotriene metabolism following systemic uptake is rapid and occurs in the liver. The primary metabolites of calcipotriene are less potent than the parent compound.Calcipotriene is metabolized to MC1046 (the alpha,beta-unsaturated ketone analogue of calcipotriene), which is metabolized further to MC1080 (a saturated ketone analogue). MC1080 is the major metabolite in plasma. MC1080 is slowly metabolized to calcitroic acid.
Betamethasone dipropionate:
Betamethasone dipropionate is metabolized to betamethasone 17-propionate and betamethasone, including the 6beta-hydroxy derivatives of those compounds by hydrolysis. Betamethasone 17-propionate (B17P) is the primary metabolite. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
When calcipotriene was applied topically to mice for up to 24 months at dosages of 3, 10 and 30 mcg/kg/day (9, 30 and 90 mcg/m2/day, respectively), no significant changes in tumor incidence were observed when compared to control.
A 104-week oral carcinogenicity study was conducted with calcipotriene in male and female rats at doses of 1, 5 and 15 mcg/kg/day (6, 30, and 90 mcg/m2/day, respectively). Beginning week 71, the dosage for high-dose animals of both genders was reduced to 10 mcg/kg/day (60 mcg/m2/day). A treatment-related increase in benign C-cell adenomas was observed in the thyroid of females that received 15 mcg/kg/day. A treatment-related increase in benign pheochromocytomas was observed in the adrenal glands of males that received 15 mcg/kg/day. No other statistically significant differences in tumor incidence were observed when compared to control. The relevance of these findings to patients is unknown.
When betamethasone dipropionate was applied topically to CD-1 mice for up to 24 months at dosages approximating 1.3, 4.2 and 8.5 mcg/kg/day in females, and 1.3, 4.2, and 12.9 mcg/kg/day in males (up to 26 mcg/m2/day and 39 mcg/m2/day, in females and males, respectively), no significant changes in tumor incidence were observed when compared to control.
When betamethasone dipropionate was administered via oral gavage to male and female Sprague Dawley rats for up to 24 months at dosages of 20, 60, and 200 mcg/kg/day (120, 360, and 1200 mcg/m2/day, respectively), no significant changes in tumor incidence were observed when compared to control.
Calcipotriene did not elicit any genotoxic effects in the Ames mutagenicity assay, the mouse lymphoma TK locus assay, the human lymphocyte chromosome aberration test, or the mouse micronucleus test. Betamethasone dipropionate did not elicit any genotoxic effects in the Ames mutagenicity assay, the mouse lymphoma TK locus assay, or in the rat micronucleus test.
Studies in rats with oral doses of up to 54 mcg/kg/day (324 mcg/m2/day) of calcipotriene indicated no impairment of fertility or general reproductive performance. Studies in male rats at oral doses of up to 200 mcg/kg/day (1200 mcg/m2/day), and in female rats at oral doses of up to 1000 mcg/kg/day (6000 mcg/m2/day), of betamethasone dipropionate indicated no impairment of fertility.
-
14 CLINICAL STUDIES
Clinical Trials Conducted in Subjects 18 years and older with Plaque Psoriasis
In an international, multi-center, double-blind, vehicle- and active-controlled, parallel-group trial, 1603 subjects with mild to very severe plaque psoriasis on trunk and limbs were treated once daily for 4 weeks. Subjects were randomized to one of four treatment arms: Taclonex® Ointment, calcipotriene hydrate 50 mcg/g in the same vehicle, betamethasone dipropionate 0.64 mg/g in the same vehicle, and vehicle alone. The mean age of the subjects was 48.4 years and 60.5% were male. Most subjects had disease of moderate severity at baseline.
Efficacy was assessed as the proportion of subjects with absent or very mild disease according to the Investigator's Global Assessment of Disease Severity at end of treatment (4 weeks). "Absent" disease was defined as no evidence of redness, thickness, or scaling. "Very mild disease" was defined as controlled disease, but not entirely cleared: lesions with some discoloration with absolutely minimal thickness, i.e. the edges to the lesion(s) could just be felt. Table 3 contains the response rates for this trial.
Table 3
Percentage of Subjects with Absent or Very Mild Disease According to the Investigator's Global Assessment of Disease Severity at End of Treatment (4 weeks)*
Taclonex® Ointment Calcipotriene Betamethasone dipropionate Vehicle N = 490 N = 480 N = 476 N = 157 Absent or very mild disease 48.0% 16.5% 26.3% 7.6% * Subjects with mild disease at baseline were required to have "Absent" disease to be considered a success.
In addition to the pivotal trial (N = 490), four randomized, double-blind, vehicle- or active-controlled, parallel-group trials were conducted and provided supportive evidence of efficacy. These trials included a total of 1058 subjects treated with Taclonex® Ointment once daily for up to 4 weeks.Clinical Trial Conducted in Subjects 12 to 17 years with Plaque Psoriasis
A prospective, uncontrolled trial (N=33) was conducted in pediatric subjects ages 12 to 17 years with plaque psoriasis involving 5-30% of the body surface area. Approximately 91% of subjects had moderate disease at baseline. Subjects were treated once daily for up to 4 weeks with Taclonex® Ointment. All subjects were evaluated for safety including calcium metabolism (N=33) and 32 subjects were evaluated for HPA axis suppression [see Clinical Pharmacology (12.2)].
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information).
Inform patients of the following:
- Instruct adult patients (18 years and older) not to use more than 100 g per week.
- Instruct pediatric patients (12 to 17 years) not to use more than 60 g per week.
- Discontinue therapy when control is achieved unless directed otherwise by the physician.
- Avoid use of Taclonex® Ointment on the face, underarms, groin or eyes. If this medicine gets on face or in eyes, wash area right away.
- Do not occlude the treatment area with a bandage or other covering unless directed by the physician.
- Note that local reactions and skin atrophy are more likely to occur with occlusive use, prolonged use or use of higher potency corticosteroids.
- Wash hands after application.
- Advise patients to report any visual symptoms to their healthcare providers.
- Advise a woman to use Taclonex® Ointment on the smallest area of skin and for the shortest duration possible while pregnant or breastfeeding. Advise breastfeeding women not to apply Taclonex® Ointment directly to the nipple and areola to avoid direct infant exposure.
- Instruct patients not to use other products containing calcipotriene or a corticosteroid should not be used with Taclonex® Ointment without first talking to the physician.
- Instruct patients who use Taclonex® Ointment to avoid excessive exposure to either natural or artificial sunlight (including tanning booths, sun lamps, etc.).

Manufactured by:
LEO Laboratories Ltd. (LEO Pharma)
Dublin 12, Ireland
Distributed by:
LEO Pharma Inc.
Seven Giralda Farms
Madison, NJ 07940, USA
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
Taclonex® (TAK-lo-NEKS)
(calcipotriene and betamethasone dipropionate)Ointment, 0.005%/0.064%
Read the Patient Information that comes with Taclonex® Ointment before you start using it and each time you refill your prescription. There may be new information. This leaflet does not take the place of talking with your doctor about your condition or treatment.
Important information: Taclonex® Ointment is for use on the skin only (topical use only). Do not use Taclonex® Ointment on the face, under arms or on groin area. Do not swallow Taclonex® Ointment. Another product, Taclonex® Topical Suspension contains the same medicine that is in Taclonex® Ointment and is used to treat plaque psoriasis on the scalp. If you use both medicines to treat your plaque psoriasis, be sure to follow your doctor's directions carefully so that you do not use too much of one or both of these medications.
What is Taclonex® Ointment?
Taclonex® Ointment is a prescription medicine that is for use on the skin only (a topical medicine). Taclonex® Ointment is used to treat plaque psoriasis in patients 12 years of age and older.
Taclonex® Ointment has not been studied in patients under the age of 12 years.Who should not use Taclonex® Ointment?
Do not use Taclonex® Ointment if you:
- have thin skin (atrophy) at the site to be treated
- are allergic to anything in Taclonex® Ointment. See the end of this leaflet for a complete list of ingredients.
What should I tell my doctor before using Taclonex® Ointment?
Tell your doctor about all of your health conditions, including if you:
- have a skin infection. Your skin infection should be treated before starting Taclonex® Ointment.
- have a calcium metabolism disorder
- have one of the following types of psoriasis:
• erythrodermic psoriasis
• exfoliative psoriasis
• pustular psoriasis - are getting phototherapy treatments (light therapy) for your psoriasis
- are pregnant or planning to become pregnant. It is not known if Taclonex® Ointment can harm your unborn baby. You and your doctor will have to decide if Taclonex® Ointment is right for you while pregnant.
- are breastfeeding or plan to breastfeed. It is not known if Taclonex® Ointment passes into your milk and if it can harm your baby. If you use Taclonex® Ointment while breastfeeding, use Taclonex® Ointment on the smallest area of skin and for the shortest time needed. If you use Taclonex® Ointment, do not apply Taclonex® Ointment to your nipple or areola to avoid getting Taclonex® Ointment into your baby's mouth.
Tell your doctor about all the medicines you take, including prescription, and nonprescription medicines, vitamins and herbal supplements.
Taclonex® Ointment and some other medicines can interact with each other. Especially tell your doctor if you use:
- other corticosteroid medicines
- other medicines for your psoriasis
How should I use Taclonex® Ointment?
- Use Taclonex® Ointment exactly as prescribed by your doctor.
- If you are 18 years of age or older, you should not use more than 100 grams of Taclonex® Ointment in 1 week.
- If you are 12 to 17 years of age, you should not use more than 60 grams of Taclonex® Ointment in 1 week.
- Apply Taclonex® Ointment once a day to the areas of your skin affected by psoriasis. Gently rub Taclonex® Ointment into your affected skin areas.
- Only use Taclonex® Ointment as directed by your doctor. Taclonex® Ointment is recommended for up to 4 weeks of treatment. Do not use Taclonex® Ointment for more than 4 weeks unless prescribed by your doctor.
- Do not use Taclonex® Ointment on the face, under arms or on groin area. If you accidentally get Taclonex® Ointment on the face or in the eyes wash the area with water right away.
- If you forget to use Taclonex® Ointment, use it as soon as you remember. Then go on as before.
- Wash your hands well after applying Taclonex® Ointment.
- If you are breastfeeding, do not use Taclonex® Ointment on the breast while nursing.
Using Taclonex® Ointment:
Do not bandage or tightly cover the treated skin area.Remove the cap and check that the aluminum seal covers the tube before the first use. To break the seal, turn the cap over and punch through the seal.
What should I avoid while using Taclonex® Ointment?
Avoid spending a long time in the sunlight. Avoid tanning booths and sunlamps. Use sunscreen if you have to be in the sunlight. Talk to your doctor if you get a sunburn.What are the possible side effects of Taclonex® Ointment?
The most common side effects are:- itching
- rash
Other less common side effects with Taclonex® Ointment include:
- redness of the skin
- skin irritation
- skin burning
- inflamed hair pores (folliculitis)
- change of skin color (at the site of application)
- rash with pus-filled papules
- thinning of the skin (atrophy)
- swollen fine blood vessels (this makes your skin appear red at the site of application)
Taclonex® Ointment may cause serious side effects. Serious side effects are more likely to happen if you use too much Taclonex® Ointment, use it for too long, or use it with other topical medicines that contain corticosteroids, calcipotriene, or certain other ingredients. Check with your doctor before using other topical medicines. Taclonex® Ointment can pass through your skin. Serious side effects may include:
- too much calcium in your blood or urine
- adrenal gland problems
Your doctor may do special blood and urine tests to check your calcium levels and adrenal gland function while you are using Taclonex® Ointment.
Call your doctor about any side effect that bothers you or that does not go away.These are not all of the side effects with Taclonex® Ointment. Ask your doctor or pharmacist for more information.
Vision problems. Taclonex® Ointment may increase your chance of developing cataract(s) and glaucoma. Tell your healthcare provider if you develop blurred vision or other vision problems during treatment with Taclonex® Ointment.
How should I store Taclonex® Ointment?
- Store Taclonex® Ointment at room temperature, 68°F - 77°F (20°C - 25°C); Make sure the cap on the tube is tightly closed.
- Taclonex® Ointment has an expiration date (exp.) marked on the tube. Do not use the ointment after this date.
- Keep Taclonex® Ointment and all medicines out of the reach of children and pets.
General information about Taclonex® Ointment
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use Taclonex® Ointment for a condition for which it was not prescribed. Do not give Taclonex® Ointment to other people, even if they have the same symptoms you have. It may harm them.This leaflet summarizes the most important information about Taclonex® Ointment. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about Taclonex® Ointment that is written for health professionals.
What are the ingredients in Taclonex® Ointment?
Active ingredients: calcipotriene hydrate, betamethasone dipropionate.
Inactive ingredients: butylhydroxytoluene, mineral oil, polyoxypropylene stearyl ether, all-rac-alpha-tocopherol, white petrolatum.Manufactured by:
LEO Laboratories Ltd. (LEO Pharma)
Dublin 12, Ireland
Distributed by:
LEO Pharma Inc.
Seven Giralda Farms
Madison, NJ 07940, USA
Revised: 01/2023
- PRINCIPAL DISPLAY PANEL - 60 g Tube Carton
-
INGREDIENTS AND APPEARANCE
TACLONEX
calcipotriene and betamethasone dipropionate ointmentProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50222-227 Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CALCIPOTRIENE MONOHYDRATE (UNII: S7499TYY6G) (CALCIPOTRIENE - UNII:143NQ3779B) CALCIPOTRIENE 50 ug in 1 g BETAMETHASONE DIPROPIONATE (UNII: 826Y60901U) (BETAMETHASONE - UNII:9842X06Q6M) BETAMETHASONE 0.5 mg in 1 g Inactive Ingredients Ingredient Name Strength .ALPHA.-TOCOPHEROL, DL- (UNII: 7QWA1RIO01) MINERAL OIL (UNII: T5L8T28FGP) PETROLATUM (UNII: 4T6H12BN9U) POLYPROPYLENE GLYCOL 11 STEARYL ETHER (UNII: S4G2J0Y0LG) BUTYLATED HYDROXYTOLUENE (UNII: 1P9D0Z171K) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50222-227-04 1 in 1 CARTON 06/01/2008 10/31/2024 1 60 g in 1 TUBE; Type 0: Not a Combination Product 2 NDC:50222-227-81 1 in 1 CARTON 06/01/2008 10/31/2022 2 100 g in 1 TUBE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021852 06/01/2008 10/31/2024 Labeler - LEO Pharma Inc. (832692615) Registrant - LEO Pharma A/S (306218108) Establishment Name Address ID/FEI Business Operations LEO Laboratories Ltd. 219532322 ANALYSIS(50222-227) , MANUFACTURE(50222-227) , PACK(50222-227) , LABEL(50222-227)
