Label: CEFOTAXIME injection

- NDC Code(s): 0143-9935-01
- Packager: Hikma Pharmaceuticals USA Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated April 11, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
SPL UNCLASSIFIED SECTION
Rx ONLY
PHARMACY BULK PACKAGE – NOT FOR DIRECT INFUSION To reduce the development of drug-resistant bacteria and maintain the effectiveness of Cefotaxime for Injection, USP (cefotaxime sodium) and other antibacterial drugs, Cefotaxime for Injection, USP should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria.
-
DESCRIPTION
Sterile cefotaxime sodium is a semisynthetic, broad spectrum cephalosporin antibiotic for parenteral administration. It is the sodium salt of 7-[2-(2-amino-4-thiazolyl) glyoxylamido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo [4.2.0] oct-2-ene-2-carboxylate 72 (Z)-(o-methyloxime), acetate (ester). Cefotaxime for Injection, USP contains approximately 50.5 mg (2.2 mEq) of sodium per gram of cefotaxime activity. Solutions of Cefotaxime for Injection, USP range from very pale yellow to light amber depending on the concentration and the diluent used. The pH of the injectable solutions usually ranges from 5.0 to 7.5. The CAS Registry Number is 64485-93-4.
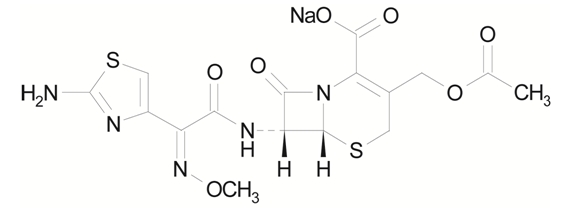
C16H16N5NaO7S2 MW 477.45
Cefotaxime for Injection, USP is supplied as a dry powder in Pharmacy Bulk Packages containing cefotaxime sodium equivalent to 10 g of cefotaxime. FURTHER DILUTION IS REQUIRED BEFORE USE. A Pharmacy Bulk Package is a container of a sterile preparation for parenteral use, which contains many single doses. This Pharmacy Bulk Package is for use in a pharmacy admixture service; it provides many singles doses of cefotaxime for addition to suitable parenteral fluids in the preparation of admixtures for intravenous infusion. (See DOSAGE AND ADMINISTRATION and DIRECTIONS FOR PROPER USE OF PHARMACY BULK PACKAGE.)
-
CLINICAL PHARMACOLOGY
Following IM administration of a single 500 mg or 1 g dose of cefotaxime to normal volunteers, mean peak serum concentrations of 11.7 and 20.5 mcg/mL respectively were attained within 30 minutes and declined with an elimination half-life of approximately 1 hour. There was a dose‑dependent increase in serum levels after the IV administration of 500 mg, 1 g, and 2 g of cefotaxime (38.9, 101.7, and 214.4 mcg/mL respectively) without alteration in the elimination half-life. There is no evidence of accumulation following repetitive IV infusion of 1 g doses every 6 hours for 14 days as there are no alterations of serum or renal clearance. About 60% of the administered dose was recovered from urine during the first 6 hours following the start of the infusion.
Approximately 20-36% of an intravenously administered dose of 14C-cefotaxime is excreted by the kidney as unchanged cefotaxime and 15-25% as the desacetyl derivative, the major metabolite. The desacetyl metabolite has been shown to contribute to the bactericidal activity. Two other urinary metabolites (M2 and M3) account for about 20-25%. They lack bactericidal activity.
A single 50 mg/kg dose of cefotaxime was administered as an intravenous infusion over a 10- to 15-minute period to 29 newborn infants grouped according to birth weight and age. The mean half-life of cefotaxime in infants with lower birth weights (≤1500 grams), regardless of age, was longer (4.6 hours) than the mean half-life (3.4 hours) in infants whose birth weight was greater than 1500 grams. Mean serum clearance was also smaller in the lower birth weight infants. Although the differences in mean half-life values are statistically significant for weight, they are not clinically important. Therefore, dosage should be based solely on age. (See DOSAGE AND ADMINISTRATION section.)
Drug Interactions
A single intravenous dose and oral dose of probenecid (500 mg each) followed by two oral doses of probenecid 500 mg at approximately hourly intervals administered to three healthy male subjects receiving a continuous infusion of cefotaxime increased the steady-state plasma concentration of cefotaxime by approximately 80%. In another study, administration of oral probenecid 500 mg every 6 hours to six healthy male subjects with cefotaxime 1 gram infused over 5 minutes decreased the total clearance of cefotaxime by approximately 50%.
Additionally, no disulfiram-like reactions were reported in a study conducted in 22 healthy volunteers administered cefotaxime and ethanol.
Microbiology
Mechanism of Action
Cefotaxime sodium is a bactericidal agent that acts by inhibition of bacterial cell wall synthesis. Cefotaxime has activity in the presence of some beta-lactamases, both penicillinases and cephalosporinases, of Gram-negative and Gram-positive bacteria.Mechanism of Resistance
Resistance to cefotaxime is primarily through hydrolysis by beta-lactamase, alteration of penicillin-binding proteins (PBPs), and decreased permeability.Susceptibility to cefotaxime will vary geographically and may change over time; local susceptibility data should be consulted, if available. Cefotaxime has been shown to be active against most isolates of the following bacteria both in vitro and in clinical infections as described in the INDICATIONS AND USAGE section:
Gram-positive bacteria
Enterococcus spp. a
Staphylococcus aureus (methicillin-susceptible isolates only)
Staphylococcus epidermidis
Streptococcus pneumoniae
Streptococcus pyogenes (Group A beta-hemolytic streptococci)
Streptococcus spp. (Viridans group streptococci)Gram-negative bacteria
Acinetobacter spp.
Citrobacter spp. b
Enterobacter spp. b
Escherichia coli b
Haemophilus influenzae
Haemophilus parainfluenzae
Klebsiella spp. (including Klebsiella pneumoniae) b
Morganella morganii b
Neisseria gonorrhoeae (including beta-lactamase-positive and negative strains)
Neisseria meningitidis
Proteus mirabilis b
Proteus vulgaris b
Providencia rettgeri b
Providencia stuartii b
Serratia marcescens baEnterococcus species may be intrinsically resistant to cefotaxime.
b Most extended spectrum beta-lactamase (ESBL)-producing and carbapenemase-producing isolates are resistant to cefotaxime.Anaerobic bacteria
Bacteroides spp., including some isolates of Bacteroides fragilis
Clostridium spp. (most isolates of Clostridium difficile are resistant)
Fusobacterium spp. (including Fusobacterium nucleatum)
Peptococcus spp.
Peptostreptococcus spp.The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following microorganisms exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to 1 mcg/mL. However, the efficacy of cefotaxime in treating clinical infections due to these microorganisms has not been established in adequate and well-controlled clinical trials.
Gram-negative bacteria
Providencia spp.
Salmonella spp. (including Salmonella typhi)
Shigella spp. -
INDICATIONS AND USAGE
Treatment
Cefotaxime for Injection, USP is indicated for the treatment of patients with serious infections caused by susceptible strains of the designated microorganisms in the diseases listed below.
(1) Lower respiratory tract infections, including pneumonia, caused by Streptococcus pneumoniae (formerly Diplococcus pneumoniae), Streptococcus pyogenes* (Group A streptococci) and other streptococci (excluding enterococci, e.g., Enterococcus faecalis), Staphylococcus aureus (penicillinase and non-penicillinase producing), Escherichia coli, Klebsiella species, Haemophilus influenzae (including ampicillin resistant strains), Haemophilus parainfluenzae, Proteus mirabilis, Serratia marcescens*, Enterobacter species, indole positive Proteus and Pseudomonas species (including P. aeruginosa).
(2) Genitourinary infections. Urinary tract infections caused by Enterococcus species, Staphylococcus epidermidis, Staphylococcus aureus*, (penicillinase and non-penicillinase producing), Citrobacter species, Enterobacter species, Escherichia coli, Klebsiella species, Proteus mirabilis, Proteus vulgaris*, Providencia stuartii, Morganella morganii*, Providencia rettgeri*, Serratia marcescens and Pseudomonas species (including P. aeruginosa). Also, uncomplicated gonorrhea (cervical/urethral and rectal) caused by Neisseria gonorrhoeae, including penicillinase producing strains.
(3) Gynecologic infections, including pelvic inflammatory disease, endometritis and pelvic cellulitis caused by Staphylococcus epidermidis, Streptococcus species, Enterococcus species, Enterobacter species*, Klebsiella species*, Escherichia coli, Proteus mirabilis, Bacteroides species (including Bacteroides fragilis*), Clostridium species, and anaerobic cocci (including Peptostreptococcus species and Peptococcus species) and Fusobacterium species (including F. nucleatum*).
Cefotaxime for Injection, USP, like other cephalosporins, has no activity against Chlamydia trachomatis. Therefore, when cephalosporins are used in the treatment of patients with pelvic inflammatory disease and C. trachomatis is one of the suspected pathogens, appropriate anti‑chlamydial coverage should be added.
(4) Bacteremia/Septicemia caused by Escherichia coli, Klebsiella species, and Serratia marcescens, Staphylococcus aureus and Streptococcus species (including S. pneumoniae).
(5) Skin and skin structure infections caused by Staphylococcus aureus (penicillinase and non-penicillinase producing), Staphylococcus epidermidis, Streptococcus pyogenes (Group A streptococci) and other streptococci, Enterococcus species, Acinetobacter species*, Escherichia coli, Citrobacter species (including C. freundii*), Enterobacter species, Klebsiella species, Proteus mirabilis, Proteus vulgaris*, Morganella morganii, Providencia rettgeri*, Pseudomonas species, Serratia marcescens, Bacteroides species, and anaerobic cocci (including Peptostreptococcus* species and Peptococcus species).
(6) Intra-abdominal infections including peritonitis caused by Streptococcus species*, Escherichia coli, Klebsiella species, Bacteroides species, and anaerobic cocci (including Peptostreptococcus* species and Peptococcus* species) Proteus mirabilis*, and Clostridium species*.
(7) Bone and/or joint infections caused by Staphylococcus aureus (penicillinase and non‑penicillinase producing strains), Streptococcus species (including S. pyogenes*), Pseudomonas species (including P. aeruginosa*), and Proteus mirabilis*.
(8) Central nervous system infections, e.g., meningitis and ventriculitis, caused by Neisseria meningitidis, Haemophilus influenzae, Streptococcus pneumoniae, Klebsiella pneumoniae* and Escherichia coli*.
(*) Efficacy for this organism, in this organ system, has been studied in fewer than 10 infections.
Although many strains of enterococci (e.g., S. faecalis) and Pseudomonas species are resistant to cefotaxime sodium in vitro, Cefotaxime for Injection, USP has been used successfully in treating patients with infections caused by susceptible organisms.
Specimens for bacteriologic culture should be obtained prior to therapy in order to isolate and identify causative organisms and to determine their susceptibilities to cefotaxime. Therapy may be instituted before results of susceptibility studies are known; however, once these results become available, the antibiotic treatment should be adjusted accordingly.
In certain cases of confirmed or suspected gram-positive or gram-negative sepsis or in patients with other serious infections in which the causative organism has not been identified, Cefotaxime for Injection, USP may be used concomitantly with an aminoglycoside. The dosage recommended in the labeling of both antibiotics may be given and depends on the severity of the infection and the patient’s condition. Renal function should be carefully monitored, especially if higher dosages of the aminoglycosides are to be administered or if therapy is prolonged, because of the potential nephrotoxicity and ototoxicity of aminoglycoside antibiotics. It is possible that nephrotoxicity may be potentiated if Cefotaxime for Injection, USP is used concomitantly with an aminoglycoside.
Prevention
The administration of Cefotaxime for Injection, USP preoperatively reduces the incidence of certain infections in patients undergoing surgical procedures (e.g., abdominal or vaginal hysterectomy, gastrointestinal and genitourinary tract surgery) that may be classified as contaminated or potentially contaminated.
In patients undergoing cesarean section, intraoperative (after clamping the umbilical cord) and postoperative use of Cefotaxime for Injection, USP may also reduce the incidence of certain postoperative infections. See DOSAGE AND ADMINISTRATION section.
Effective use for elective surgery depends on the time of administration. To achieve effective tissue levels, Cefotaxime for Injection, USP should be given 1/2 or 1 1/2 hours before surgery. See DOSAGE AND ADMINISTRATION section.
For patients undergoing gastrointestinal surgery, preoperative bowel preparation by mechanical cleansing as well as with a non- absorbable antibiotic (e.g., neomycin) is recommended.
If there are signs of infection, specimens for culture should be obtained for identification of the causative organism so that appropriate therapy may be instituted.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of Cefotaxime for Injection, USP and other antibacterial drugs, Cefotaxime for Injection, USP should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
- CONTRAINDICATIONS
-
WARNINGS
BEFORE THERAPY WITH CEFOTAXIME IS INSTITUTED, CAREFUL INQUIRY SHOULD BE MADE TO DETERMINE WHETHER THE PATIENT HAS HAD PREVIOUS HYPERSENSITIVITY REACTIONS TO CEFOTAXIME SODIUM, CEPHALOSPORINS, PENICILLINS, OR OTHER DRUGS. THIS PRODUCT SHOULD BE GIVEN WITH CAUTION TO PATIENTS WITH TYPE I HYPERSENSITIVITY REACTIONS TO PENICILLIN. ANTIBIOTICS SHOULD BE ADMINISTERED WITH CAUTION TO ANY PATIENT WHO HAS DEMONSTRATED SOME FORM OF ALLERGY, PARTICULARLY TO DRUGS. IF AN ALLERGIC REACTION TO CEFOTAXIME OCCURS, DISCONTINUE TREATMENT WITH THE DRUG. SERIOUS HYPERSENSITIVITY REACTIONS MAY REQUIRE EPINEPHRINE AND OTHER EMERGENCY MEASURES.
During post-marketing surveillance, a potentially life-threatening arrhythmia was reported in each of six patients who received a rapid (less than 60 seconds) bolus injection of cefotaxime through a central venous catheter. Therefore, cefotaxime should only be administered as instructed in the DOSAGE AND ADMINISTRATION section.
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including cefotaxime, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
-
PRECAUTIONS
General
Prescribing cefotaxime in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
Cefotaxime should be prescribed with caution in individuals with a history of gastrointestinal disease, particularly colitis.
Because high and prolonged serum antibiotic concentrations can occur from usual doses in patients with transient or persistent reduction of urinary output because of renal insufficiency, the total daily dosage should be reduced when cefotaxime is administered to such patients. Continued dosage should be determined by degree of renal impairment, severity of infection, and susceptibility of the causative organism.
Although there is no clinical evidence supporting the necessity of changing the dosage of cefotaxime sodium in patients with even profound renal dysfunction, it is suggested that, until further data are obtained, the dose of cefotaxime sodium be halved in patients with estimated creatinine clearances of less than 20 mL/min/1.73 m2.
When only serum creatinine is available, the following formula5 (based on sex, weight, and age of the patient) may be used to convert this value into creatinine clearance. The serum creatinine should represent a steady state of renal function.
Weight (kg) x (140 - age)
Males: 72 × serum creatinine
Females: 0.85 × above value
As with other antibiotics, prolonged use of cefotaxime may result in overgrowth of nonsusceptible organisms. Repeated evaluation of the patient’s condition is essential. If superinfection occurs during therapy, appropriate measures should be taken.
Leukopenia, neutropenia, granulocytopenia and, more rarely, bone marrow failure, pancytopenia, or agranulocytosis may develop during treatment with cefotaxime. For courses of treatment lasting longer than 10 days, blood counts should therefore be monitored and treatment discontinuation should be considered in case of abnormal results.
Cefotaxime, like other parenteral anti-infective drugs, may be locally irritating to tissues. In most cases, perivascular extravasation of cefotaxime responds to changing of the infusion site. In rare instances, extensive perivascular extravasation of cefotaxime may result in tissue damage and require surgical treatment. To minimize the potential for tissue inflammation, infusion sites should be monitored regularly and changed when appropriate.
Information for patients
Patients should be counseled that antibacterial drugs including cefotaxime should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When cefotaxime is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by cefotaxime or other antibacterial drugs in the future.
Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
Drug Interactions
As with other cephalosporins, cefotaxime may potentiate the nephrotoxic effects of nephrotoxic drugs such as aminoglycosides, NSAIDs and furosemide.
Probenecid interferes with the renal tubular transfer of cefotaxime, decreasing the total clearance of cefotaxime by approximately 50% and increasing the plasma concentrations of cefotaxime. Administration of cefotaxime in excess of 6 grams/day should be avoided in patients receiving probenecid (see CLINICAL PHARMACOLOGY, Drug Interactions).
Drug/Laboratory Test Interactions
Cephalosporins, including cefotaxime sodium, are known to occasionally induce a positive direct Coombs’ test.
A false-positive reaction for glucose in the urine may occur with copper reduction tests (Benedict’s or Fehling’s solution or with CLINITEST® tablets), but not with enzyme-based tests for glycosuria (e.g., CLINISTIX® or TesTape®). There are no reports in published literature that link elevations of plasma glucose levels to the use of cefotaxime.
Carcinogenesis, Mutagenesis
Lifetime studies in animals to evaluate carcinogenic potential have not been conducted. Cefotaxime was not mutagenic in the mouse micronucleus test or in the Ames test. Cefotaxime did not impair fertility to rats when administered subcutaneously at doses up to 250 mg/kg/day (0.2 times the maximum recommended human dose based on mg/m2) or in mice when administered intravenously at doses up to 2000 mg/kg/day (0.7 times the recommended human dose based on mg/m2).
Pregnancy: Teratogenic Effects
Reproduction studies have been performed in pregnant mice given cefotaxime intravenously at doses up to 1200 mg/kg/day (0.4 times the recommended human dose based on mg/m2) or in pregnant rats when administered intravenously at doses up to 1200 mg/kg/day (0.8 times the recommended human dose based on mg/m2). No evidence of embryotoxicity or teratogenicity was seen in these studies. Although cefotaxime has been reported to cross the placental barrier and appear in cord blood, the effect on the human fetus is not known. There are no well-controlled studies in pregnant women. Because animal reproductive studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nonteratogenic Effects
Use of the drug in women of child-bearing potential requires that the anticipated benefit be weighed against the possible risks.
In perinatal and postnatal studies with rats, the pups in the group given 1200 mg/kg/day of cefotaxime were significantly lighter in weight at birth and remained smaller than pups in the control group during the 21 days of nursing.
Nursing Mothers
Cefotaxime is excreted in human milk in low concentrations. Caution should be exercised when cefotaxime is administered to a nursing woman.
Geriatric Use
Of the 1409 subjects in clinical studies of cefotaxime, 632 (45%) were 65 and over, while 258 (18%) were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (see PRECAUTIONS, General).
-
ADVERSE REACTIONS
Clinical Trials Experience
Cefotaxime is generally well tolerated. The most common adverse reactions have been local reactions following IM or IV injection.
Other adverse reactions have been encountered infrequently.
The most frequent adverse reactions (greater than 1%) are:
Local (4.3%) - Injection site inflammation with IV administration. Pain, induration, and tenderness after IM injection.
Hypersensitivity (2.4%) - Rash, pruritus, fever, eosinophilia.
Gastrointestinal (1.4%) - Colitis, diarrhea, nausea, and vomiting.
Symptoms of pseudomembranous colitis can appear during or after antibiotic treatment. Nausea and vomiting have been reported rarely.
Less frequent adverse reactions (less than 1%) are:
Hematologic System - Neutropenia, leukopenia, have been reported. Some individuals have developed positive direct Coombs Tests during treatment with cefotaxime and other cephalosporin antibiotics.
Genitourinary System - Moniliasis, vaginitis.
Central Nervous System - Headache.
Liver - Transient elevations in AST, ALT, serum LDH, and serum alkaline phosphatase levels have been reported.
Kidney - As with some other cephalosporins, transient elevations of BUN have been occasionally observed with cefotaxime.
Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of cefotaxime. Because these reactions were reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiovascular System - Potentially life-threatening arrhythmias following rapid (less than 60 seconds) bolus administration via central venous catheter have been observed.
Central Nervous System - Administration of high doses of beta-lactam antibiotics, including cefotaxime, particularly in patients with renal insufficiency may result in encephalopathy (e.g. impairment of consciousness, abnormal movements and convulsions). Dizziness has also been reported.
Cutaneous - As with other cephalosporins, isolated cases of toxic epidermal necrolysis,
Stevens‑Johnson syndrome, and erythema multiforme have been reported. Acute generalized exanthematous pustulosis (AGEP) has also been reported.
General disorders and administration site conditions - Inflammatory reactions at the injection site, including phlebitis/thrombophlebitis.
Hematologic System - Hemolytic anemia, agranulocytosis, thrombocytopenia, pancytopenia, bone marrow failure.Hypersensitivity - Anaphylaxis (e.g., angioedema, bronchospasm, malaise possibly culminating in shock), urticaria.
Kidney - Interstitial nephritis, transient elevations of creatinine, acute renal failure.
Liver - Hepatitis, jaundice, cholestasis, elevations of gamma GT and bilirubin.
Cephalosporin Class Labeling
In addition to the adverse reactions listed above which have been observed in patients treated with cefotaxime sodium, the following adverse reactions and altered laboratory tests have been reported for cephalosporin class antibiotics: allergic reactions, hepatic dysfunction including cholestasis, aplastic anemia, hemorrhage, and false-positive test for urinary glucose.
Several cephalosporins have been implicated in triggering seizures, particularly in patients with renal impairment when the dosage was not reduced. See DOSAGE AND ADMINISTRATION and OVERDOSAGE. If seizures associated with drug therapy occur, the drug should be discontinued. Anticonvulsant therapy can be given if clinically indicated.
To report SUSPECTED ADVERSE REACTIONS, contact West-Ward Pharmaceutical Corp. at 1-877‑233-2001 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
OVERDOSAGE
The acute toxicity of cefotaxime was evaluated in neonatal and adult mice and rats. Significant mortality was seen at parenteral doses in excess of 6000 mg/kg/day in all groups. Common toxic signs in animals that died were a decrease in spontaneous activity, tonic and clonic convulsions, dyspnea, hypothermia, and cyanosis. Cefotaxime sodium overdosage has occurred in patients. Most cases have shown no overt toxicity. The most frequent reactions were elevations of BUN and creatinine. There is a risk of reversible encephalopathy in cases of administration of high doses of beta-lactam antibiotics including cefotaxime. No specific antidote exists. Patients who receive an acute overdosage should be carefully observed and given supportive treatment.
-
DOSAGE AND ADMINISTRATION
The intent of this Pharmacy Bulk Package is for the preparation of solutions for intravenous infusion only.
Adults
Dosage and route of administration should be determined by susceptibility of the causative organisms, severity of the infection, and the condition of the patient (see table for dosage guideline). Cefotaxime may be administered IV after reconstitution. The maximum daily dosage should not exceed 12 grams.
GUIDELINES FOR DOSAGE OF CEFOTAXIME FOR INJECTION Type of Infection Daily Dose
(grams)Frequency and Route Gonococcal urethritis/cervicitis in males and females 0.5 0.5 gram IM (single dose) Rectal gonorrhea in females 0.5 0.5 gram IM (single dose) Rectal gonorrhea in males 1 1 gram IM (single dose) Uncomplicated infections 2 1 gram every 12 hours IM or IV Moderate to severe infections 3-6 1-2 grams every 8 hours IM or IV Infections commonly needing antibiotics in higher dosage (e.g., septicemia) 6-8 2 grams every 6-8 hours IV Life-threatening infections up to 12 2 grams every 4 hours IV
If C. trachomatis is a suspected pathogen, appropriate anti-chlamydial coverage should be added, because cefotaxime sodium has no activity against this organism.
To prevent postoperative infection in contaminated or potentially contaminated surgery, the recommended dose is a single 1 gram IV administered 30 to 90 minutes prior to start of surgery.
Cesarean Section Patients
The first dose of 1 gram is administered intravenously as soon as the umbilical cord is clamped. The second and third doses should be given as 1 gram intravenously at 6 and 12 hours after the first dose.
Neonates, Infants, and Children
The following dosage schedule is recommended:
Neonates (birth to 1 month):
0-1 week of age 50 mg/kg per dose every 12 hours IV
1-4 weeks of age 50 mg/kg per dose every 8 hours IV
It is not necessary to differentiate between premature and normal-gestational age infants.
Infants and Children (1 month to 12 years):
For body weights less than 50 kg, the recommended daily dose is 50 to 180 mg/kg IV body weight divided into four to six equal doses. The higher dosages should be used for more severe or serious infections, including meningitis. For body weights 50 kg or more, the usual adult dosage should be used; the maximum daily dosage should not exceed 12 grams.
Geriatric Use
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. (See PRECAUTIONS, General and PRECAUTIONS, Geriatric Use.)
Impaired Renal Function
See PRECAUTIONS, General.
NOTE: As with antibiotic therapy in general, administration of cefotaxime should be continued for a minimum of 48 to 72 hours after the patient defervesces or after evidence of bacterial eradication has been obtained; a minimum of 10 days of treatment is recommended for infections caused by Group A beta-hemolytic streptococci in order to guard against the risk of rheumatic fever or glomerulonephritis; frequent bacteriologic and clinical appraisal is necessary during therapy of chronic urinary tract infection and may be required for several months after therapy has been completed; persistent infections may require treatment of several weeks and doses smaller than those indicated above should not be used.
DIRECTIONS FOR PROPER USE OF PHARMACY BULK PACKAGE:
The 10 gram Pharmacy Bulk Package should be penetrated ONLY ONE TIME utilizing a suitable sterile transfer device or dispensing set and should be reconstituted with 47 mL of diluent for an approximate concentration of 200 mg/mL (withdrawable volume 52 mL) or 97 mL of diluent for an approximate concentration of 100 mg/mL withdrawable volume 102 mL), in a suitable work area such as a laminar flow hood, using aseptic technique. Shake to dissolve; inspect for particulate matter and discoloration prior to use. Stock solutions may be further diluted for IV infusion with diluents as listed in COMPATIBILITY AND STABILITY section. The reconstituted content of the 10 g Pharmacy Bulk Package should be withdrawn immediately, using a suitable sterile transfer device or dispensing set which allows measured dispensing of the contents. However, if it is not possible, aliquoting operations must be completed within four hours of reconstitution. Discard the reconstituted stock solution 4 hours after initial entry. Solutions of cefotaxime range from very pale yellow to light amber, depending on concentration, diluent used, and length and condition of storage.
Reconstituted Bulk Solutions Should Not Be Used For Direct Infusion
NOTE: Solutions of cefotaxime must not be admixed with aminoglycoside solutions. If cefotaxime and aminoglycosides are to be administered to the same patient, they must be administered separately and not as mixed injection.
A SOLUTION OF 1 G CEFOTAXIME IN 14 ML OF STERILE WATER FOR INJECTION IS ISOTONIC.
IV Administration: The IV route is preferable for patients with bacteremia, bacterial septicemia, peritonitis, meningitis, or other severe or life-threatening infections, or for patients who may be poor risks because of lowered resistance resulting from such debilitating conditions as malnutrition, trauma, surgery, diabetes, heart failure, or malignancy, particularly if shock is present or impending.
For intermittent IV administration, a solution containing 1 gram or 2 grams in 10 mL of Sterile Water for Injection can be injected over a period of three to five minutes. Cefotaxime should not be administered over a period of less than three minutes. (See WARNINGS). With an infusion system, it may also be given over a longer period of time through the tubing system by which the patient may be receiving other IV solutions. However, during infusion of the solution containing cefotaxime, it is advisable to discontinue temporarily the administration of other solutions at the same site.
For the administration of higher doses by continuous IV infusion, a solution of cefotaxime may be added to IV bottles containing the solutions discussed below.
COMPATIBILITY AND STABILITY
Solutions of cefotaxime reconstituted as described above (DIRECTIONS FOR PROPER USE OF PHARMACY BULK PACKAGE) should be discarded 4 hours after initial entry.
Reconstituted solutions may be further diluted up to 1000 mL with the following solutions and maintain satisfactory potency for 24 hours at or below 22°C, and at least 5 days under refrigeration (at or below 5°C): 0.9% Sodium Chloride Injection; 5 or 10% Dextrose Injection; 5% Dextrose and 0.9% Sodium Chloride Injection, 5% Dextrose and 0.45% Sodium Chloride Injection; 5% Dextrose and 0.2% Sodium Chloride Injection; Lactated Ringer’s Solution; Sodium Lactate Injection (M/6); 10% Invert Sugar Injection, 8.5% TRAVASOL® (Amino Acid) Injection without Electrolytes.
NOTE: Cefotaxime solutions exhibit maximum stability in the pH 5-7 range. Solutions of cefotaxime should not be prepared with diluents having a pH above 7.5, such as Sodium Bicarbonate Injection.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
-
HOW SUPPLIED
Sterile Cefotaxime for Injection, USP, in Pharmacy Bulk Packages, is a dry off-white to pale yellow crystalline powder containing cefotaxime sodium as follows:
10 g cefotaxime (free acid equivalent) in bottles, packages of 1 (NDC 0143-9935-01); NOT FOR DIRECT ADMINISTRATION.
NOTE: Cefotaxime for Injection, USP in the dry state should be stored at 20º to 25°C (68º to 77°F) [See USP Controlled Room Temperature].
The dry material as well as solutions tend to darken depending on storage conditions and should be protected from elevated temperatures and excessive light.
-
REFERENCES
- Cockcroft, D.W. and Gault, M.H.: Prediction of Creatinine Clearance from Serum Creatinine, Nephron 16:31–41, 1976.
Travasol® is manufactured by Clintec.
Manufactured by:
HIKMA FARMACÊUTICA (PORTUGAL), S.A.
Estrada do Rio da Mó, 8, 8A e 8B - Fervença - 2705 – 906 Terrugem SNT, PORTUGAL
Distributed by:
WEST-WARD
A HIKMA COMPANY
Eatontown, NJ 07724 USA
Revised: May 2020
PIN106-WES/6
-
PRINCIPAL DISPLAY PANEL
NDC 0143-9935-01
CEFOTAXIME
FOR INJECTION, USPPHARMACY BULK PACKAGE
NOT FOR DIRECT INFUSION10 grams* per Pharmacy Bulk Package
NOT TO BE DISPENSED AS A UNIT
FOR IV USE ONLY
Rx ONLYThis Pharmacy Bulk Package is intended for preparing IV admixtures only. See
insert for complete dosage information and proper use of the container.*Each vial contains sterile cefotaxime sodium equivalent to 10 g of cefotaxime. The
sodium content is approximately 50.5 mg (2.2 mEq) of sodium per gram of cefotaxime.USUAL DOSAGE: See package insert. Reconstitute with suitable diluents as listed
in the package insert. Shake to dissolve.Storage: Cefotaxime for Injection, USP in the dry state should be stored at 20º to
25ºC (68º to 77ºF) [See USP Controlled Room Temperature]. PROTECT FROM LIGHT.RETAIN IN CARTON UNTIL TIME OF USE.
Withdraw reconstituted contents immediately. However, if it is not possible,
aliquoting operations must be completed within 4 hours of reconstitution.
Discard reconstituted stock solution 4 hours after initial entry.Concentration of the Solution Amount of Diluent 1 g/5 mL 47 mL 1 g/10 mL 97 mL Date Prepared: _________________ Do Not Use After: _____________________________ 
-
INGREDIENTS AND APPEARANCE
CEFOTAXIME
cefotaxime injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0143-9935 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CEFOTAXIME SODIUM (UNII: 258J72S7TZ) (CEFOTAXIME - UNII:N2GI8B1GK7) CEFOTAXIME 10 g Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0143-9935-01 1 in 1 PACKAGE; Type 0: Not a Combination Product 11/20/2002 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065071 11/20/2002 Labeler - Hikma Pharmaceuticals USA Inc. (001230762) Establishment Name Address ID/FEI Business Operations HIKMA FARMACEUTICA (PORTUGAL), S.A 452742943 analysis(0143-9935) , label(0143-9935) , manufacture(0143-9935) , pack(0143-9935)