Label: JOENJA- leniolisib tablet, film coated

- NDC Code(s): 71274-170-60
- Packager: Pharming Healthcare Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated June 5, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use JOENJA safely and effectively. See full prescribing information for JOENJA.
JOENJA® (leniolisib) tablets, for oral use
Initial U.S. Approval: 2023RECENT MAJOR CHANGES
Warnings and Precautions, Hypersensitivity Reactions, Including Anaphylaxis (5.3) 05/2025 INDICATIONS AND USAGE
JOENJA is a kinase inhibitor indicated for the treatment of activated phosphoinositide 3-kinase delta (PI3Kδ) syndrome (APDS) in adult and pediatric patients 12 years of age and older. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 70 mg leniolisib (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Embryo-Fetal Toxicity: JOENJA may cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.1, 8.1, 8.3)
- Vaccinations: Live, attenuated vaccinations may be less effective if administered during JOENJA treatment. (5.2)
- Risk of Hypersensitivty Reactions Including Anaphylaxis: Hypersensitivity reactions including anaphlyaxis have been reported in the postmarketing setting. If hypersensitivity reactions occur, discontinue JOENJA and institute appropriate therapy. (5.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence > 10%) were headache, sinusitis, and atopic dermatitis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pharming Healthcare Inc. at 1-800-930-5221 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to Treatment with JOENJA
2.2 Recommended Dosage and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
5.2 Vaccinations
5.3 Risk of Hypersensitivity Reactions, Including Anaphylaxis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on JOENJA
7.2 Effects of JOENJA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to Treatment with JOENJA
Verify pregnancy status in females of reproductive potential prior to initiating treatment with JOENJA [see Warnings and Precautions (5.1), and Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage and Administration
The recommended dosage of JOENJA in adult and pediatric patients 12 years of age and older weighing 45 kg or greater is 70 mg administered orally twice daily approximately 12 hours apart, with or without food. There is no recommended dosage for patients weighing less than 45 kg.
Advise patients that if a dose is missed by more than 6 hours, wait and take the next dose at the usual time.
Advise patients that if vomiting occurs within 1 hour after taking JOENJA, take JOENJA as soon as possible. If vomiting occurs more than 1 hour after dosing, wait and take the next dose at the usual time.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
Based on findings in animals, JOENJA may cause fetal harm when administered to a pregnant woman. Administration of leniolisib to rats and rabbits during the period of organogenesis caused embryo-fetal toxicity including malformations at exposures that were 2-6 times higher than the maximum recommended human dose (MRHD) in APDS patients based on AUC comparisons. Verify the pregnancy status of patients of reproductive potential prior to starting treatment. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use highly effective methods of contraception during treatment and for 1 week after the last dose [see Dosage and Administration (2.1), Use in Specific Populations (8.1, 8.3)].
5.2 Vaccinations
Live, attenuated vaccinations may be less effective if administered during JOENJA treatment.
5.3 Risk of Hypersensitivity Reactions, Including Anaphylaxis
Hypersensitivity reaction(s), including anaphylaxis, have been reported in postmarketing setting. If a clinically significant hypersensitivity reaction occurs, discontinue JOENJA and institute appropriate therapy [see Adverse Reactions (6.2)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Risk of Hypersensitivity Reactions, Including Anaphylaxis [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of JOENJA reflects exposure based on 38 adult and pediatric patients 12 years of age and older with activated phosphoinositide 3-kinase delta (PI3Kδ) syndrome (APDS) from the placebo-controlled portion of Study 2201 [see Clinical Studies (14)] and additional open-label clinical safety data. Thirty-seven of 38 patients received JOENJA 70 mg orally twice daily for at least 25 weeks and 66% were exposed for 96 weeks or longer. Median duration of JOENJA treatment was approximately 2 years, and 4 patients had more than 5 years of JOENJA exposure.
The data below are based on the 12-week, placebo-controlled portion of Study 2201 in which either JOENJA 70 mg (N=21) or placebo (N=10) was administered twice daily to patients with APDS. Demographics of the patients who participated in this study are summarized in Clinical Studies [see Clinical Studies (14)]. Table 1 presents the number of patients and incidence, rounded to the nearest percent, of adverse reactions that occurred in 2 or more patients treated with JOENJA and for which the incidence in patients treated with JOENJA was greater than the incidence in patients treated with placebo.
The most common adverse reactions (> 10%) were headache, sinusitis, and atopic dermatitis.
Table 1 Adverse Reactions Reported by 2 or More JOENJA-Treated Patients and More Frequently than Placebo Adverse Reactions JOENJA
N=21
n (%)Placebo
N=10
n (%)1Dermatitis atopic: including dermatitis atopic and eczema
2Tachycardia: including tachycardia and sinus tachycardia
Headache 5 (24) 2 (20) Sinusitis 4 (19) 0 Dermatitis atopic1 3 (14) 0 Tachycardia2 2 (10) 0 Diarrhea 2 (10) 0 Fatigue 2 (10) 1 (10) Pyrexia 2 (10) 0 Back pain 2 (10) 0 Neck pain 2 (10) 0 Alopecia 2 (10) 0 6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of JOENJA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: hypersensitivity (anaphylaxis)
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on JOENJA
Strong CYP3A4 Inhibitors
Concomitant use of JOENJA with strong CYP3A4 inhibitors should be avoided.
JOENJA is a substrate of CYP3A4. Leniolisib exposure was increased 2-fold when co-administered with itraconazole, a strong CYP3A4 inhibitor [see Clinical Pharmacology (12.3)].
Strong and Moderate CYP3A4 Inducers
Concomitant use of JOENJA with strong and moderate CYP3A4 inducers should be avoided.
JOENJA is a substrate of CYP3A4. Concomitant use of strong and moderate CYP3A4 inducers may result in reduced leniolisib exposure and thus reduced leniolisib efficacy [see Clinical Pharmacology (12.3)].
7.2 Effects of JOENJA on Other Drugs
BCRP, OATP1B1, and OATP1B3 Substrates
Concomitant use of JOENJA with BCRP, OATP1B1, and OATP1B3 substrates should be avoided.
JOENJA is an inhibitor of BCRP, OATP1B1, and OATP1B3 transporters. Administration of JOENJA increases exposure of BCRP, OATP1B1, and OATP1B3 substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
JOENJA can cause fetal harm based on findings from animal studies. There are no available data on JOENJA use in pregnant women to inform a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
In animal reproduction studies, oral administration of leniolisib to pregnant rats and rabbits during the period of organogenesis at exposures approximately 2-6 times the MRHD on an AUC basis, produced embryofetal toxicity including malformations (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage of clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Leniolisib was administered orally to pregnant rats at doses of 10, 30, and 120 mg/kg/day during the period of organogenesis from gestation Day 6 to Day 17. Leniolisib at a dose of 120 mg/kg/day was associated with decreased fetal body weight, visceral and skeletal variations, and external, visceral, and skeletal malformations (eye bulge, microphthalmia, anophthalmia, and reduction in orbital socket size) in the presence of maternal toxicity (decrease in body weight gain) at exposures approximately 6 times the MRHD on an AUC basis. No developmental toxicity was observed in rats at an exposure approximately 2 times the MRHD (on an AUC basis at a maternal oral dose of 30 mg/kg/day).
Leniolisib was administered orally to pregnant rabbits at doses of 10, 30, and 100 mg/kg/day during the period of organogenesis from gestation Day 7 to Day 20. Leniolisib at a dose of 100 mg/kg/day was associated with skeletal variations as well as visceral and skeletal malformations (microphthalmia and reduction in orbital socket size) in the presence of maternal toxicity (decrease in body weight gain) at exposures approximately 2 times the MRHD on an AUC basis. No developmental toxicity was observed in rabbits at an exposure approximately 0.3 times the MRHD (on an AUC basis at a maternal oral dose of 30 mg/kg/day).
In a pre- and postnatal developmental toxicity study, leniolisib was administered orally to pregnant rats at oral doses of 10, 30, and 90 mg/kg/day from gestation day 7 through postnatal day 21. Leniolisib at a dose of 90 mg/kg/day (approximately 5 times the exposure at the MRHD on an AUC basis) was associated with a slight decrease in the percentage of pups born that survived 21 days postpartum (lactation index) and decreased pup body weights were observed prior to weaning.
8.2 Lactation
Risk Summary
There are no data on the presence of leniolisib or its metabolites in human milk or the effects on the breastfed infant or milk production. Leniolisib is present in rat milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions from leniolisib in the breastfed child, advise women not to breastfeed during treatment with JOENJA and for 1 week after the last dose.
Data
Animal Data
Leniolisib was present in the milk of lactating rats administered oral doses of 10, 30, and 90 mg/kg/day from gestation day 7 through postnatal day 21. Leniolisib concentrations were approximately 2- to 3-fold higher in milk than in maternal plasma. The concentration of leniolisib in animal milk does not necessarily predict the concentration of drug in human milk.
8.3 Females and Males of Reproductive Potential
Based on findings from animal studies, JOENJA may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of JOENJA for the treatment of activated phosphoinositide 3-kinase delta syndrome have been established in pediatric patients 12 years of age and older. Use of JOENJA for this indication is supported by evidence from an adequate and well-controlled study in adult and pediatric patients 12 years of age and older [see Clinical Studies (14)]. There is no recommended dosage for pediatric patients 12 years of age and older who weigh less than 45 kg [see Dosage and Administration (2.2)].
The safety and effectiveness of JOENJA have not been established in pediatric patients below the age of 12 years.
Juvenile Animal Toxicity Data
Studies were conducted with leniolisib in juvenile rats starting at postnatal day (PND) 7 (the equivalent of a human newborn) to PND 77 (the equivalent of a human adult). Death was observed in juvenile rats that received 90 mg/kg/day, approximately 2-4 times the MRHD on an AUC basis and occurred primarily during the pre-weaning period (PND 9 to PND 15). Changes in the onset of puberty (delays in males and accelerations in females) were observed in juvenile rats at equal to or greater than 30 mg/kg/day leniolisib, which is one half to equivalent to, the MRHD on an AUC basis. A no effect dose level was identified at an exposure approximately 0.2 times the MRHD on an AUC basis.
8.5 Geriatric Use
Because clinical studies of JOENJA did not include any patients 65 years of age and older, it cannot be determined whether they respond differently from younger adult patients.
8.6 Hepatic Impairment
Leniolisib is extensively (60%) metabolized by the liver. The effect of hepatic impairment on the pharmacokinetics of leniolisib has not been studied. The use of JOENJA in patients with moderate to severe hepatic impairment is not recommended [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
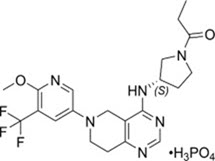
Leniolisib is a kinase inhibitor. The chemical name for leniolisib phosphate is 1-[(3S)-3-[[5,6,7,8-Tetrahydro-6-[6-methoxy-5-(trifluoromethyl)-3-pyridinyl]pyrido[4,3-d]pyrimidin-4-yl]amino]-1-pyrrolidinyl]-1-propanone phosphate (1:1).
Leniolisib phosphate has the following structural formula:
The molecular formula is C21H25F3N6O2•H3PO4 and the molecular weight is 450.47 g/mol for the free base, 548.46 g/mol for the phosphate salt.
Leniolisib phosphate is a white to yellowish to yellowish-greenish powder. The aqueous solubility of leniolisib phosphate is pH dependent with decreasing solubility observed with increasing pH.
JOENJA film-coated tablets are for oral administration. Each tablet contains 70 mg of leniolisib (equivalent to 85.26 mg leniolisib phosphate) with the following inactive ingredients: colloidal silicon dioxide, hydroxypropyl methylcellulose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The tablet filmcoating contains hydroxypropyl methylcellulose, iron oxide red, iron oxide yellow, polyethylene glycol, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Leniolisib inhibits PI3K-delta by blocking the active binding site of PI3K-delta. In cell-free isolated enzyme assays, leniolisib was selective for PI3K-delta over PI3K-alpha (28-fold), PI3K-beta (43-fold), and PI3K-gamma (257-fold), as well as the broader kinome. In cell-based assays, leniolisib reduced pAkt pathway activity and inhibited proliferation and activation of B and T cell subsets. Gain-of-function variants in the gene encoding the p110-delta catalytic subunit or loss of function variants in the gene encoding the p85-alpha regulatory subunit each cause hyperactivity of PI3K-delta. Leniolisib inhibits the signalling pathways that lead to increased production of PIP3, hyperactivity of the downstream mTOR/Akt pathway, and to the dysregulation of B and T cells.
12.2 Pharmacodynamics
Ex vivo pharmacodynamics of leniolisib [proportion of phosphorylated Akt (pAkt)-positive B cells] were assessed intra-individually at 10, 30, and 70 mg twice daily for 4 weeks at each dose level in patients with APDS. Within the explored dose range, higher leniolisib plasma concentrations were generally associated with higher reduction of pAkt-positive B cells and higher doses were associated with a slightly higher peak reduction as well as more sustained reduction. Treatment with JOENJA 70 mg twice a day at steady state is estimated to produce time-averaged reduction of pAkt-positive B cells by approximately 80%.
12.3 Pharmacokinetics
The systemic drug exposure (AUC and Cmax) of leniolisib increased dose proportionally within the studied range of doses (20 to 140 mg twice a day dosing and single doses of 10 to 400 mg). During twice daily dosing approximately 12 hours apart, leniolisib accumulates approximately 1.4-fold (range of 1.0 to 2.2) in achieving steady-state, consistent with an effective half-life (t1/2) of approximately 7 hours. Steady state drug concentrations can be expected to be reached after approximately 2 to 3 days of JOENJA treatment. The pharmacokinetics of leniolisib are similar between healthy participants and APDS patients.
Absorption
In a placebo controlled, single and multiple ascending dose study in healthy participants, leniolisib median time to maximum plasma concentration (Tmax) occurred at about 1 hour postdose. Tmax appeared independent of dose and was not altered after multiple oral doses. Food is unlikely to have a clinically meaningful effect on the systemic exposure of leniolisib during JOENJA treatment.
Distribution
The systemic decay in leniolisib plasma concentration over time is bi-exponential, indicating a distribution delay towards peripheral tissues. The apparent terminal elimination t1/2 is approximately 10 hours. The volume of distribution of leniolisib is estimated to be 28.5 L in patients with APDS. Leniolisib was highly bound (94.5%) to plasma proteins.
Elimination
The mean recovery of total 14C-radioactivity following a single oral dose of 70 mg 14C-leniolisib was 92.5% (67.0% and 25.5% recovered via feces and urine, respectively) 168 hours postdose. Unchanged leniolisib (6.32%) was the predominant drug-related material recovered in urine.
Metabolism
Leniolisib was 60% metabolized by the liver, with CYP3A4 being the most predominant enzyme involved (95.4%) in the primary oxidative metabolism of leniolisib with minor contribution from other enzymes (3.5% CYP3A5, 0.7% CYP1A2 and 0.4% CYP2D6). Intestinal secretion by BCRP as well as extrahepatic CYP1A1 cannot be excluded as excretion routes.
Specific Populations
Pediatric Patients
Following a single 70 mg oral dose of leniolisib in APDS patients, leniolisib systemic exposures were comparable between pediatric patients (12 to 17 years of age) and adults (≥ 18 years of age), with median Tmax (ranging from 1 to 5 hours) reached approximately 3 hours post-dose in patients 12 to 17 years of age. The observed difference in the median Tmax between pediatric patients (12 to 17 years of age) and adults (≥ 18 years of age) is not clinically relevant given the PK variability and comparable concentration-time profiles between the two age groups.
Patients with Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of leniolisib has not been evaluated. As leniolisib is metabolized to a large extent by the liver (60%), use of JOENJA is not recommended in patients with moderate to severe hepatic impairment [see Specific Populations (8.6)].
Drug Interaction Studies
Strong CYP3A4 Inhibitors: Leniolisib-exposure was increased approximately 2-fold when administered with itraconazole (strong CYP3A4 inhibitor).
Moderate CYP3A4 Inhibitors: Physiological based pharmacokinetic (PBPK) model-based simulations predicted a maximum increase of 75% in leniolisib AUC0-12 with erythromycin (moderate CYP3A4 inhibitor).
CYP3A4 Inducers: PBPK model-based simulations predicted a maximum decrease of 78% and 58% in leniolisib AUC0-12 with rifampin (strong CYP3A4 inducer) and efavirenz (moderate CYP3A4 inducer), respectively.
CYP2D6 and P-gp inhibitors: Quinidine (strong P-gp and CYP2D6 inhibitor) had no effect on leniolisib systemic exposure. Leniolisib is not a sensitive substrate of P-gp and CYP2D6.
Oral Contraceptives: When combined with a monophasic oral contraceptive containing levonorgestrel and ethinylestradiol, leniolisib increased ethinylestradiol exposure by approximately 25 to 30% in terms of both AUC and Cmax, but did not affect the Cmax or AUC of levonorgestrel. Efficacy of a combined oral contraceptive composed of ethinylestradiol and levonorgestrel is not expected to be compromised by concomitant use with leniolisib.
Gastric Acid Reducing Agents: Leniolisib exhibits pH-dependent solubility (pH range of 1.2 to 4), with low solubility at higher pH values (≥ 5). However, PK results from APDS patients did not indicate that acid reducing agents (e.g., H2-antagonists, proton pump inhibitors) have a clinically relevant effect on leniolisib systemic exposure.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Leniolisib was not carcinogenic in the 6-month carcinogenicity study in Tg.rasH2 transgenic mice at doses up to 80 mg/kg/day.
Leniolisib was not genotoxic in the in vitro Ames assay, in vitro chromosomal aberration assay in human lymphocytes, or micronucleus assays in TK6 cells (in vitro) and rats (in vivo).
In a fertility study, male rats had decreased round spermatids and decreased spermatocytes in the testis at an oral dose of 90 mg/kg/day (approximately 2 times the MRHD on an AUC basis). Leniolisib had no effect on fertility in female rats at oral doses up to 90 mg/kg/day (approximately 4 times the MRHD on an AUC basis). No effects on male or female fertility and reproductive performance indices were observed up to the maximum dose administered of 90 mg/kg/day (approximately 2-4 times the MRHD on an AUC basis).
-
14 CLINICAL STUDIES
The efficacy of JOENJA was evaluated in the placebo-controlled portion of Study 2201 (NCT02435173), a 12-week blinded, randomized, placebo-controlled study in adult and pediatric patients 12 years of age and older with confirmed APDS-associated genetic PI3Kδ mutation with a documented variant in either PIK3CD or PIK3R1. Baseline patient demographics are shown in Table 2.
Table 2 Baseline Demographic and Disease Characteristics in Patients with APDS (Study 2201) Demographics and Disease Characteristics JOENJA
(N=21)Placebo
(N=10)Demographics 1Patient age from study Day -4 up to initial JOENJA dosing
Age1(Years) Mean (SD) 22.2 (10.00) 26.7 (13.43) Age Categories
< 18, n (%)
(Min, Max)
≥ 18, n (%)
(Min, Max)
8 (38)
(12, 17)
13 (62)
(18, 54)
4 (40)
(15, 17)
6 (60)
(18, 48)Sex, n (%)
Male
Female
11 (52)
10 (48)
4 (40)
6 (60)Race, n (%)
Asian
Black
White
Other
1 (5)
1 (5)
18 (86)
1 (5)
1 (10)
1 (10)
7 (70)
1 (10)Ethnicity, n (%)
Hispanic or Latino
Not Hispanic or Latino
Not reported
0
14 (67)
7 (33)
1 (10)
7 (70)
2 (20)Disease Characteristics APDS 1 (PIK3CD variant), n (%) 16 (76) 9 (90) APDS 2 (PIK3R1 variant), n (%) 5 (24) 1 (10) Concomitant glucocorticoids, n (%) 12 (57) 6 (60) Concomitant immunoglobulin G (IgG), n (%) 14 (67) 7 (70) Previous rapamycin/sirolimus use, n (%) 4 (19) 3 (30) Patients had nodal and/or extranodal lymphoproliferation, as measured by index nodal lesion selected by the Cheson methodology on CT or MRI and clinical findings and manifestations compatible with APDS (e.g., history of repeated oto-sino-pulmonary infections, organ dysfunction). Immunosuppressive medications or PI3Kδ inhibitors (selective or non-selective) were prohibited within 6 weeks of baseline (Day -1 and the visit prior to first study drug administration) and throughout the study. In addition, patients who had previous or concurrent B cell depleters (e.g., rituximab) within 6 months of baseline were excluded from the study, unless absolute B lymphocytes in the blood were normal. B cell depleters were prohibited throughout the study.
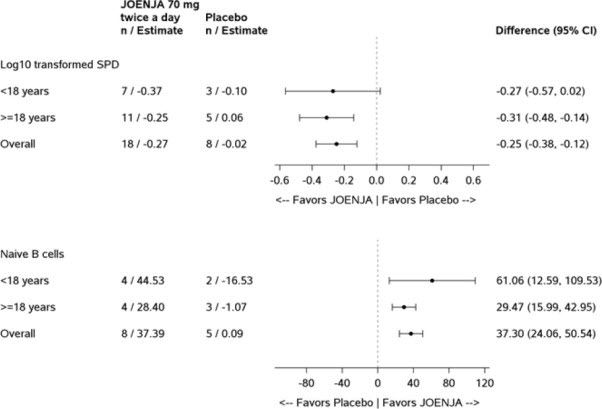
Thirty-one patients were randomized 2:1 to receive either JOENJA 70 mg (N=21) or placebo (N=10) twice a day for 12 weeks. The co-primary efficacy endpoints were improvement in lymphoproliferation as measured by a change from baseline in lymphadenopathy measured by the log10-transformed sum of product diameters and the normalization of immunophenotype as measured by the percentage of naïve B cells out of total B cells. Both co-primary efficacy endpoints were statistically significant (Table 3).
Table 3 Co-primary Endpoints in Placebo-Controlled Portion of Study 2201 at Week 12 (Day 85) JOENJA
(N=21)Placebo
(N=10)CI=confidence interval; SD= Standard deviation; SE=standard error; SPD=sum of product diameters; vs=versus; LS Mean: Least-squares mean
Note: The LS mean change from baseline, difference in LS mean change from baseline between JOENJA and placebo and its p-value were obtained from an Analysis of Covariance model with treatment, glucocorticoids use and immunoglobulin replacement therapy at baseline, and baseline measurement as covariates.
aChange in index lesion size was measured using the log10 transformed sum of the product of diameters (SPD) of the largest lymph nodes (maximum of 6) identified as per the Cheson criteria on CT/MRI.
bThe analysis excluded 2 patients from each treatment group due to protocol deviations and 1 JOENJA patient having complete resolution of the index lesion identified at baseline.
cCell surface markers used to distinguish naïve B cells on flow cytometry were CD19+ CD27- CD10-.
dThe analysis excluded 2 patients from each treatment group due to protocol deviations, 5 JOENJA patients and 3 placebo patients with more than or equal to 48% naïve B cells at baseline, 5 JOENJA patients with no Day 85 measurement, and 1 JOENJA patient with no baseline measurement.
eBaseline is defined as the arithmetic mean of the Baseline and Day 1 values when both were available, and if either value was missing, the existing value was used.
Log10-Transformed SPD of Index Lesions (Excluding Patients with 0 Lesions at Baseline)a nb 18 8 Baseline Mean (SD) 3.03 (0.42) 3.05 (0.39) Change from Baseline, LS Mean (SE) -0.27 (0.04) -0.02 (0.05) Difference vs. Placebo (95% CI) -0.25 (-0.38, -0.12) p-value 0.0006 Percentage of Naïve B Cells out of Total B Cells (Patients with < 48% of Naïve B Cells at Baseline)c nd 8 5 Baselinee Mean (SD) 27.16 (13.16) 30.51 (7.97) Change from Baseline, LS Mean (SE) 37.39 (5.34) 0.09 (6.66) Difference vs. Placebo (95% CI) 37.30 (24.06, 50.54) p-value 0.0002 Figure 1 represents the co-primary endpoints grouped by age (< 18 years of age vs ≥ 18 years of age).
Figure 1 Difference from Baseline in Log10 Transformed SPD of Index Lesions and Percentage of Naïve B Cells out of Total B Cells (Patients with < 48% of Naïve B Cells at Baseline) by Age Group
-
16 HOW SUPPLIED/STORAGE AND HANDLING
JOENJA is available in 70 mg tablet: yellow, oval-shaped, biconvex, bevelled edge film-coated tablet debossed with "70" on one side and "LNB" on the other side. It is supplied in bottles with a child-resistant cap of 60 tablets (NDC 71274-170-60).
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Do not refrigerate.
-
17 PATIENT COUNSELING INFORMATION
Embryo-Fetal Toxicity
- Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy. [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1, 8.3)].
- Advise females of reproductive potential to use highly effective contraceptive during treatment with JOENJA and for 1 week after the last dose [see Warnings and Precautions (5.1) and Use in Specific Populations (8.3)].
Vaccinations
Instruct patients to inform the healthcare providers that they are taking JOENJA prior to a potential vaccination [see Warnings and Precautions (5.2)].
Risk of Hypersensitivity Reactions, Including Anaphylaxis
Advise patients to discontinue JOENJA and to seek immediate medical attention if they develop any signs and symptoms of serious allergic reactions [see Warnings and Precautions (5.3)].
Lactation
Advise women not to breastfeed during treatment with JOENJA and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Administration
Advise patients JOENJA may be taken with or without food [see Dosage and Administration (2.2)].
Instruct patients that if they miss a dose by more than 6 hours, skip the missed dose. Advise patients to take the next dose as scheduled [see Dosage and Administration (2.2)].
Instruct patients that if vomiting occurs within 1 hour after taking JOENJA, take a dose as soon as possible [see Dosage and Administration (2.2)].
Manufactured for:
Pharming Healthcare Inc.,
10 Independence Blvd.
Warren, NJ USA 07059JOENJA is a trademark of Pharming Intellectual Property B.V.
©2025 Pharming Technologies B.V. All rights reserved.
AC2004703US-0850
- PRINCIPAL DISPLAY PANEL - 70 mg Tablet Bottle Carton
-
INGREDIENTS AND APPEARANCE
JOENJA
leniolisib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71274-170 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Leniolisib phosphate (UNII: 3700M1H39Q) (Leniolisib - UNII:L22772Z9CP) Leniolisib 70 mg Product Characteristics Color yellow (yellow) Score no score Shape OVAL (OVAL) Size 16mm Flavor Imprint Code LNB;70 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71274-170-60 1 in 1 CARTON 03/29/2023 1 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA217759 03/29/2023 Labeler - Pharming Healthcare Inc. (073255908)