Label: ERIVEDGE- vismodegib capsule

- NDC Code(s): 50242-140-01, 50242-140-86
- Packager: Genentech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated December 2, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ERIVEDGE safely and effectively. See full prescribing information for ERIVEDGE.
ERIVEDGE® (vismodegib) capsules, for oral use
Initial U.S. Approval: 2012WARNING: EMBRYO-FETAL TOXICITY
See full prescribing information for complete boxed warning.
- ERIVEDGE can cause embryo-fetal death or severe birth defects when administered to a pregnant woman. ERIVEDGE is embryotoxic, fetotoxic, and teratogenic in animals. Teratogenic effects included severe midline defects, missing digits, and other irreversible malformations. (5.1, 8.1)
- Verify the pregnancy status of females of reproductive potential within 7 days prior to initiating ERIVEDGE. Advise pregnant women of the potential risks to a fetus. Advise females of reproductive potential to use effective contraception during and after ERIVEDGE. (2.1, 5.1, 8.1, 8.3)
- Advise males of the potential risk of ERIVEDGE exposure through semen and to use condoms with a pregnant partner or a female partner of reproductive potential. (5.1, 8.3)
INDICATIONS AND USAGE
ERIVEDGE® (vismodegib) is a hedgehog pathway inhibitor indicated for the treatment of adults with metastatic basal cell carcinoma, or with locally advanced basal cell carcinoma that has recurred following surgery or who are not candidates for surgery and who are not candidates for radiation. (1)
DOSAGE AND ADMINISTRATION
The recommended dosage is 150 mg orally once daily. (2)
DOSAGE FORMS AND STRENGTHS
150 mg capsules. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
- The most common adverse reactions (incidence of ≥ 10%) are muscle spasms, alopecia, dysgeusia, weight loss, fatigue, nausea, diarrhea, decreased appetite, constipation, arthralgias, vomiting, and ageusia.
To report SUSPECTED ADVERSE REACTIONS, contact Genentech, Inc. at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: EMBRYO-FETAL TOXICITY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Safety Information
2.2 Recommended Dosage
2.3 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
5.2 Severe Cutaneous Adverse Reactions
5.3 Musculoskeletal Adverse Reactions
5.4 Premature Fusion of the Epiphyses
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: EMBRYO-FETAL TOXICITY
- ERIVEDGE can cause embryo-fetal death or severe birth defects when administered to a pregnant woman. ERIVEDGE is embryotoxic, fetotoxic, and teratogenic in animals. Teratogenic effects included severe midline defects, missing digits, and other irreversible malformations [see Warnings and Precautions (5.1), Use in Specific Populations (8.1)].
- Verify the pregnancy status of females of reproductive potential within 7 days prior to initiating ERIVEDGE. Advise pregnant women of the potential risks to a fetus. Advise females of reproductive potential to use effective contraception during and after ERIVEDGE [see Dosage and Administration (2.1), Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
- Advise males of the potential risk of ERIVEDGE exposure through semen and to use condoms with a pregnant partner or a female partner of reproductive potential [see Warnings and Precautions (5.1), Use in Specific Populations (8.3)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Safety Information
Verify pregnancy status of females of reproductive potential within 7 days prior to initiating ERIVEDGE [see Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage
The recommended dosage of ERIVEDGE is 150 mg taken orally once daily, with or without food, until disease progression or until unacceptable toxicity.
Swallow capsules whole. Do not open or crush capsules.
If a dose of ERIVEDGE is missed, resume dosing with the next scheduled dose.
2.3 Dosage Modifications for Adverse Reactions
Withhold ERIVEDGE for up to 8 weeks for intolerable adverse reactions until improvement or resolution. Treatment durations shorter than 8 weeks prior to interruptions have not been studied.
Permanently discontinue ERIVEDGE if patients experience severe cutaneous adverse reactions (SCARs) including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), or drug reaction with eosinophilia and systemic symptoms (DRESS) [see Warnings and Precautions (5.2)].
Interrupt ERIVEDGE for severe or intolerable musculoskeletal adverse reactions. Permanently discontinue ERIVEDGE for recurrent, severe or intolerable musculoskeletal adverse reactions [see Warnings and Precautions (5.3)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
Based on its mechanism of action, ERIVEDGE can cause embryo-fetal death or severe birth defects when administered to a pregnant woman. In animal reproduction studies, vismodegib was embryotoxic, fetotoxic, and teratogenic at maternal exposures lower than the human exposures at the recommended dose of 150 mg once daily [see Use in Specific Populations (8.1)].
Females of Reproductive Potential
Verify the pregnancy status of females of reproductive potential within 7 days prior to initiating ERIVEDGE. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during therapy with ERIVEDGE and for 24 months after the final dose [see Use in Specific Populations (8.1, 8.3)].
Males
Vismodegib is present in semen. It is not known if the amount of vismodegib in semen can cause embryo-fetal harm. Advise males to use condoms, even after a vasectomy, to avoid potential drug exposure in pregnant partners and female partners of reproductive potential during therapy and for 3 months after the final dose of ERIVEDGE. Advise male patients not to donate semen during and for 3 months after the final dose of ERIVEDGE [see Use in Specific Populations (8.3)].
5.2 Severe Cutaneous Adverse Reactions
Severe cutaneous adverse reactions (SCARs), including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS), which can be life-threatening or fatal, have been reported during treatment with ERIVEDGE [see Adverse Reactions (6.2)].
Permanently discontinue ERIVEDGE in patients with these reactions [see Dosage and Administration (2.3)].
5.3 Musculoskeletal Adverse Reactions
Musculoskeletal adverse reactions, which may be accompanied by serum creatine phosphokinase (CPK) elevations, have occurred with ERIVEDGE and other drugs which inhibit the hedgehog (Hh) pathway. In the pooled safety population in clinical trials of ERIVEDGE, musculoskeletal and connective tissue adverse reactions occurred in 78% of patients treated, with 7% (9/138) reported as Grade 3. The most frequent manifestations of musculoskeletal and connective tissue adverse reactions (all grades) reported were muscle spasms (72%) and arthralgias (16%). In a post-approval clinical trial of 1232 patients, Grade 3 or 4 elevations in serum CPK laboratory values occurred in 2.4% of the 453 patients who had any CPK measurement [see Adverse Reactions (6.1)].
Obtain baseline serum creatine phosphokinase (CPK) and creatinine levels and as clinically indicated (e.g., if muscle symptoms are reported). Depending on the severity of symptoms, temporary dose interruption or discontinuation may be required for musculoskeletal adverse reactions or serum CPK elevation [see Dosage and Administration (2.3)].
5.4 Premature Fusion of the Epiphyses
Premature fusion of the epiphyses has been reported in pediatric patients exposed to ERIVEDGE. In some cases, fusion progressed after drug discontinuation [see Use in Specific Populations (8.4)]. ERIVEDGE is not indicated for pediatric patients.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.1)]
- Severe Cutaneous Adverse Reactions [see Warnings and Precautions (5.2)]
- Musculoskeletal Adverse Reactions [see Warnings and Precautions (5.3)]
- Premature Fusion of the Epiphyses [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety data described below reflect exposure to ERIVEDGE in 138 patients with advanced basal cell carcinoma (BCC) who received ERIVEDGE at doses ≥ 150 mg orally daily in four open-label, uncontrolled, dose-ranging or fixed single dose clinical trials [Study SHH3925g, SHH4437g, SHH4476g and SHH4610g]. The median age of these patients was 61 years (range 21 to 101 years), 100% were White (including Hispanics), and 64% were male. The median duration of treatment was approximately 10 months (range 21 days to 36 months); 111 patients received ERIVEDGE for 6 months or longer.
The most common adverse reactions (≥ 10%) were muscle spasms, alopecia, dysgeusia, weight loss, fatigue, nausea, diarrhea, decreased appetite, constipation, arthralgias, vomiting, and ageusia (Table 1).
Table 1: Adverse Reactions Occurring in ≥ 10% of Patients with Advanced Basal Cell Carcinoma Adverse Reaction ERIVEDGE
(N = 138)All Grades* (%) Grade 3 (%) Grade 4 (%) - *
- Grading according to National Cancer Institute-Common Terminology Criteria for Adverse Events version 3.0.
Gastrointestinal Nausea 30% 0.7% - Diarrhea 29% 0.7% - Constipation 21% - - Vomiting 14% - - General Fatigue 40% 5% 0.7% Investigations Weight loss 45% 7% - Metabolism and nutrition Decreased appetite 25% 2.2% - Musculoskeletal and connective tissue Muscle spasms 72% 3.6% - Arthralgias 16% 0.7% Nervous system Dysgeusia 55% - - Ageusia 11% - - Skin and subcutaneous tissue Alopecia 64% - - Amenorrhea
Among patients from the clinical trials included in the pooled safety data analysis, 30% of 10 pre-menopausal women developed amenorrhea while receiving ERIVEDGE.
Laboratory Abnormalities
Grade 3 laboratory abnormalities observed in clinical trials were hyponatremia (4%), azotemia (2%) and hypokalemia (1%).
Additionally, in a post-approval clinical trial conducted in 1232 patients with locally advanced or metastatic BCC treated with ERIVEDGE, a subset of 29 patients had baseline values for blood creatine phosphokinase (CPK) reported. Within this subset of patients, 38% had a shift from baseline, including Grade 3 (3%) increased CPK. Grade 3 or 4 increased CPK occurred in 2.4% of the 453 patients across the entire study population with any CPK measurement.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ERIVEDGE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hepatobiliary disorders: Drug-induced liver injury
Skin and subcutaneous tissue disorders: Stevens-Johnson syndrome/toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms [see Warnings and Precautions (5.2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ERIVEDGE during pregnancy. Report pregnancies to Genentech at 1-888-835-2555.
Risk Summary
Based on its mechanism of action and findings from animal reproduction studies, ERIVEDGE can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. In animal reproduction studies, oral administration of vismodegib during organogenesis at doses below the 150 mg clinical dose resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats (see Data). There are no human data on the use of ERIVEDGE in pregnant women. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal toxicity study, pregnant rats were administered vismodegib orally at doses of 10, 60, or 300 mg/kg/day during the period of organogenesis. Pre- and post-implantation loss were increased at doses of ≥ 60 mg/kg/day [approximately 2 times the human exposure at the 150 mg clinical dose based on area under the curve (AUC)], which included early resorption of 100% of the fetuses. A dose of 10 mg/kg/day [approximately 0.2 times the human exposure (AUC) at the recommended 150 mg clinical dose] resulted in malformations (including missing and/or fused digits, open perineum and craniofacial anomalies) and retardations or variations (including dilated renal pelvis, dilated ureter, and incompletely or unossified sternal elements, centra of vertebrae, or proximal phalanges and claws).
8.2 Lactation
No data are available regarding the presence of vismodegib in human milk, the effects of the drug on the breastfed child, or the effects of the drug on milk production. Because of the potential for serious adverse reactions in breastfed infants from ERIVEDGE, advise women that breastfeeding is not recommended during therapy with ERIVEDGE and for 24 months after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential within 7 days prior to initiating ERIVEDGE.
Contraception
Based on its mechanism of action and animal data, ERIVEDGE can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise females of reproductive potential to use effective contraception during therapy with ERIVEDGE and for 24 months after the final dose.
Males
Vismodegib is present in semen [see Clinical Pharmacology (12.3)]. It is not known if the amount of vismodegib in semen can cause embryo-fetal harm. Advise male patients to use condoms, even after a vasectomy, to avoid drug exposure to pregnant partners and female partners of reproductive potential during therapy with and for 3 months after the final dose of ERIVEDGE. Advise males of the potential risk to an embryo or fetus if a female partner of reproductive potential is exposed to ERIVEDGE. Advise males not to donate semen during therapy with ERIVEDGE and for 3 months after the final dose.
Infertility
Females
Amenorrhea can occur in females of reproductive potential. Reversibility of amenorrhea is unknown [see Adverse Reactions (6.1)].
8.4 Pediatric Use
The safety and effectiveness of ERIVEDGE have not been established in pediatric patients.
Premature fusion of the epiphyses [see Warnings and Precautions (5.3)] and precocious puberty have been reported in pediatric patients exposed to ERIVEDGE. In some cases, epiphyseal fusion progressed after drug discontinuation.
Juvenile Animal Toxicity Data
In repeat-dose toxicology studies in rats, administration of oral vismodegib resulted in toxicities in bone and teeth. Effects on bone consisted of closure of the epiphyseal growth plate when oral vismodegib was administered for 26 weeks at ≥ 50 mg/kg/day (approximately ≥ 0.4 times the human exposure (AUC) at the 150 mg clinical dose). Abnormalities in growing incisor teeth (including degeneration/necrosis of odontoblasts, formation of fluid-filled cysts in the dental pulp, ossification of the root canal, and hemorrhage resulting in breakage or loss of teeth) were observed after administration of oral vismodegib at ≥ 15 mg/kg/day (approximately ≥ 0.2 times the human exposure (AUC) at the 150 mg clinical dose).
8.5 Geriatric Use
Clinical studies of ERIVEDGE did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients.
8.6 Hepatic Impairment
No dose adjustment is required in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dose adjustment is required in patients with renal impairment [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
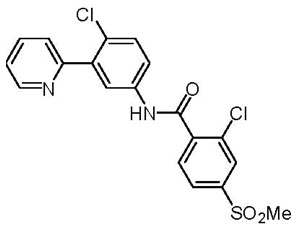
Vismodegib is a hedgehog (Hh) pathway inhibitor, which is described chemically as 2-Chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide. The molecular formula is C19H14Cl2N2O3S. The molecular weight is 421.3 g/mol and the structural formula is:
Vismodegib is a crystalline free base with a pKa (pyridinium cation) of 3.8, appearing as a white to tan powder. The solubility of vismodegib is pH dependent with 0.1 µg/mL at pH 7 and 0.99 mg/mL at pH 1. The partition coefficient (log P) is 2.7.
ERIVEDGE (vismodegib) for oral administration is supplied in capsules containing 150 mg vismodegib and the following inactive ingredients: microcrystalline cellulose, lactose monohydrate, sodium lauryl sulfate, povidone, sodium starch glycolate, talc, and magnesium stearate (non-bovine). The capsule shell contains gelatin, titanium dioxide, red iron oxide, and black iron oxide. The black printing ink contains shellac and black iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Vismodegib is an inhibitor of the Hedgehog pathway. Vismodegib binds to and inhibits Smoothened, a transmembrane protein involved in Hedgehog signal transduction.
12.3 Pharmacokinetics
The pharmacokinetics of vismodegib were studied in healthy subjects and patients. Following daily oral dosing, the pharmacokinetics of vismodegib appear to be nonlinear with steady state achieved within 7 days. Increasing the dose from 150 mg to 540 mg (1 to 3.6 times the recommended dose) does not result in higher steady state plasma concentrations. Average plasma concentration of vismodegib at steady state (Css,avg) is ~23 µM following 150 mg once daily dose.
Absorption
The single dose absolute bioavailability of vismodegib is 32%. Absorption is saturable as evidenced by the lack of dose proportional increase in exposure after a single dose of 270 mg or 540 mg vismodegib.
Distribution
The volume of distribution of vismodegib ranges from 16.4 to 26.6 L. Vismodegib plasma protein binding in patients is > 99%. Vismodegib binds to both human serum albumin and alpha-1-acid glycoprotein (AAG) and binding to AAG is saturable.
Male patients had an average concentration of vismodegib in semen on day 8 that was 6.5% of the average steady state concentration (Css) observed in plasma.
Elimination
The estimated elimination half-life (t1/2) of vismodegib is 4 days after continuous once-daily dosing and 12 days after a single dose.
Metabolism
Greater than 98% of the total circulating drug-related components are the parent drug. Metabolic pathways of vismodegib in humans include oxidation, glucuronidation, and pyridine ring cleavage. The two most abundant oxidative metabolites recovered in feces are produced in vitro by recombinant CYP2C9 and CYP3A.
Specific Populations
Weight (41-140 kg), age (26-89 years), sex, mild to moderate renal impairment (creatinine clearance of 30 to 79 mL/min), mild hepatic impairment (normal total bilirubin and aspartate transaminase (AST) > upper limit of normal (ULN) or total bilirubin > 1 to 1.5 times ULN), moderate hepatic impairment (total bilirubin > 1.5 to 3 times ULN), or severe hepatic impairment (total bilirubin > 3 to 10 times ULN) had no clinically relevant effects on the systemic exposure of vismodegib. The impact of severe renal impairment on the pharmacokinetics of vismodegib is unknown.
Drug Interaction Studies
Clinical Studies
No clinically significant differences in vismodegib pharmacokinetics were observed when used concomitantly with fluconazole (moderate CYP2C9 and CYP3A4 inhibitor), itraconazole (strong CYP3A4 inhibitor and P-gp inhibitor) and rabeprazole (gastric acid reducing agent, proton pump inhibitor).
No clinically significant differences in the pharmacokinetics of the following drugs were observed when used concomitantly with vismodegib: rosiglitazone (a CYP2C8 substrate) or ethinyl estradiol and norethindrone (oral contraceptive).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies were performed in mice and rats. No carcinogenic potential was identified in either species. Vismodegib was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay and was not clastogenic in the in vitro human chromosomal aberration assay in human peripheral blood lymphocytes or in the in vivo rat bone marrow micronucleus assay.
In a dedicated 26-week rat fertility study, no effects on male reproductive organs or fertility endpoints were observed at vismodegib doses of 100 mg/kg/day [approximately 1.3 times the human exposure (steady state AUC0-24hr) at the 150 mg clinical dose] either at the end of dosing or following a 16-week recovery phase. While there were increased numbers of degenerating germ cells and hypospermia in sexually immature dogs observed at ≥ 50 mg/kg/day in the 4-week general toxicity study, there were no effects on male reproductive organs in sexually mature rats and dogs, in the vismodegib general toxicity studies of up to 26-weeks.
In a female fertility study, treatment of rats with vismodegib at 100 mg/kg/day [approximately 1.2-times the human exposure (steady state AUC0-24hr) at the 150 mg clinical dose] for 26-weeks prior to mating resulted in decreased implantations, increased percent preimplantation loss, and decreased numbers of dams with viable embryos. No vismodegib-related changes in fertility were observed following a 16-week recovery period. In a 26-week general toxicity study in rats, decreased numbers of corpora lutea were observed at 100 mg/kg/day; the effect was not reversed by the end of an 8-week recovery period.
13.2 Animal Toxicology and/or Pharmacology
Neurologic effects characterized as limb or body tremors or twitching were observed in rats administered oral vismodegib for 4 weeks or longer at ≥ 50 mg/kg/day (approximately ≥ 0.4 times the human exposure (AUC) at the 150 mg clinical dose). These observations resolved upon discontinuation of dosing and were not associated with microscopic findings.
-
14 CLINICAL STUDIES
A single, international, single-arm, multi-center, open-label, 2-cohort trial [SHH4476g (NCT00833417)] was conducted in 104 patients with either metastatic basal cell carcinoma (mBCC) (n = 33) or locally advanced BCC (laBCC) (n = 71). Patients with laBCC were required to have lesions that had recurred after radiotherapy, unless radiotherapy was contraindicated or inappropriate (e.g. Gorlin syndrome; limitations because of location of tumor or cumulative prior radiotherapy dose), and where the lesions were either unresectable or surgical resection would result in substantial deformity. Patients were to receive ERIVEDGE 150 mg orally once daily until disease progression or unacceptable toxicity.
The major efficacy outcome measure was objective response rate (ORR) as assessed by an independent review facility (IRF). In the mBCC cohort, tumor response was assessed according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0. In the laBCC cohort, tumor response evaluation included measurement of externally assessable tumor (including scar) and assessment for ulceration in photographs, radiographic assessment of target lesions (if appropriate), and tumor biopsy. An objective response in laBCC required at least one of the following criteria and absence of any criterion for disease progression: (1) ≥ 30% reduction in lesion size [sum of the longest diameter (SLD)] from baseline in target lesions by radiographic assessment; (2) ≥ 30% reduction in SLD from baseline in externally visible dimension of target lesions; (3) complete resolution of ulceration in all target lesions. Complete response was defined as objective response (as defined above) with no residual BCC on sampling tumor biopsy. Disease progression was defined as any of the following: (1) ≥ 20% increase in the SLD from nadir in target lesions (either by radiography or by externally visible dimension); (2) new ulceration of target lesions persisting without evidence of healing for at least 2 weeks; (3) new lesions by radiographic assessment or physical examination; (4) progression of non-target lesions by RECIST.
Of the 104 patients enrolled, 96 patients were evaluable for ORR. Twenty-one percent of patients carried a diagnosis of Gorlin syndrome. The median age of the efficacy evaluable population was 62 years (46% were at least 65 years old), 61% male and 100% White. For the mBCC cohort (n = 33), 97% of patients had prior therapy including surgery (97%), radiotherapy (58%), and systemic therapies (30%). For the laBCC cohort (n = 63), 94% of patients had prior therapies including surgery (89%), radiotherapy (27%), and systemic/topical therapies (11%). The median duration of treatment was 10.2 months (range 0.7 to 18.7 months).
The efficacy results are presented in Table 2.
Table 2: Efficacy Results for Evaluable Patients in Study SHH4476g* mBCC
(n = 33)laBCC
(n = 63)- *
- Patients who received at least one dose of ERIVEDGE with independent pathologist-confirmed diagnosis of BCC
- †
- IRF = Independent Review Facility
- ‡
- For laBCC, complete response was defined as objective response with no residual BCC on sampling tumor biopsy.
- §
- CI = Confidence Interval
- ¶
- NE = Not estimable
IRF†-Confirmed ORR, n (%) 10 (30.3) 27 (42.9) (95% CI) (15.6, 48.2) (30.5, 56.0) Complete response‡ 0 (0.0) 13 (20.6) Partial response 10 (30.3) 14 (22.2) Median Response Duration (months) 7.6 7.6 (95% CI§) (5.6, NE¶) (5.7, 9.7) - 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Embryo-Fetal Toxicity [see Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)]
Females of Reproductive Potential
- Advise women of the potential risk to a fetus. Advise female patients and female partners of male patients to contact their healthcare provider with a known or suspected pregnancy.
- Inform females there is a Pregnancy Exposure Registry that monitors pregnancy outcomes in females exposed to ERIVEDGE during pregnancy and that they can contact the Pregnancy Exposure Registry by calling 1-888-835-2555.
- Advise females of reproductive potential to use effective contraception during therapy with ERIVEDGE and for 24 months after the final dose.
Males
- Advise males, even those with prior vasectomy, to use condoms to avoid potential drug exposure in both pregnant partners and female partners of reproductive potential during therapy with ERIVEDGE and for 3 months after the final dose.
- Advise males not to donate semen during therapy with ERIVEDGE and for 3 months after the final dose.
Lactation
- Advise women not to breastfeed during therapy with ERIVEDGE and for 24 months after the final dose [see Use in Specific Populations (8.2)].
Severe Cutaneous Reactions
- Inform patients of the signs and symptoms of severe cutaneous reactions. Advise patients to contact their healthcare provider immediately for signs and symptoms of severe cutaneous reactions [see Warnings and Precautions (5.2)].
Muscle-related adverse reactions
- Advise patients starting therapy with ERIVEDGE of the risk of muscle-related adverse reactions. Advise patients to contact their healthcare provider immediately for any new unexplained muscle pain, tenderness, or weakness occurring during treatment or that persists after discontinuing ERIVEDGE [see Warnings and Precautions (5.3)]
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: March 2023 MEDICATION GUIDE
ERIVEDGE® (EH-rih-vej)
(vismodegib)
CapsuleWhat is the most important information I should know about ERIVEDGE?
ERIVEDGE can cause your baby to die before it is born (be stillborn) or cause your baby to have severe birth defects.
For females who can become pregnant:- You should talk with your healthcare provider about the risks of ERIVEDGE to your unborn child.
- Your healthcare provider will do a pregnancy test within 7 days before you start taking ERIVEDGE.
- In order to avoid pregnancy, you should use birth control during treatment and for 24 months after your final dose of ERIVEDGE. Talk with your healthcare provider about what birth control method is right for you during this time.
- Talk to your healthcare provider right away if you have unprotected sex or if you think that your birth control has failed.
- Tell your healthcare provider right away if you become pregnant or think that you may be pregnant.
- ERIVEDGE is present in semen. Do not donate semen while you are taking ERIVEDGE and for 3 months after your final dose.
- You should always use a condom, even if you have had a vasectomy, during sex with female partners who are pregnant or who are able to become pregnant, during treatment with ERIVEDGE and for 3 months after your final dose to protect your female partner from being exposed to ERIVEDGE.
- Tell your healthcare provider right away if your partner becomes pregnant or thinks she is pregnant while you are taking ERIVEDGE.
There is a Pregnancy Exposure Registry for females taking ERIVEDGE who become pregnant. The purpose of this registry is to monitor the health of you and your unborn baby. If you think that you or your female partner may have been exposed to ERIVEDGE during pregnancy, talk to your healthcare provider right away. If you become pregnant during treatment with ERIVEDGE, you or your healthcare provider should report your pregnancy to Genentech at 1-888-835-2555.What is ERIVEDGE?
ERIVEDGE is a prescription medicine used to treat adults with a type of skin cancer, called basal cell carcinoma, that has spread to other parts of the body, or that has come back after surgery or that your healthcare provider decides cannot be treated with surgery or radiation.
It is not known if ERIVEDGE is safe and effective in children.Before taking ERIVEDGE, tell your healthcare provider about all of your medical conditions, including if you: - are pregnant or plan to become pregnant. See "What is the most important information I should know about ERIVEDGE?"
- are breastfeeding or plan to breastfeed. It is not known if ERIVEDGE passes into your breast milk. You should not breastfeed during treatment and for 24 months after your final dose of ERIVEDGE. Talk to your healthcare provider about the best way to feed your baby during this time.
How should I take ERIVEDGE? - Take ERIVEDGE exactly as your healthcare provider tells you.
- You can take ERIVEDGE with or without food.
- Swallow ERIVEDGE capsules whole. Do not open or crush the capsules.
- Take ERIVEDGE 1 time each day.
- If you miss a dose, skip the missed dose. Just take your next scheduled dose.
- Your healthcare provider will perform certain tests to check you for side effects before and during treatment with ERIVEDGE.
- Your healthcare provider may temporarily or permanently stop treatment with ERIVEDGE if you have certain side effects. Do not stop taking ERIVEDGE unless your healthcare provider tells you to.
What should I avoid while taking ERIVEDGE? - Do not donate blood or blood products while you are taking ERIVEDGE and for 24 months after your final dose.
- For Males: Do not donate semen while you are taking ERIVEDGE and for 3 months after your final dose.
What are the possible side effects of ERIVEDGE?
ERIVEDGE can cause serious side effects, including:-
Severe skin reactions. Severe skin reactions have happened in some people taking ERIVEDGE. You may need to be treated in a hospital because these severe skin reactions can be life-threatening or lead to death.
Tell your healthcare provider right away if you develop any of the following signs or symptoms of a severe skin reaction, including:
- blisters or peeling of your skin
- blisters on your lip, or around your mouth or eyes
- mouth sores or genital sores
- high fever or flu-like symptoms
- enlarged lymph nodes
- skin pain and burning
- Your healthcare provider may permanently stop ERIVEDGE if you develop a severe skin reaction.
- Muscle Problems. Muscle problems are common with ERIVEDGE, but can also sometimes be serious. ERIVEDGE can increase your risk of muscle spasms or muscle pain. Tell your healthcare provider right away if you develop any new or worsening muscle pain, tenderness, or weakness during or after treatment with ERIVEDGE. Your healthcare provider should do a blood test to check for muscle problems and to check your kidney function before you start taking ERIVEDGE, and as needed during treatment if you develop muscle problems.
- Bone growth problems. Bone growth problems have happened in children who have been exposed to ERIVEDGE. These problems may continue even after stopping treatment with ERIVEDGE.
- muscle spasms
- hair loss
- change in how things taste or loss of taste
- weight loss
- tiredness
- nausea
- diarrhea
- decreased appetite
- constipation
- joint pain
- vomiting
ERIVEDGE can cause absence of menstrual periods (amenorrhea) in females who are able to become pregnant. It is not known if amenorrhea is permanent. Talk to your healthcare provider if you have concerns about fertility.
These are not all the possible side effects of ERIVEDGE.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Genentech, Inc. at 1-888-835-2555.How should I store ERIVEDGE? - Store ERIVEDGE at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep the bottle tightly closed to protect ERIVEDGE from moisture.
General information about the safe and effective use of ERIVEDGE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ERIVEDGE for a condition for which it was not prescribed. Do not give ERIVEDGE to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ERIVEDGE that is written for health professionals.What are the ingredients in ERIVEDGE?
Active ingredient: vismodegib
Inactive ingredients: microcrystalline cellulose, lactose monohydrate, sodium lauryl sulfate, povidone, sodium starch glycolate, talc, and magnesium stearate (non-bovine). The capsule shell contains gelatin, titanium dioxide, red iron oxide, and black iron oxide. The black printing ink contains shellac and black iron oxide.
Manufactured by: Patheon, Inc. Mississauga, Canada
Distributed by: Genentech USA, Inc. A Member of the Roche Group 1 DNA Way South San Francisco, CA 94080-4990
ERIVEDGE is a registered trademark of Genentech, Inc.
©2023 Genentech, Inc.
For more information, call 1-855-737-4833 or go to www.erivedge.com -
SPL UNCLASSIFIED SECTION
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
- PRINCIPAL DISPLAY PANEL - 150 mg Capsule Bottle Carton
-
INGREDIENTS AND APPEARANCE
ERIVEDGE
vismodegib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-140 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VISMODEGIB (UNII: 25X868M3DS) (VISMODEGIB - UNII:25X868M3DS) VISMODEGIB 150 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SODIUM LAURYL SULFATE (UNII: 368GB5141J) POVIDONE K30 (UNII: U725QWY32X) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TALC (UNII: 7SEV7J4R1U) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color GRAY (gray opaque) , PINK (pink opaque) Score no score Shape CAPSULE (hard capsule) Size 19mm Flavor Imprint Code VISMO;150;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-140-01 1 in 1 CARTON 01/30/2012 1 28 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 2 NDC:50242-140-86 1 in 1 CARTON 04/09/2013 2 28 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203388 01/30/2012 Labeler - Genentech, Inc. (080129000) Registrant - Patheon Inc. (240769596) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche AG 482242971 ANALYSIS(50242-140) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd. 485244961 ANALYSIS(50242-140) , LABEL(50242-140) , PACK(50242-140)
