Label: ALLOPURINOL tablet


-
NDC Code(s):
29300-349-01,
29300-349-05,
29300-349-10,
29300-350-01, view more29300-350-05, 29300-350-10
- Packager: Unichem Pharmaceuticals (USA), Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated February 9, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ALLOPURINOL TABLETS safely and effectively. See full prescribing information for ALLOPURINOL TABLETS.
ALLOPURINOL tablets, for oral use.
Initial U.S. Approval:1966INDICATIONS AND USAGE
Allopurinol is a xanthine oxidase inhibitor indicated for the management of:
- Adult patients with signs and symptoms of primary or secondary gout (acute attacks, tophi, joint destruction, uric acid lithiasis, and/or nephropathy) (1)
- Adult and pediatric patients with leukemia, lymphoma and solid tumor malignancies who are receiving cancer therapy which causes elevations of serum and urinary uric acid levels (1)
- Adult patients with recurrent calcium oxalate calculi whose daily uric acid excretion exceeds 800 mg/day in male patients and 750 mg/day in female patients, despite lifestyle changes (1)
Limitations of Use
Allopurinol tablets are not recommended for the treatment of asymptomatic hyperuricemia. (1)
DOSAGE AND ADMINISTRATION
-
Gout: Prior to initiating treatment assess serum uric acid level, complete blood count, chemistry panel, liver and kidney function tests. Prophylactic treatment for gout flares is recommended. (2.1,2.2)
- Patients with normal kidney function: Initial dosage is 100 mg orally daily. Increase by 100 mg weekly increments until serum uric acid of 6 mg/dl or less is reached (maximum 800 mg daily). (2.3)
- Patients with impaired kidney function: The initial dosage is 50 mg orally daily. Follow recommendations for titration in patients with renal impairment until target serum uric acid level is reached. (2.6)
- See complete information in the Full Prescribing Information (FPI).
- Hyperuricemia Associated with Cancer Therapy: The recommended dosage is:
DOSAGE FORMS AND STRENGTHS
Tablets: 100 mg, and 300 mg, functionally scored (3)
CONTRAINDICATIONS
Known hypersensitivity to allopurinol or to any of the ingredients of allopurinol tablets. (4)
WARNINGS AND PRECAUTIONS
- Skin Rash and Hypersensitivity: Allopurinol has been associated with serious and sometimes fatal dermatological reactions. Discontinue allopurinol at the first appearance of skin rash or other signs of hypersensitivity reaction. (5.1)
- Gout Flares: May occur during initiation of treatment. Concurrent prophylactic treatment with colchicine or anti-inflammatory agents is recommended. (5.2)
- Nephrotoxicity: Allopurinol may affect kidney function. Patients with decreased kidney function require lower doses of allopurinol. (5.3)
- Hepatoxicity: Cases of reversible hepatotoxicity have occurred. If signs and symptoms of hepatotoxicity develop, evaluate liver function. (5.4)
- Myelosuppression: Bone marrow suppression has been reported with allopurinol. (5.5)
- Potential Effect on Driving and Use of Machinery: Drowsiness, somnolence and dizziness have been reported in patients taking allopurinol. (5.6)
ADVERSE REACTIONS
Most common adverse reactions (incidence > 1%) are nausea, diarrhea, and increase in liver function tests. (6)
To report SUSPECTED ADVERSE REACTIONS, Unichem Pharmaceuticals (USA), Inc., at 1-866-562-4616 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- The following drugs may increase the risk of serious skin reactions: bendamustine, thiazide diuretics, ampicillin and amoxicillin. (7.1)
- Capecitabine: Avoid concomitant use. (7.2)
- Mercaptopurine or Azathioprine: Reduce mercaptopurine or azathioprine dose as recommended in the respective prescribing information. (7.2)
- Pegloticase: Discontinue and refrain from initiating treatment with allopurinol. (7.2)
- See FPI for complete list of significant drug interactions. (7.2)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Testing Prior to Treatment Initiation
2.2 Recommended Prophylaxis for Gout Flares
2.3 Recommended Dosage for Gout
2.4 Recommended Dosage for Hyperuricemia Associated with Cancer Therapy
2.5 Recommended Dosage for Management of Recurrent Calcium Oxalate Calculi in Hyperuricosuric Patients
2.6 Recommended Dosage in Patients with Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Skin Rash and Hypersensitivity
5.2 Gout Flares
5.3 Nephrotoxicity
5.4 Hepatotoxicity
5.5 Myelosuppression
5.6 Potential Effect on Driving and Use of Machinery
6 ADVERSE REACTIONS
7 DRUG INTERACTIONS
7.1 Drugs Known to Affect the Occurrence of Skin Rash and Hypersensitivity
7.2 Drugs Known to Have Clinically Important Drug Interactions with Allopurinol
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Allopurinol tablets are indicated for:
- The management of adults with signs and symptoms of primary or secondary gout (acute attacks, tophi, joint destruction, uric acid lithiasis, and/or nephropathy)
- The management of adult and pediatric patients with leukemia, lymphoma and solid tumor malignancies who are receiving cancer therapy which causes elevations of serum and urinary uric acid levels
- The management of adult patients with recurrent calcium oxalate calculi whose daily uric acid excretion exceeds 800 mg/day in male patients and 750 mg/day in female patients, despite lifestyle changes (such as reduction of dietary sodium, non-dairy animal protein, oxylate rich foods, refined sugars and increases in oral fluids and fruits and vegetables)
Allopurinol tablets are not recommended for the treatment of asymptomatic hyperuricemia.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Testing Prior to Treatment Initiation
Prior to initiating treatment with allopurinol tablets in patients with gout, assess the following baseline tests: serum uric acid level, complete blood count, chemistry panel, liver function tests (serum alanine aminotransferase [ALT], aspartate aminotransferase [AST], alkaline phosphatase, and total bilirubin), kidney function tests (serum creatinine and eGFR).
2.2 Recommended Prophylaxis for Gout Flares
Gout flares may occur after initiation of allopurinol tablets due to changing serum uric acid levels resulting in mobilization of urate from tissue deposits. Flare prophylaxis with colchicine or an anti- inflammatory agent according to practice guidelines is recommended upon initiation of allopurinol tablets. While adjusting the dosage of allopurinol tablets in patients who are being treated with colchicine and/or anti-inflammatory agents, continue flare prophylaxis drugs until serum uric acid has been normalized and the patient has been free of gout flares for several months. If a gout flare occurs during allopurinol tablets treatment, allopurinol tablets need not be discontinued. Manage the gout flare concurrently, as appropriate for the individual patient [see Warnings and Precautions (5.2)].
2.3 Recommended Dosage for Gout
The initial recommended dosage for the management of gout is 100 mg orally daily, with weekly increments of 100 mg, until a serum uric acid level of 6 mg/dL or less is reached. Initiating treatment with lower dosages of allopurinol tablets and titrating slowly, decreases the risk of gout flares and drug induced serious adverse reactions.
In patients with renal impairment the initial dosage is 50 mg orally daily with lower dose increases until serum uric acid level of 6 mg/dL or less is reached. For complete dosage recommendations for patients with renal impairment see Table 1 [see Dosage and Administration (2.6)].
The minimal effective dosage is 100 mg to 200 mg daily and the maximal recommended dosage is 800 mg daily. The appropriate dosage may be administered in divided doses or as a single equivalent dose with the 300 mg tablet. Doses in excess of 300 mg should be administered in divided doses. Monitor patients' kidney function during the early stages of administration of allopurinol tablets and decrease the dosage or withdraw the drug if persistent abnormalities in kidney function occur [see Dosage and Administration (2.6), Warnings and Precautions (5.3), Use in Specific Populations (8.6)].
The dosage of allopurinol tablets to achieve control of gout varies with the severity of the disease. In general, gout control is achieved with 200 mg to 300 mg daily in patients with mild gout, and with 400 mg to 600 mg daily in patients with moderate to severe tophaceous gout. Gout attacks usually become shorter and less severe after several months of therapy.
If a dose of allopurinol tablets is missed, there is no need to double the dose at the next scheduled time. Allopurinol tablets are generally better tolerated if taken following meals. A fluid intake sufficient to yield a daily urinary output of at least 2 liters and the maintenance of a neutral or preferably, slightly alkaline urine are desirable.
Inform patients of the possibility of gout flares [see Warnings and Precautions (5.2)]. Instruct them to remain on allopurinol tablets if this occurs and to increase fluid intake during therapy to prevent kidney stones.
Concurrent Use of Uricosuric Agents
Some patients, may benefit using uricosuric agents concurrently, to reduce serum uric acid to target levels.
When transferring a patient from a uricosuric agent to allopurinol tablets, reduce the dose of the uricosuric agent over a period of several weeks and increase the dose of allopurinol tablets gradually to the required dose needed to maintain target serum uric acid level.
2.4 Recommended Dosage for Hyperuricemia Associated with Cancer Therapy
Initiate therapy with allopurinol tablets 24 hours to 48 hours before the start of chemotherapy known to cause tumor cell lysis. Administer fluids sufficient to yield a daily urinary output of at least 2 liters in adults (at least 100 mL/m2/hour in pediatric patients) with a neutral or, preferably, slightly alkaline urine.
The recommended dosage of allopurinol tablets is:
- Adult patients – 300 mg to 800 mg orally daily.
- Pediatric patients - 100 mg/m2orally every 8 hours to 12 hours (10 mg/kg/day, maximum 800 mg/day).In patients with body surface area < 0.5 m2, consider using an alternative allopurinol formulation.
The dosage of allopurinol tablets to maintain normal or near-normal serum uric acid varies with the severity of the disease. Monitor serum uric acid levels at least daily and administer allopurinol tablets at a dose and frequency to maintain the serum uric acid within the normal range. Discontinue allopurinol tablets when the risk of tumor lysis has abated (2 days to 3 days from start of chemotherapy). For complete dosage recommendations for patients with renal impairment, see Table 2 [see Dosage and Administration (2.6)].
2.5 Recommended Dosage for Management of Recurrent Calcium Oxalate Calculi in Hyperuricosuric Patients
The recommended dosage for the management of recurrent calcium oxalate stones in hyperuricosuric patients is 200 mg to 300 mg orally daily in divided doses or as the single equivalent. This dose may be adjusted depending upon the resultant control of the hyperuricosuria based upon subsequent 24-hour urinary urate determinations.
2.6 Recommended Dosage in Patients with Renal Impairment
The recommended initial dosages of allopurinol tablets in adult patients with renal impairment are shown in Tables 1 and 2 [see Use in Specific Populations (8.6)].
Patients with Gout
The recommended initial dosages in adult patients with gout with impaired kidney function are shown in Table 1 [see Use in Specific Populations (8.6)].
Initiate treatment with a lower dose of allopurinol tablets and increase the dose gradually in 50 mg/day increments every 2 weeks to 4 weeks in patients with renal impairment to decrease the risk of drug induced serious adverse reactions. Use the lowest dose possible to achieve the desired effect on serum and/or urine uric acid. Monitor kidney function in gout patients with chronic kidney disease closely when initiating treatment with allopurinol tablets and decrease or withdraw the drug if increased abnormalities in kidney function appear and persist.
Table 1. Recommended Initial Dosage in Adult Patients with Gout eGFR
Initial Dosage
> 60 mL/minute
No dosage modification
> 30 to 60 mL/minute
50 mg daily
> 15 to 30 mL/minute
50 mg every other day
5 to 15 mL/minute
50 mg twice weekly
< 5 mL/minute
50 mg once weekly
The maximum dosage that should be used in patients with various levels of renal impairment is not defined at different eGFR levels.
Patients with Recurrent Calcium Oxalate Calculi
Data are insufficient to provide dosage recommendations for the treatment of recurrent calcium oxalate calculi in patients with renal impairment. Allopurinol and its metabolites are excreted by the kidney, and accumulation of the drug can occur in renal failure [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)].
Hyperuricemia Associated with Cancer Therapy
The recommended dosage of allopurinol tablets for the management of hyperuricemia associated with cancer therapy in adult patients with renal impairment is shown in Table 2 [see Use in Specific Populations (8.6)].
Table 2. Recommended Dosage of Allopurinol Tablets in Adult Patients for Management ofHyperuricemia Associated with Cancer Therapy with Renal Impairment eGFR
Recommended Dosage
> 20 mL/min to 60 mL/min
No dosage modification
10 mL/min to 20 mL/min
200 mg/day
< 10 mL/min
100 mg/day
On dialysis
50 mg every 12 hours, or
100 mg every 24 hours
Treatment with allopurinol tablets has not been studied in pediatric patients with severe renal impairment (eGFR < 20 mL/min) or on dialysis. There is insufficient information to establish dosing for allopurinol tablets in pediatric patients with renal impairment. In these patients, consider the risks and potential benefits before initiating treatment with allopurinol tablets [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)].
-
3 DOSAGE FORMS AND STRENGTHS
Allopurinol tablets, USP have functional scoring and are available in the following strengths:
- 100 mg: A white to off-white, round, flat, beveled, uncoated tablets debossed with "349" above breakline, "U" below breakline on one side and plain on the other side.
- 300 mg: A white to off-white, round, flat, beveled, uncoated tablets debossed with "350" above breakline, "U" below breakline on one side and plain on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Skin Rash and Hypersensitivity
Serious and sometimes fatal dermatologic reactions, including toxic epidermal necrolysis (TEN), Stevens-Johnson syndrome (SJS), and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported in patients taking allopurinol [see Adverse Reactions (6)]. These reactions occur in approximately 5 in 10,000 (0.05%) patients taking allopurinol. Other serious hypersensitivity reactions that have been reported include exfoliative, urticarial and purpuric lesions, generalized vasculitis, and irreversible hepatotoxicity. Discontinue allopurinol permanently at the first appearance of skin rash or other signs which may indicate a hypersensitivity reaction.
The HLA-B*58:01 allele is a genetic marker for severe skin reactions indicative of hypersensitivity to allopurinol. Patients who carry the HLA-B*58:01 allele are at a higher risk of allopurinol hypersensitivity syndrome (AHS), but hypersensitivity reactions have been reported in patients who do not carry this allele. The frequency of this allele is higher in individuals of African, Asian (e.g., Han Chinese, Korean, Thai), and Native Hawaiian/Pacific Islander ancestry [see Clinical Pharmacology (12.5)]. The use of allopurinol is not recommended in HLA- B*58:01 positive patients unless the benefits clearly outweigh the risks.
Consider screening for HLA-B*5801 before starting treatment with allopurinol in patients from populations in which the prevalence of this HLA-B*5801 allele is known to be high. Screening is generally not recommended in patients from populations in which the prevalence of HLA-B*58:01 is low, or in current allopurinol users, as the risk of SJS/TEN/DRESS is largely confined to the first few months of therapy, regardless of HLA- B*58:01 status.
Hypersensitivity reactions to allopurinol may be increased in patients with decreased kidney function receiving thiazide diuretics and allopurinol concurrently. Concomitant use of the following drugs may also increase the risk of skin rash, which may be severe: bendamustine, ampicillin and amoxicillin [see Drug Interactions (7.1)].
Discontinue allopurinol immediately if a skin rash develops. Instruct patients to stop taking allopurinol immediately and seek medical attention promptly if they develop a rash.
5.2 Gout Flares
Gout flares have been reported during initiation of treatment with allopurinol, even when normal or subnormal serum uric acid levels have been attained due to the mobilization of urates from tissue deposits. Even with adequate therapy with allopurinol, it may require several months to deplete the uric acid pool sufficiently to achieve control of the flares. The flares typically become shorter and less severe after several months of therapy.
In order to prevent gout flares when treatment with allopurinol is initiated, concurrent prophylactic treatment with colchicine or an anti-inflammatory agent is recommended [see Dosage and Administration (2.2)]. Advise patients to continue allopurinol and prophylactic treatment even if gout flares occur, as it may take months to achieve control of gout flares.
5.3 Nephrotoxicity
Treatment with allopurinol may result in acute kidney injury due to formation of xanthine calculi or due to precipitation of urates in patients receiving concomitant uricosuric agents. Patients with pre-existing kidney disease, including chronic kidney disease or history of kidney stones, may be at increased risk for worsening of kidney function or acute kidney injury due to xanthine calculi while receiving treatment with allopurinol.
In patients receiving allopurinol for the management of gout or the management of recurrent calcium oxalate calculi, monitor kidney function frequently during the early stages of allopurinol administration. Maintain fluid intake sufficient to yield a urinary output of at least 2 liters per day of neutral or, preferably, slightly alkaline urine to avoid the possibility of formation of xanthine calculi and help prevent renal precipitation of urates in patients receiving concomitant uricosuric agents.
In patients receiving allopurinol for the management of tumor lysis syndrome, monitor kidney function at least daily during the early stages of allopurinol administration. Maintain fluid intake sufficient to yield a urinary output of at least 2 liters per day in adults and at least 2 liters/m2/day (or at least 100 mL/m2/hour) in pediatric patients [see Dosage and Administration (2.4)].
5.4 Hepatotoxicity
Cases of reversible clinical hepatotoxicity have occurred in patients taking allopurinol, and in some patients, asymptomatic rises in serum alkaline phosphatase or serum transaminase have been observed. If anorexia, weight loss, or pruritus develop in patients on allopurinol, evaluate liver enzymes. In patients with pre-existing liver disease, monitor liver enzymes periodically. Discontinue allopurinol in patients with elevated liver enzymes.
5.5 Myelosuppression
Myelosuppression, manifested by anemia, leukopenia or thrombocytopenia, has been reported in patients receiving allopurinol. The cytopenias have occurred as early as 6 weeks up to 6 years after the initiation of therapy of allopurinol. Concomitant use of allopurinol with cytotoxic drugs associated with myelosuppression may increase the risk of myelosuppression. Monitor blood counts more frequently when cytotoxic drugs are used concomitantly [Drug Interactions (7.2)].
Concomitant use with allopurinol increases the exposure of either mercaptopurine or azathioprine which may increase the risk of myelosuppression. Reduce the dosage of mercaptopurine or azathioprine as recommended in their respective prescribing information when used concomitantly with allopurinol [see Drug Interactions (7.2)].
5.6 Potential Effect on Driving and Use of Machinery
Drowsiness, somnolence and dizziness have been reported in patients taking allopurinol [see Adverse Reactions (6)]. Inform patients also that the central nervous system depressant effects of allopurinol may be additive to those of alcohol and other CNS depressants.
Advise patients to avoid operation of automobiles or other dangerous machinery and activities made hazardous by decreased alertness when starting allopurinol or increasing the dose, until they know how the drug affects them.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Skin Rash and Hypersensitivity [see Warnings and Precautions (5.1)]
- Nephrotoxicity [see Warnings and Precautions (5.3)]
- Hepatoxicity [see Warnings and Precautions (5.4)]
- Myelosuppression [see Warnings and Precautions (5.5)]
- Potential Effect on Driving and Use of Machinery [see Warnings and Precautions (5.6)]
The following adverse reactions associated with the use of allopurinol were identified in literature, unpublished clinical trials or postmarketing reports. Because some of these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The most frequent adverse reaction to allopurinol is skin rash.
Most Common Adverse Reactions (≥ 1%)
Gastrointestinal: Diarrhea, nausea, alkaline phosphatase increase, AST/ALT increase.
Metabolic and Nutritional: Acute attacks of gout. Skin and Appendages: Rash, maculopapular rash.
Less Common Adverse Reactions (< 1%)
Body As a Whole: Ecchymosis, fever, headache, malaise.
Cardiovascular: Necrotizing angiitis, vasculitis, pericarditis, peripheral vascular disease, thrombophlebitis, bradycardia, vasodilation.
Gastrointestinal: Hepatic necrosis, granulomatous hepatitis, hepatomegaly, hyperbilirubinemia, cholestatic jaundice, vomiting, intermittent abdominal pain, gastritis, dyspepsia, hemorrhagic pancreatitis, gastrointestinal bleeding, stomatitis, salivary gland swelling, hyperlipidemia, tongue edema, anorexia.
Hemic and Lymphatic: Thrombocytopenia, eosinophilia, leukocytosis, leukopenia, aplastic anemia, agranulocytosis, eosinophilic fibrohistiocytic lesion of bone marrow, pancytopenia, prothrombin decrease, anemia, hemolytic anemia, reticulocytosis, lymphadenopathy, lymphocytosis.
Musculoskeletal: Myopathy, arthralgias, myalgia.
Nervous: Peripheral neuropathy, neuritis, paresthesia, somnolence, optic neuritis, confusion, dizziness, vertigo, foot drop, decrease in libido, depression, amnesia, tinnitus, asthenia, insomnia.
Respiratory: Epistaxis, bronchospasm, asthma, pharyngitis, rhinitis.
Skin and Appendages: Erythema multiforme exudativum (Stevens-Johnson syndrome), toxic epidermal necrolysis (Lyell's syndrome), hypersensitivity vasculitis, purpura, vesicular bullous dermatitis, exfoliative dermatitis, eczematoid dermatitis, pruritus, urticaria, alopecia, onycholysis, lichen planus, furunculosis, facial edema, sweating, skin edema.
Special Senses: Taste loss/perversion, cataracts, macular retinitis, iritis, conjunctivitis, amblyopia.
Urogenital: Renal failure, uremia, nephritis, impotence, primary hematuria, albuminuria.
Endocrine: Infertility (male), hypercalcemia, gynecomastia (male).
-
7 DRUG INTERACTIONS
7.1 Drugs Known to Affect the Occurrence of Skin Rash and Hypersensitivity
Concomitant use of the following drugs may increase the risk of skin rash, which may be severe: bendamustine, thiazide diuretics, ampicillin and amoxicillin. Renal impairment may further increase risk with concomitant use of thiazide diuretics [see Warnings and Precautions (5.1, 5.2) and Clinical Pharmacology (12.2)].
Monitor kidney function and reduce the dose of allopurinol in patients with concomitant thiazide diuretic use and impaired renal function [see Dosage and Administration (2.6), Warnings and Precautions (5.1)].
Discontinue allopurinol at the first appearance of skin rash or other signs which may indicate a hypersensitivity reaction when use concomitantly with these drugs [see Warnings and Precautions (5.1)].
7.2 Drugs Known to Have Clinically Important Drug Interactions with Allopurinol
Table 3: Interventions for Clinically Important Drug Interactions with Allopurinol Capecitabine
Clinical Impact
Concomitant use with allopurinol may decrease concentration of capecitabine's active metabolites, which may decrease capecitabine efficacy.
Intervention
Avoid the use of allopurinol during treatment with capecitabine
Chlorpropamide
Clinical Impact
Allopurinol prolongs the half-life of chlorpropamide as both compete for renal tubular excretion. In patients with renal insufficiency, the risk of hypoglycemia may be increased due to this mechanism.
Intervention
Monitor patients with renal insufficiency for hypoglycemia when administering chlorpropamide and allopurinol concomitantly.
Cyclosporine
Clinical Impact
Concomitant use of allopurinol increases cyclosporine concentrations, which may increase the risk of adverse reactions.
Intervention
Increase frequency of monitoring cyclosporine concentrations as reflected in its prescribing information and modify the dosage of cyclosporine as appropriate when used concomitantly with allopurinol.
Cyclophosphamide and Other Cytotoxic Agents
Clinical Impact
Concomitant use of allopurinol with cyclophosphamide and other cytotoxic agents (doxorubicin, bleomycin, procarbazine, mechloroethamine) increases bone marrow suppression among patients with neoplastic disease, except leukemia.
Intervention
Blood count monitoring and regular physician follow-up are recommended.
Dicumarol
Clinical Impact
Allopurinol prolongs the half-life of the anticoagulant, dicumarol. The mechanism of this drug interaction has not been established but should be noted when allopurinol is given to patients already on dicumarol therapy.
Intervention
Monitor prothrombin time. Adjust the dosage of dicumarol accordingly when allopurinol is added to anticoagulant therapy.
Fluorouracil
Clinical Impact
Based on non-clinical data, allopurinol may decrease anti-tumor activity due to suppression of phosphorylation of 5-fluorouracil.
Intervention
Concomitant administration with fluorouracil should be avoided.
Mercaptopurine or Azathioprine
Clinical Impact
Allopurinol inhibits xanthine oxidase mediated metabolism of mercaptopurine and azathioprine. Concomitant use of allopurinol increases the exposure of either mercaptopurine or azathioprine which may increase the risk of their adverse reactions, including myelosuppression [see Warnings and Precautions 5.5].
Intervention
In patients receiving mercaptopurine or azathioprine, the concomitant administration of 300 mg to 600 mg of allopurinol per day will require a reduction in dose to approximately one third to one fourth of the usual dose of mercaptopurine or azathioprine. Subsequent adjustment of doses of mercaptopurine or azathioprine should be made on the basis of therapeutic response and the appearance of toxic effects.
Pegloticase
Clinical Impact
Concomitant use of allopurinol and pegloticase may potentially blunt the rise of serum uric acid levels and increase the risk of pegloticase related anaphylaxis in patients whose uric acid level increase to above 6 mg/dL.
Intervention
Discontinue and do not institute allopurinol therapy during treatment with pegloticase.
Theophylline
Clinical Impact
Concomitant use of allopurinol doses greater than or equal to 600 mg/day may decrease the clearance of theophylline
Intervention
Monitor and adjust theophylline doses as reflected in the prescribing information.
Uricosuric Drugs
Clinical Impact
Uricosuric agents increase the excretion of the active allopurinol metabolite oxypurinol. Concomitant use with uricosuric agents decreases oxypurinol exposure which may reduce the inhibition of xanthine oxidase by oxypurinol and increases the urinary excretion of uric acid.
The net effect of such combined therapy may be useful in some patients in achieving minimum serum uric acid levels provided the total urinary uric acid load does not exceed the competence of the patient's kidney function.
Intervention
Monitor uric acid levels due to the increased chance of hypouricemic effects.
Warfarin
Clinical Impact
Allopurinol may inhibit the metabolism of warfarin, possibly enhancing its anticoagulant effect.
Intervention
Monitor patients on concomitant therapy for excessive anticoagulation. Assess INR frequently and adjust warfarin dosage accordingly when allopurinol is added to warfarin therapy.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Based on findings in animals, allopurinol may cause fetal harm when administered to a pregnant woman. Adverse developmental outcomes have been described in exposed animals (see Data). Allopurinol and its metabolite oxypurinol have been shown to cross the placenta following administration of maternal allopurinol.
Available limited published data on allopurinol use in pregnant women do not demonstrate a clear pattern or increase in frequency of adverse developmental outcomes. Among approximately 50 pregnancies described in published literature, 2 infants with major congenital malformations have been reported with following maternal allopurinol exposure. Advise pregnant women of the potential risk to a fetus.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown.In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
Experience with allopurinol during human pregnancy has been limited partly because women of reproductive age rarely require treatment with allopurinol. A case report published in 2011 described the outcome of a full-term pregnancy in a 35-year-old woman who had recurrent kidney stones since age 18 who took allopurinol throughout the pregnancy. The child had multiple complex birth defects and died at 8 days of life. A second report in 2013 provided data on 31 prospectively ascertained pregnancies involving mothers exposed to allopurinol for varying durations during the first trimester. The overall rate of major fetal malformations and spontaneous abortions was reported to be within the normal expected range; however, one child had severe malformations similar to those described in the cited earlier case report.
Animal Data
There was no evidence of fetotoxicity or teratogenicity in rats or rabbits treated during the period of organogenesis with oral allopurinol at doses up to 200 mg/kg/day and up to 100 mg/kg/day, respectively (about 2.4 times the human dose on a mg/m2 basis). However, there is a published report in pregnant mice that single intraperitoneal doses of 50 mg/kg or 100 mg/kg (about 0.3 or 0.6 times the human dose on a mg/m2 basis) of allopurinol on gestation days 10 or 13 produced significant increases in fetal deaths and teratogenic effects (cleft palate, harelip, and digital defects). It is uncertain whether these findings represented a fetal effect or an effect secondary to maternal toxicity.
8.2 Lactation
Allopurinol and oxypurinol are present in human milk. Based on information from a single case report, allopurinol and its active metabolite, oxypurinol, were detected in the milk of a mother receiving 300 mg of allopurinol daily at 5 weeks postpartum. The estimated relative infant dose were 0.14 mg/kg and 0.2 mg/kg of allopurinol and between 7.2 mg/kg to 8 mg/kg of oxypurinol daily. There was no report of effects of allopurinol on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatments with allopurinol and for one week after the last dose.
8.4 Pediatric Use
Hyperuricemia Associated with Cancer Therapy
The safety and effectiveness of allopurinol for the management of pediatric patients with leukemia, lymphoma and solid tumor malignancies who are receiving cancer therapy which causes elevations of serum and urinary uric acid levels have been established in approximately 200 pediatric patients. The efficacy and safety profile observed in this patient population were similar to that observed in adults.
Primary or Secondary Gout
The safety and effectiveness of allopurinol have not been established for the treatment of signs and symptoms of primary or secondary gout in pediatric patients.
Recurrent Calcium Oxalate Calculi
The safety and effectiveness of allopurinol have not been established for the management of pediatric patients with recurrent calcium oxalate calculi.
Inborn Errors of Metabolism
The safety and effectiveness of allopurinol have not been established in pediatric patients with rare inborn errors of purine metabolism.
8.6 Renal Impairment
Allopurinol and its primary active metabolite, oxipurinol, are eliminated by the kidneys; therefore, changes in renal function have a profound effect on exposure. In patients with decreased renal function or who have concurrent illnesses which can affect renal function, perform periodic laboratory parameters of renal function and reassess the patient's dosage of allopurinol [see Dosage and Administration (2.6), Warnings and Precautions (5.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
Allopurinol, USP is a xanthine oxidase inhibitor. It has the following structural formula:
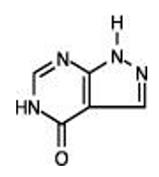
Allopurinol, USP is known chemically as 1, 5-dihydro-4H-pyrazolo [3, 4-d]pyrimidin-4-one and it has a molecular weight of 136.11 g/mol. Its solubility in water at 37°C is 80.0 mg/dL and is greater in an alkaline solution. It is a xanthine oxidase inhibitor which is administered orally.
Each white to off-white scored tablet contains 100 mg or 300 mg of allopurinol, USP and the inactive ingredients croscarmellose sodium, lactose monohydrate, magnesium stearate, pregelatinized starch and povidone.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Allopurinol is a structural analogue of the natural purine base, hypoxanthine.
Allopurinol acts on purine catabolism, without disrupting the biosynthesis of purines. It reduces the production of uric acid by inhibiting the biochemical reactions immediately preceding its formation. It is an inhibitor of xanthine oxidase, the enzyme responsible for the conversion of hypoxanthine to xanthine and of xanthine to uric acid, the end product of purine metabolism in humans. Allopurinol is metabolized to the corresponding xanthine analogue, oxypurinol (alloxanthine), which also is an inhibitor of xanthine oxidase.
12.2 Pharmacodynamics
Allopurinol reduces the production of uric acid by inhibiting the biochemical reactions immediately preceding its formation in a dose dependent manner. The pharmacological action of allopurinol is generally believed to be mediated by its oxypurinol metabolite.
Effect on Hypoxanthine and Xanthine
Reutilization of both hypoxanthine and xanthine for nucleotide and nucleic acid synthesis is markedly enhanced when their oxidations are inhibited by allopurinol and oxipurinol. This reutilization does not disrupt normal nucleic acid anabolism, however, because feedback inhibition is an integral part of purine biosynthesis. As a result of xanthine oxidase inhibition, the serum concentration of hypoxanthine plus xanthine in patients receiving allopurinol for treatment of hyperuricemia is usually in the range of 0.3 mg/dL to 0.4 mg/dL compared to a normal level of approximately 0.15 mg/dL. A maximum of 0.9 mg/dL of these oxypurines has been reported when the serum urate was lowered to less than 2 mg/dL by high doses of allopurinol. These values are far below the saturation levels at which point their precipitation would be expected to occur (above 7 mg/dL). The increased xanthine and hypoxanthine in the urine in patients who were treated with oral allopurinol have not been accompanied by problems of nephrolithiasis; however, there are isolated case reports of xanthine crystalluria.
Drug Interaction Studies
Fluorouracil: Based on non-clinical data, allopurinol may decrease anti-tumor activity due to suppression of phosphorylation of 5-fluorouracil.
Pegloticase: Concomitant use of allopurinol and pegloticase may potentially blunt the rise of serum uric acid levels required for monitoring the safe use of pegloticase.
Cytotoxic Agents: Enhanced bone marrow suppression by cyclophosphamide and other cytotoxic agents has been reported among patients with neoplastic disease, except leukemia, in the presence of allopurinol.
Thiazide Diuretics: Reports that the concomitant administration of allopurinol and thiazide diuretics contributed to increased allopurinol toxicity were reviewed; however, a causal mechanism or cause-and-effect relationship was not found.
12.3 Pharmacokinetics
Allopurinol is approximately 90% absorbed from the gastrointestinal tract. Peak plasma levels generally occur at 1.5 hours and 4.5 hours for allopurinol and oxipurinol respectively. After a single oral dose of 300 mg allopurinol, maximum plasma levels of about 3 mcg/mL of allopurinol and 6.5 mcg/mL of oxipurinol are produced.
Elimination
The half-life of allopurinol and oxipurinol are approximately 1 hour to 2 hours and 15 hours following oral dose of allopurinol, respectively.
Metabolism
Allopurinol is metabolized to the corresponding xanthine analogue, oxypurinol (alloxanthine), which also is an inhibitor of xanthine oxidase.
Excretion
Allopurinol and its primary active metabolite, oxipurinol, are eliminated by the kidneys. Approximately 20% of the ingested allopurinol is excreted in the feces. Oxipurinol is primarily eliminated unchanged in urine by glomerular filtration and tubular reabsorption.
Drug Interaction Studies
Capecitabine: Concomitant use with allopurinol may decrease concentration of capecitabine's active metabolites, which may decrease capecitabine efficacy.
Cyclosporine: Concomitant use of allopurinol increases cyclosporine concentrations which may increase the risk of adverse reactions.
Mercaptopurine or Azathioprine: Allopurinol inhibits xanthine oxidase mediated metabolism of mercaptopurine and azathioprine. Concomitant use of allopurinol increases the exposure of either mercaptopurine or azathioprine which may increase the risk of their adverse reactions including myelosuppression.
Theophylline: Concomitant use of allopurinol doses greater than or equal to 600 mg/day may decrease the clearance of theophylline.
Uricosuric Agents: Uricosuric agents increase the excretion of the active allopurinol metabolite oxypurinol. Concomitant use with uricosuric agents decreases oxypurinol exposure which may reduce the inhibition of xanthine oxidase by oxypurinol and increases the urinary excretion of uric acid.
Warfarin: Allopurinol may inhibit the metabolism of warfarin, possibly enhancing its anticoagulant effect.
12.5 Pharmacogenomics
The HLA-B*5801 allele is a genetic marker that has shown to be associated with risk of developing allopurinol related hypersensitivity syndrome (DRESS) and SJS/TEN. The frequency of the HLA- B*58:01 allele ranges from 8 to 10% in Han Chinese populations, about 8% in Thai populations, and about 6% in Korean populations based upon published literature and available databases. The frequency of the HLA-B*58:01 allele is about 4% in Blacks, about 1 % to 2 % in indigenous peoples of the Americas and Hispanic populations, and < 1% in people from European descent and Japanese.
Stevens-Johnson syndrome (SJS)/Toxic epidermal necrolysis (TEN) can still occur in patients who are found to be negative for HLA-B*5801 irrespective of ethnic origin.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of tumorigenicity was observed in male or female mice or rats that received oral allopurinol for the majority of their life spans (greater than 88 weeks) at doses up to 20 mg/kg/day (0.1 and 0.2 times the MRHD on a mg/m2 basis in mice and rats, respectively).
Allopurinol tested negative in the following genotoxicity assays: the in vitro Ames assay, in vitro mouse lymphoma assay, and in vivo rat bone marrow micronucleus assay. Allopurinol administered intravenously to rats (50 mg/kg) was not incorporated into rapidly replicating intestinal DNA. No evidence of clastogenicity was observed in lymphocytes taken from patients treated with allopurinol (mean duration of treatment 40 months), or in an in vitro assay with human lymphocytes.
Allopurinol oral doses of 20 mg/kg/day had no effect on male or female fertility in rats or rabbits (approximately 0.2 or 0.5 times the MRHD on a mg/m2 basis, respectively).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Allopurinol tablets, USP are available as follows:
The 100 mg tablets are white to off-white, round, flat, beveled, uncoated tablets debossed with "349" above breakline, "U" below breakline on one side and plain on the other side. They are available as:
Bottles of 100: NDC 29300-349-01
Bottles of 500: NDC 29300-349-05
Bottles of 1,000: NDC 29300-349-10
The 300 mg tablets are white to off-white, round, flat, beveled, uncoated tablets debossed with "350" above breakline, "U" below breakline on one side and plain on the other side. They are available as:
Bottles of 100: NDC 29300-350-01
Bottles of 500: NDC 29300-350-05
Bottles of 1,000: NDC 29300-350-10
Storage and Handling
Store at 20°C to 25°C (USP Controlled Room Temperature) (68°F to 77°F) in a dry place. Dispense in a tight container as defined in the USP.
-
17 PATIENT COUNSELING INFORMATION
Advise patients to take allopurinol after meals to minimize gastric irritation. If a single dose of allopurinol is occasionally forgotten, there is no need to double the dose at the next scheduled time.
Skin Rash and Hypersensitivity
Inform patients that allopurinol may increase the risk of serious and sometimes fatal dermatologic reactions. Instruct patients to discontinue allopurinol and to seek medical attention immediately, at the first sign of a skin rash, blisters, fever, painful urination, blood in the urine, irritation of the eyes, swelling of the lips or mouth, or other signs and symptoms of hypersensitivity reactions [see Warnings and Precautions (5.1)].
Gout Flares During Treatment With Allopurinol
Inform patients that gout flares may occur during initiation of treatment with allopurinol, even when their serum uric acid is normal. Concurrent use of additional medications such as colchicine or other anti-inflammatory agents can prevent gout flares. Advise patients to continue treatment with both, allopurinol and the prophylactic therapy as prescribed, even if gout flares occur. Reassure them that it may take months to achieve control of the flares but the flares typically become shorter and less severe after several months of therapy [see Warnings and Precautions (5.2)].
Nephrotoxicity
Inform patients that allopurinol may affect kidney function. Advise them to increase fluid intake during therapy (i.e., for adults, at least 2 liters of liquids per day) and to stay well hydrated to prevent kidney stones [see Warnings and Precautions (5.3)].
Hepatotoxicity
Inform patients of the risk of hepatotoxicity and to report to their healthcare provider any signs and symptoms of liver failure, including jaundice, pruritus, bleeding, bruising, or anorexia [see Warnings and Precautions (5.4)].
Myelosuppression
Advise patients of the risk of myelosuppression and to report any signs and symptoms of infection, fever, bleeding, shortness of breath, or significant fatigue to their healthcare provider [see Warnings and Precautions (5.5)].
Potential Effect on Driving and Use of Machinery
Inform patients that drowsiness, somnolence and dizziness have been reported in patients taking allopurinol. Inform also that the central nervous system depressant effects of allopurinol may be additive to those of alcohol and other CNS depressants. Advise patients to avoid operation of automobiles or other dangerous machinery and activities made hazardous by decreased alertness when starting allopurinol or increasing the dose, until they know how the drug affects them [see Warnings and Precautions (5.6)].
Risks Associated with Use of Concomitant Medications
Inform patients that there are risks of adverse effects when allopurinol are used with the following drugs: dicumarol, warfarin, sulfinpyrazone, mercaptopurine, azathioprine, ampicillin, amoxicillin, pegloticase, theophylline, and thiazide diuretics. Advise patients to disclose all medications in use and they should follow the instructions of their physician [see Drug Interactions (7.2)].
Pregnancy
Advise pregnant women of the potential risk to a fetus. Advise women to notify their healthcare provider if they become pregnant or intend to become pregnant during treatment with allopurinol [see Use in Specific Populations (8.1)].
Lactation
Advise women not to breastfeed during treatment with allopurinol and for one week after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
UNICHEM LABORATORIES LTD.
Ind. Area, Meerut Road, Ghaziabad – 201 003, India
and
UNICHEM LABORATORIES LTD.
Pilerne Ind. Estate, Pilerne, Bardez, Goa 403 511, India.
Manufactured for:

13015364
08-R-02/2024
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ALLOPURINOL
allopurinol tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:29300-349 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALLOPURINOL (UNII: 63CZ7GJN5I) (ALLOPURINOL - UNII:63CZ7GJN5I) ALLOPURINOL 100 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE K30 (UNII: U725QWY32X) STARCH, CORN (UNII: O8232NY3SJ) Product Characteristics Color WHITE (White to Off White) Score 2 pieces Shape ROUND Size 8mm Flavor Imprint Code 349;U Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:29300-349-01 100 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/12/2019 2 NDC:29300-349-05 500 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/12/2019 3 NDC:29300-349-10 1000 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/12/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA211820 03/12/2019 ALLOPURINOL
allopurinol tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:29300-350 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALLOPURINOL (UNII: 63CZ7GJN5I) (ALLOPURINOL - UNII:63CZ7GJN5I) ALLOPURINOL 300 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE K30 (UNII: U725QWY32X) STARCH, CORN (UNII: O8232NY3SJ) Product Characteristics Color WHITE (White to Off White) Score 2 pieces Shape ROUND Size 11mm Flavor Imprint Code 350;U Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:29300-350-01 100 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/12/2019 2 NDC:29300-350-05 500 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/12/2019 3 NDC:29300-350-10 1000 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/12/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA211820 03/12/2019 Labeler - Unichem Pharmaceuticals (USA), Inc. (181620514)

