Label: PRECOSE- acarbose tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 54868-5831-0, 54868-5831-1 - Packager: Physicians Total Care, Inc.
- This is a repackaged label.
- Source NDC Code(s): 50419-863
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated May 23, 2012
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
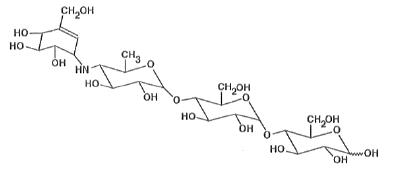
PRECOSE® (acarbose tablets) is an oral alpha-glucosidase inhibitor for use in the management of type 2 diabetes mellitus. Acarbose is an oligosaccharide which is obtained from fermentation processes of a microorganism, Actinoplanes utahensis, and is chemically known as O-4,6-dideoxy- 4-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-2-cyclohexen-1-yl]amino]-α-D-glucopyranosyl-(1 → 4)-O-α-D-glucopyranosyl-(1 → 4)-D-glucose. It is a white to off-white powder with a molecular weight of 645.6. Acarbose is soluble in water and has a pKa of 5.1. Its empirical formula is C25H43NO18 and its chemical structure is as follows:
PRECOSE is available as 25 mg, 50 mg and 100 mg tablets for oral use. The inactive ingredients are starch, microcrystalline cellulose, magnesium stearate, and colloidal silicon dioxide.
-
CLINICAL PHARMACOLOGY
Acarbose is a complex oligosaccharide that delays the digestion of ingested carbohydrates,
thereby resulting in a smaller rise in blood glucose concentration following meals. As a consequence of plasma glucose reduction, PRECOSE reduces levels of glycosylated hemoglobin in patients with type 2 diabetes mellitus. Systemic non-enzymatic protein glycosylation, as reflected by levels of glycosylated hemoglobin, is a function of average blood glucose concentration over time.Mechanism of Action:
In contrast to sulfonylureas, PRECOSE does not enhance insulin secretion. The antihyperglycemic action of acarbose results from a competitive, reversible inhibition of pancreatic alpha-amylase and membrane-bound intestinal alpha-glucoside hydrolase enzymes. Pancreatic alpha-amylase hydrolyzes complex starches to oligosaccharides in the lumen of the small intestine, while the membrane-bound intestinal alpha-glucosidases hydrolyze oligosaccharides, trisaccharides, and disaccharides to glucose and other monosaccharides in the brush border of the small intestine. In diabetic patients, this enzyme inhibition results in a delayed glucose absorption and a lowering of postprandial hyperglycemia.
Because its mechanism of action is different, the effect of PRECOSE to enhance glycemic control is additive to that of sulfonylureas, insulin or metformin when used in combination. In addition, PRECOSE diminishes the insulinotropic and weight-increasing effects of sulfonylureas.
Acarbose has no inhibitory activity against lactase and consequently would not be expected to induce lactose intolerance.
Pharmacokinetics:
Absorption
In a study of 6 healthy men, less than 2% of an oral dose of acarbose was absorbed as active drug, while approximately 35% of total radioactivity from a 14C-labeled oral dose was absorbed. An average of 51% of an oral dose was excreted in the feces as unabsorbed drug-related radioactivity within 96 hours of ingestion. Because acarbose acts locally within the gastrointestinal tract, this low systemic bioavailability of parent compound is therapeutically desired. Following oral dosing of healthy volunteers with 14C-labeled acarbose, peak plasma concentrations of radioactivity were attained 14–24 hours after dosing, while peak plasma concentrations of active drug were attained at approximately 1 hour. The delayed absorption of acarbose-related radioactivity reflects the absorption of metabolites that may be formed by either intestinal bacteria or intestinal enzymatic hydrolysis.
Metabolism
Acarbose is metabolized exclusively within the gastrointestinal tract, principally by intestinal bacteria, but also by digestive enzymes. A fraction of these metabolites (approximately 34% of the dose) was absorbed and subsequently excreted in the urine. At least 13 metabolites have been separated chromatographically from urine specimens. The major metabolites have been identified as 4-methylpyrogallol derivatives (that is, sulfate, methyl, and glucuronide conjugates). One metabolite (formed by cleavage of a glucose molecule from acarbose) also has alpha-glucosidase inhibitory activity. This metabolite, together with the parent compound, recovered from the urine, accounts for less than 2% of the total administered dose.
Excretion
The fraction of acarbose that is absorbed as intact drug is almost completely excreted by the kidneys. When acarbose was given intravenously, 89% of the dose was recovered in the urine as active drug within 48 hours. In contrast, less than 2% of an oral dose was recovered in the urine as active (that is, parent compound and active metabolite) drug. This is consistent with the low bioavailability of the parent drug. The plasma elimination half-life of acarbose activity is approximately 2 hours in healthy volunteers. Consequently, drug accumulation does not occur with three times a day (t.i.d.) oral dosing.
Special Populations
The mean steady-state area under the curve (AUC) and maximum concentrations of acarbose were approximately 1.5 times higher in elderly compared to young volunteers; however, these differences were not statistically significant. Patients with severe renal impairment (Clcr < 25 mL/min/1.73m2) attained about 5 times higher peak plasma concentrations of acarbose and 6 times larger AUCs than volunteers with normal renal function. No studies of acarbose pharmacokinetic parameters according to race have been performed. In U.S. controlled clinical studies of PRECOSE in patients with type 2 diabetes mellitus, reductions in glycosylated hemoglobin levels were similar in Caucasians (n=478) and African-Americans (n=167), with a trend toward a better response in Latinos (n=132).
Drug-Drug Interactions
Studies in healthy volunteers have shown that PRECOSE has no effect on either the pharmacokinetics or pharmacodynamics of nifedipine, propranolol, or ranitidine. PRECOSE did not interfere with the absorption or disposition of the sulfonylurea glyburide in diabetic patients. PRECOSE may affect digoxin bioavailability and may require dose adjustment of digoxin by 16% (90% confidence interval: 8-23%), decrease mean Cmaxof digoxin by 26% (90% confidence interval: 16–34%) and decreases mean trough concentrations of digoxin by 9% (90% confidence limit: 19% decrease to 2% increase). (See PRECAUTIONS, Drug Interactions.)
The amount of metformin absorbed while taking PRECOSE was bioequivalent to the amount absorbed when taking placebo, as indicated by the plasma AUC values. However, the peak plasma level of metformin was reduced by approximately 20% when taking PRECOSE due to a slight delay in the absorption of metformin. There is little if any clinically significant interaction between PRECOSE and metformin.
-
CLINICAL TRIALS
Clinical Experience from Dose Finding Studies in Type 2 Diabetes Mellitus Patients on Dietary Treatment Only: Results from six controlled, fixed-dose, monotherapy studies of PRECOSE in the treatment of type 2 diabetes mellitus, involving 769 PRECOSE-treated patients, were combined and a weighted average of the difference from placebo in the mean change from baseline in glycosylated hemoglobin (HbA1c) was calculated for each dose level as presented below:
Table 1 Mean Placebo-Subtracted Change in HbA1c in Fixed-Dose Monotherapy Studies Dose of PRECOSE N Change in HbA1c
%p-Value - *
- Although studies utilized a maximum dose of 200 or 300 mg t.i.d., the maximum recommended dose for patients < 60 kg is 50 mg t.i.d.; the maximum recommended dose for patients > 60 kg is 100 mg t.i.d.
25 mg t.i.d.
110
-0.44
0.0307
50 mg t.i.d.
131
-0.77
0.0001
100 mg t.i.d.
244
-0.74
0.0001
200 mg t.i.d.*
231
-0.86
0.0001
300 mg t.i.d.*
53
-1.00
0.0001
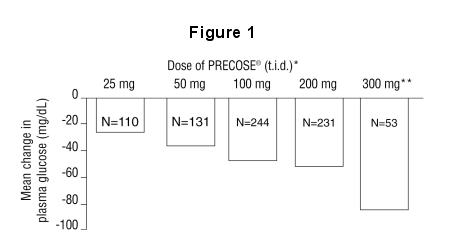
Results from these six fixed-dose, monotherapy studies were also combined to derive a weighted average of the difference from placebo in mean change from baseline for one-hour postprandial plasma glucose levels as shown in the following figure:
* PRECOSE was statistically significantly different from placebo at all doses with respect to effect on one-hour postprandial plasma glucose.
** The 300 mg t.i.d. PRECOSE regimen was superior to lower doses, but there were no statistically significant differences from 50 to 200 mg t.i.d.
Clinical Experience in Type 2 Diabetes Mellitus Patients on Monotherapy, or in Combination with Sulfonylureas, Metformin or Insulin: PRECOSE was studied as monotherapy and as combination therapy to sulfonylurea, metformin, or insulin treatment. The treatment effects on HbA1c levels and one-hour postprandial glucose levels are summarized for four placebo-controlled, double-blind, randomized studies conducted in the United States in Tables 2 and 3, respectively. The placebo-subtracted treatment differences, which are summarized below, were statistically significant for both variables in all of these studies.
Study 1 (n=109) involved patients on background treatment with diet only. The mean effect of the addition of PRECOSE to diet therapy was a change in HbA1c of -0.78%, and an improvement of one-hour postprandial glucose of -74.4 mg/dL.
In Study 2 (n=137), the mean effect of the addition of PRECOSE to maximum sulfonylurea therapy was a change in HbA1c of -0.54%, and an improvement of one-hour postprandial glucose of -33.5 mg/dL.
In Study 3 (n=147), the mean effect of the addition of PRECOSE to maximum metformin therapy was a change in HbA1c of -0.65%, and an improvement of one-hour postprandial glucose of -34.3 mg/dL.
Study 4 (n=145) demonstrated that PRECOSE added to patients on background treatment with insulin resulted in a mean change in HbA1c of -0.69%, and an improvement of one-hour postprandial glucose of -36.0 mg/dL.
A one year study of PRECOSE as monotherapy or in combination with sulfonylurea, metformin or insulin treatment was conducted in Canada in which 316 patients were included in the primary efficacy analysis (Figure 2). In the diet, sulfonylurea and metformin groups, the mean decrease in HbA1c produced by the addition of PRECOSE was statistically significant at six months, and this effect was persistent at one year. In the PRECOSE-treated patients on insulin, there was a statistically significant reduction in HbA1c at six months, and a trend for a reduction at one year.
Table 2: Effect of Precose on HbA1c HbA1c (%) Study Treatment Mean
BaselineMean change
from baselineTreatment
Differencep-Value 1
Placebo Plus Diet
8.67
+0.33
—
—
PRECOSE 100 mg t.i.d.
Plus Diet8.69
-0.45
-0.78
0.0001
2
Placebo Plus SFU*
9.56
+0.24
—
—
PRECOSE 50-300 mg t.i.d.
Plus SFU*9.64
-0.30
-0.54
0.0096
3
Placebo Plus Metformin†
8.17
+0.08
—
—
PRECOSE 50-100 mg t.i.d.
Plus Metformin†8.46
-0.57 g
-0.65
0.0001
4
Placebo Plus Insulin‡
8.69
+0.11
—
—
PRECOSE 50-100 mg t.i.d.
Plus Insulin‡8.77
-0.58
-0.69
0.0001
One-Hour Postprandial Glucose (mg/dL)
Study
Treatment
Mean
Mean change
Treatment
p-Value
Baseline
from baseline
Difference
1
Placebo Plus Diet
297.1
+31.8
—
—
PRECOSE 100 mg t.i.d.
299.1
-42.6
-74.4
0.0001
Plus Diet
2
Placebo Plus SFU*
308.6
+6.2
—
—
PRECOSE 50-300 mg t.i.d.
311.1
-27.3
-33.5
0.0017
Plus SFU*
3
Placebo Plus Metformin†
263.9
+3.3‡
—
—
PRECOSE 50-100 mg t.i.d.
283.0
-31.0‡
-34.3
0.0001
Plus Metformin†
4
Placebo Plus Insulin§
279.2
+8.0
—
—
PRECOSE 50-100 mg t.i.d.
277.8
-28.0
-36.0
0.0178
Plus Insulin§
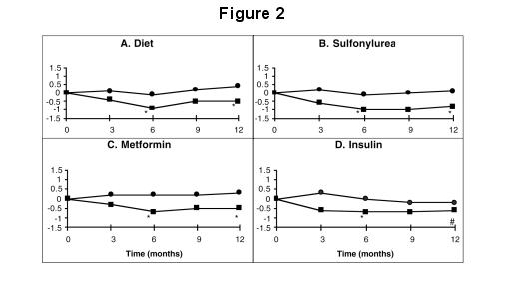
Figure 2: Effects of PRECOSE (■) and Placebo (●) on mean change in HbA1c levels from baseline throughout a one-year study in patients with type 2 diabetes mellitus when used in combination with: (A) diet alone; (B) sulfonylurea; (C) metformin; or (D) insulin. Treatment differences at 6 and 12 months were tested: * p < 0.01; # p = 0.077.
- INDICATIONS AND USAGE
-
CONTRAINDICATIONS
PRECOSE is contraindicated in patients with known hypersensitivity to the drug. Precose is contraindicated in patients with diabetic ketoacidosis or cirrhosis. PRECOSE is also contraindicated in patients with inflammatory bowel disease, colonic ulceration, partial intestinal obstruction or in patients predisposed to intestinal obstruction. In addition, PRECOSE is contraindicated in patients who have chronic intestinal diseases associated with marked disorders of digestion or absorption and in patients who have conditions that may deteriorate as a result of increased gas formation in the intestine.
-
PRECAUTIONS
General
Macrovascular Outcomes
There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with PRECOSE or any other anti-diabetic drug.
Hypoglycemia
Because of its mechanism of action, PRECOSE when administered alone should not cause hypoglycemia in the fasted or postprandial state. Sulfonylurea agents or insulin may cause hypoglycemia. Because PRECOSE given in combination with a sulfonylurea or insulin will cause a further lowering of blood glucose, it may increase the potential for hypoglycemia. Hypoglycemia does not occur in patients receiving metformin alone under usual circumstances of use, and no increased incidence of hypoglycemia was observed in patients when PRECOSE was added to metformin therapy. Oral glucose (dextrose), whose absorption is not inhibited by PRECOSE, should be used instead of sucrose (cane sugar) in the treatment of mild to moderate hypoglycemia. Sucrose, whose hydrolysis to glucose and fructose is inhibited by PRECOSE, is unsuitable for the rapid correction of hypoglycemia. Severe hypoglycemia may require the use of either intravenous glucose infusion or glucagon injection.
Elevated Serum Transaminase Levels
In long-term studies (up to 12 months, and including PRECOSE doses up to 300 mg t.i.d.) conducted in the United States, treatment-emergent elevations of serum transaminases (AST and/or ALT) above the upper limit of normal (ULN), greater than 1.8 times the ULN, and greater than 3 times the ULN occurred in 14%, 6%, and 3%, respectively, of PRECOSE-treated patients as compared to 7%, 2%, and 1%, respectively, of placebo-treated patients. Although these differences between treatments were statistically significant, these elevations were asymptomatic, reversible, more common in females, and, in general, were not associated with other evidence of liver dysfunction. In addition, these serum transaminase elevations appeared to be dose related. In US studies including PRECOSE doses up to the maximum approved dose of 100 mg t.i.d., treatment-emergent elevations of AST and/or ALT at any level of severity were similar between PRECOSE-treated patients and placebo-treated patients (p ≥ 0.496).
In approximately 3 million patient-years of international postmarketing experience with PRECOSE, 62 cases of serum transaminase elevations > 500 IU/L (29 of which were associated with jaundice) have been reported. Forty-one of these 62 patients received treatment with 100 mg t.i.d. or greater and 33 of 45 patients for whom weight was reported weighed < 60 kg. In the 59 cases where follow-up was recorded, hepatic abnormalities improved or resolved upon discontinuation of PRECOSE in 55 and were unchanged in two. Cases of fulminant hepatitis with fatal outcome have been reported; the relationship to acarbose is unclear.
Information for Patients
Patients should be told to take PRECOSE orally three times a day at the start (with the first bite) of each main meal. It is important that patients continue to adhere to dietary instructions, a regular exercise program, and regular testing of urine and/or blood glucose.
PRECOSE itself does not cause hypoglycemia even when administered to patients in the fasted state. Sulfonylurea drugs and insulin, however, can lower blood sugar levels enough to cause symptoms or sometimes life-threatening hypoglycemia. Because PRECOSE given in combination with a sulfonylurea or insulin will cause a further lowering of blood sugar, it may increase the hypoglycemic potential of these agents. Hypoglycemia does not occur in patients receiving metformin alone under usual circumstances of use, and no increased incidence of hypoglycemia was observed in patients when PRECOSE was added to metformin therapy. The risk of hypoglycemia, its symptoms and treatment, and conditions that predispose to its development should be well understood by patients and responsible family members. Because PRECOSE prevents the breakdown of table sugar, patients should have a readily available source of glucose (dextrose, D-glucose) to treat symptoms of low blood sugar when taking PRECOSE in combination with a sulfonylurea or insulin.
If side effects occur with PRECOSE, they usually develop during the first few weeks of therapy. They are most commonly mild-to-moderate gastrointestinal effects, such as flatulence, diarrhea, or abdominal discomfort, and generally diminish in frequency and intensity with time.
Laboratory Tests:
Therapeutic response to PRECOSE should be monitored by periodic blood glucose tests. Measurement of glycosylated hemoglobin levels is recommended for the monitoring of long-term glycemic control.
PRECOSE, particularly at doses in excess of 50 mg t.i.d., may give rise to elevations of serum transaminases and, in rare instances, hyperbilirubinemia. It is recommended that serum transaminase levels be checked every 3 months during the first year of treatment with PRECOSE and periodically thereafter. If elevated transaminases are observed, a reduction in dosage or withdrawal of therapy may be indicated, particularly if the elevations persist.
Renal Impairment:
Plasma concentrations of PRECOSE in renally impaired volunteers were proportionally increased relative to the degree of renal dysfunction. Long-term clinical trials in diabetic patients with significant renal dysfunction (serum creatinine > 2.0 mg/dL) have not been conducted. Therefore, treatment of these patients with PRECOSE is not recommended.
Drug Interactions
Certain drugs tend to produce hyperglycemia and may lead to loss of blood glucose control. These drugs include the thiazides and other diuretics, corticosteroids, phenothiazines, thyroid products, estrogens, oral contraceptives, phenytoin, nicotinic acid, sympathomimetics, calcium channel-blocking drugs, and isoniazid. When such drugs are administered to a patient receiving PRECOSE, the patient should be closely observed for loss of blood glucose control. When such drugs are withdrawn from patients receiving PRECOSE in combination with sulfonylureas or insulin, patients should be observed closely for any evidence of hypoglycemia.
Patients Receiving Sulfonylureas or Insulin: Sulfonylurea agents or insulin may cause hypoglycemia. PRECOSE given in combination with a sulfonylurea or insulin may cause a further lowering of blood glucose and may increase the potential for hypoglycemia. If hypoglycemia occurs, appropriate adjustments in the dosage of these agents should be made. Very rarely, individual cases of hypoglycemic shock have been reported in patients receiving PRECOSE therapy in combination with sulfonylureas and/or insulin.
Intestinal adsorbents (for example, charcoal) and digestive enzyme preparations containing carbohydrate-splitting enzymes (for example, amylase, pancreatin) may reduce the effect of PRECOSE and should not be taken concomitantly.
PRECOSE has been shown to change the bioavailability of digoxin when they are coadministered, which may require digoxin dose adjustment. (See CLINICAL PHARMACOLOGY, Drug-Drug Interactions .
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Eight carcinogenicity studies were conducted with acarbose. Six studies were performed in rats (two strains, Sprague-Dawley and Wistar) and two studies were performed in hamsters.
In the first rat study, Sprague-Dawley rats received acarbose in feed at high doses (up to approximately 500 mg/kg body weight) for 104 weeks. Acarbose treatment resulted in a significant increase in the incidence of renal tumors (adenomas and adenocarcinomas) and benign Leydig cell tumors. This study was repeated with a similar outcome. Further studies were performed to separate direct carcinogenic effects of acarbose from indirect effects resulting from the carbohydrate malnutrition induced by the large doses of acarbose employed in the studies. In one study using Sprague-Dawley rats, acarbose was mixed with feed but carbohydrate deprivation was prevented by the addition of glucose to the diet. In a 26-month study of Sprague-Dawley rats, acarbose was administered by daily postprandial gavage so as to avoid the pharmacologic effects of the drug. In both of these studies, the increased incidence of renal tumors found in the original studies did not occur. Acarbose was also given in food and by postprandial gavage in two separate studies in Wistar rats. No increased incidence of renal tumors was found in either of these Wistar rat studies. In two feeding studies of hamsters, with and without glucose supplementation, there was also no evidence of carcinogenicity.
Acarbose did not induce any DNA damage in vitro in the CHO chromosomal aberration assay, bacterial mutagenesis (Ames) assay, or a DNA binding assay. In vivo, no DNA damage was detected in the dominant lethal test in male mice, or the mouse micronucleus test.
Fertility studies conducted in rats after oral administration produced no untoward effect on fertility or on the overall capability to reproduce.
Pregnancy
Teratogenic Effects: Pregnancy Category B
The safety of PRECOSE in pregnant women has not been established. Reproduction studies have been performed in rats at doses up to 480 mg/kg (corresponding to 9 times the exposure in humans, based on drug blood levels) and have revealed no evidence of impaired fertility or harm to the fetus due to acarbose. In rabbits, reduced maternal body weight gain, probably the result of the pharmacodynamic activity of high doses of acarbose in the intestines, may have been responsible for a slight increase in the number of embryonic losses. However, rabbits given 160 mg/kg acarbose (corresponding to 10 times the dose in man, based on body surface area) showed no evidence of embryotoxicity and there was no evidence of teratogenicity at a dose 32 times the dose in man (based on body surface area). There are, however, no adequate and well-controlled studies of PRECOSE in pregnant women. Because animal reproduction studies are not always predictive of the human response, this drug should be used during pregnancy only if clearly needed. Because current information strongly suggests that abnormal blood glucose levels during pregnancy are associated with a higher incidence of congenital anomalies as well as increased neonatal morbidity and mortality, most experts recommend that insulin be used during pregnancy to maintain blood glucose levels as close to normal as possible.
Nursing Mothers:
A small amount of radioactivity has been found in the milk of lactating rats after administration of radiolabeled acarbose. It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, PRECOSE should not be administered to a nursing woman.
Geriatric Use:
Of the total number of subjects in clinical studies of PRECOSE in the United States, 27% were 65 and over, while 4% were 75 and over. No overall differences in safety and effectiveness were observed between these subjects and younger subjects. The mean steady-state area under the curve (AUC) and maximum concentrations of acarbose were approximately 1.5 times higher in elderly compared to young volunteers; however, these differences were not statistically significant.
-
ADVERSE REACTIONS
Digestive Tract
Gastrointestinal symptoms are the most common reactions to PRECOSE. In U.S. placebo-controlled trials, the incidences of abdominal pain, diarrhea, and flatulence were 19%, 31%, and 74% respectively in 1255 patients treated with PRECOSE 50–300 mg t.i.d., whereas the corresponding incidences were 9%, 12%, and 29% in 999 placebo-treated patients.
In a one-year safety study, during which patients kept diaries of gastrointestinal symptoms, abdominal pain and diarrhea tended to return to pretreatment levels over time, and the frequency and intensity of flatulence tended to abate with time. The increased gastrointestinal tract symptoms in patients treated with PRECOSE are a manifestation of the mechanism of action of PRECOSE and are related to the presence of undigested carbohydrate in the lower GI tract.
If the prescribed diet is not observed, the intestinal side effects may be intensified. If strongly distressing symptoms develop in spite of adherence to the diabetic diet prescribed, the doctor must be consulted and the dose temporarily or permanently reduced
Other Abnormal Laboratory Findings
Small reductions in hematocrit occurred more often in PRECOSE-treated patients than in placebo-treated patients but were not associated with reductions in hemoglobin. Low serum calcium and low plasma vitamin B6 levels were associated with PRECOSE therapy but are thought to be either spurious or of no clinical significance.
Postmarketing Adverse Event Reports
Additional adverse events reported from worldwide postmarketing experience include fulminant hepatitis with fatal outcome, hypersensitive skin reactions (for example rash, erythema, exanthema and uticaria), edema, ileus/subileus, jaundice and/or hepatitis and associated liver damage, thrombocytopenia, and pneumatosis cystoides intestinalis (see PRECAUTIONS).
Pneumatosis Cystoides Intestinalis
There have been rare postmarketing reports of pneumatosis cystoides intestinalis associated with the use of alpha-glucosidase inhibitors, including Precose. Pneumatosis cystoides intestinalis may present with symptoms of diarrhea, mucus discharge, rectal bleeding, and constipation. Complications may include pneumoperitoneum, volvulus, intestinal obstruction, intussusception, intestinal hemorrhage, and intestinal perforation. If pneumatosis cystoides intestinalis is suspected, discontinue Precose and perform the appropriate diagnostic imaging.
-
OVERDOSAGE
Unlike sulfonylureas or insulin, an overdose of PRECOSE will not result in hypoglycemia. An overdose may result in transient increases in flatulence, diarrhea, and abdominal discomfort which shortly subside. In cases of overdosage the patient should not be given drinks or meals containing carbohydrates (polysaccharides, oligosaccharides and disaccharides) for the next 4–6 hours.
-
DOSAGE AND ADMINISTRATION
There is no fixed dosage regimen for the management of diabetes mellitus with PRECOSE or any other pharmacologic agent. Dosage of PRECOSE must be individualized on the basis of both effectiveness and tolerance while not exceeding the maximum recommended dose of 100 mg t.i.d. PRECOSE should be taken three times daily at the start (with the first bite) of each main meal. PRECOSE should be started at a low dose, with gradual dose escalation as described below, both to reduce gastrointestinal side effects and to permit identification of the minimum dose required for adequate glycemic control of the patient. If the prescribed diet is not observed, the intestinal side effects may be intensified. If strongly distressing symptoms develop in spite of adherence to the diabetic diet prescribed, the doctor must be consulted and the dose temporarily or permanently reduced.
During treatment initiation and dose titration (see below), one-hour postprandial plasma glucose may be used to determine the therapeutic response to PRECOSE and identify the minimum effective dose for the patient. Thereafter, glycosylated hemoglobin should be measured at intervals of approximately three months. The therapeutic goal should be to decrease both postprandial plasma glucose and glycosylated hemoglobin levels to normal or near normal by using the lowest effective dose of PRECOSE, either as monotherapy or in combination with sulfonylureas, insulin or metformin.
Initial Dosage
The recommended starting dosage of PRECOSE is 25 mg given orally three times daily at the start (with the first bite) of each main meal. However, some patients may benefit from more gradual dose titration to minimize gastrointestinal side effects. This may be achieved by initiating treatment at 25 mg once per day and subsequently increasing the frequency of administration to achieve 25 mg t.i.d.
Maintenance Dosage
Once a 25 mg t.i.d. dosage regimen is reached, dosage of PRECOSE should be adjusted at 4–8 week intervals based on one-hour postprandial glucose or glycosylated hemoglobin levels, and on tolerance. The dosage can be increased from 25 mg t.i.d. to 50 mg t.i.d. Some patients may benefit from further increasing the dosage to 100 mg t.i.d. The maintenance dose ranges from 50 mg t.i.d. to 100 mg t.i.d. However, since patients with low body weight may be at increased risk for elevated serum transaminases, only patients with body weight > 60 kg should be considered for dose titration above 50 mg t.i.d. (see PRECAUTIONS). If no further reduction in postprandial glucose or glycosylated hemoglobin levels is observed with titration to 100 mg t.i.d., consideration should be given to lowering the dose. Once an effective and tolerated dosage is established, it should be maintained.
Maximum Dosage:
The maximum recommended dose for patients ≤ 60 kg is 50 mg t.i.d. The maximum recommended dose for patients > 60 kg is 100 mg t.i.d.
Patients Receiving Sulfonylureas or Insulin:
Sulfonylurea agents or insulin may cause hypoglycemia. PRECOSE given in combination with a sulfonylurea or insulin will cause a further lowering of blood glucose and may increase the potential for hypoglycemia. If hypoglycemia occurs, appropriate adjustments in the dosage of these agents should be made.
-
HOW SUPPLIED
PRECOSE is available as 25 mg, 50 mg or 100 mg round, unscored tablets. Each tablet strength is white to yellow-tinged in color. The 25 mg tablet is coded with the word “PRECOSE” on one side and “25” on the other side. The 50 mg tablet is coded with the word “PRECOSE” and “50” on the same side. The 100 mg tablet is coded with the word “PRECOSE” and “100” on the same side. PRECOSE is available in bottles of 100 and 50 mg strength in unit dose packages of 100.
Strength
NDC
Tablet
IdentificationBottles of 10:
25 mg
54868-5831-0
PRECOSE 25
Bottles of 90:
25 mg
54868-5831-1
PRECOSE 25
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
PRECOSE
acarbose tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5831(NDC:50419-863) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ACARBOSE (UNII: T58MSI464G) (ACARBOSE - UNII:T58MSI464G) ACARBOSE 25 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Product Characteristics Color WHITE (White to yellow tinged) Score no score Shape ROUND (ROUND) Size 6mm Flavor Imprint Code PRECOSE;25 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5831-0 10 in 1 BOTTLE 2 NDC:54868-5831-1 90 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020482 11/19/2007 Labeler - Physicians Total Care, Inc. (194123980) Establishment Name Address ID/FEI Business Operations Physicians Total Care, Inc. 194123980 relabel, repack