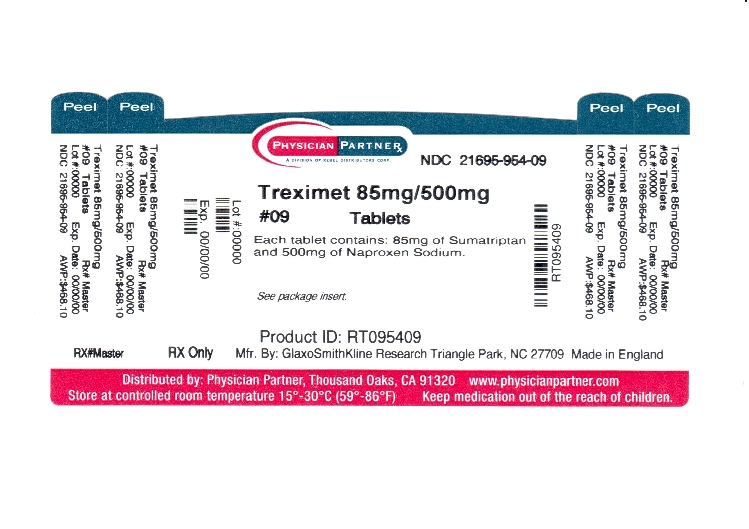
Label: TREXIMET- sumatriptan succinate and naproxen sodium tablet, film coated

-
Contains inactivated NDC Code(s)
NDC Code(s): 21695-954-09 - Packager: Rebel Distributors Corp
- This is a repackaged label.
- Source NDC Code(s): 0173-0750
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated September 13, 2011
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
WARNINGS
Cardiovascular Risk: TREXIMET may cause an increased risk of serious cardiovascular thrombotic events, myocardial infarction, and stroke, which can be fatal. This risk may increase with duration of use. Patients with cardiovascular disease or risk factors for cardiovascular disease may be at greater risk (see WARNINGS: Cardiovascular Effects).
Gastrointestinal Risk: TREXIMET contains a nonsteroidal anti-inflammatory drug (NSAID). NSAID-containing products cause an increased risk of serious gastrointestinal adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Elderly patients are at greater risk for serious gastrointestinal events (see WARNINGS: Risk of Gastrointestinal Ulceration, Bleeding, and Perforation With Nonsteroidal Anti-inflammatory Drug Therapy).
-
DESCRIPTION
TREXIMET contains sumatriptan (as the succinate), a selective 5-hydroxytryptamine1 (5-HT1) receptor subtype agonist, and naproxen sodium, a member of the arylacetic acid group of nonsteroidal anti-inflammatory drugs (NSAIDs).
Sumatriptan succinate is chemically designated as 3-[2-(dimethylamino)ethyl]-N-methyl-indole-5-methanesulfonamide succinate (1:1), and it has the following structure:
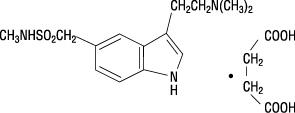
The empirical formula is C14H21N3O2S•C4H6O4, representing a molecular weight of 413.5. Sumatriptan succinate is a white to off-white powder that is readily soluble in water and in saline.
Naproxen sodium is chemically designated as (S)-6-methoxy-α-methyl-2-naphthaleneacetic acid, sodium salt, and it has the following structure:
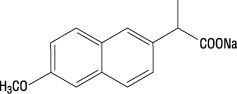
The empirical formula is C14H13NaO3, representing a molecular weight of 252.23. Naproxen sodium is a white-to-creamy white crystalline solid, freely soluble in water at neutral pH.
Each TREXIMET Tablet for oral administration contains 119 mg of sumatriptan succinate equivalent to 85 mg of sumatriptan and 500 mg of naproxen sodium. Each tablet also contains the inactive ingredients croscarmellose sodium, dextrose monohydrate, dibasic calcium phosphate, FD&C Blue No. 2, lecithin, magnesium stearate, maltodextrin, microcrystalline cellulose, povidone, sodium bicarbonate, sodium carboxymethylcellulose, talc, and titanium dioxide.
-
CLINICAL PHARMACOLOGY
Mechanism of Action
TREXIMET contains sumatriptan, a 5-HT1 receptor agonist that mediates vasoconstriction of the human basilar artery and vasculature of human dura mater, which correlates with the relief of migraine headache. It also contains naproxen, an NSAID that inhibits the synthesis of inflammatory mediators. Therefore, sumatriptan and naproxen contribute to the relief of migraine through pharmacologically different mechanisms of action.
Sumatriptan is a 5-HT1 receptor agonist that binds with high affinity to 5-HT1B and 5-HT1D receptors. Sumatriptan has only a weak affinity for 5-HT1A, 5-HT5A, and 5-HT7 receptors and no significant affinity (as measured using standard radioligand binding assays) or pharmacological activity at 5-HT2, 5-HT3, or 5-HT4 receptor subtypes or at alpha1-, alpha2-, or beta-adrenergic; dopamine1; dopamine2; muscarinic; or benzodiazepine receptors. In addition to causing vasoconstriction, experimental data from animal studies show that sumatriptan also activates 5-HT1 receptors on peripheral terminals of the trigeminal nerve innervating cranial blood vessels. Such an action may contribute to the antimigrainous effect of sumatriptan in humans. In the anesthetized dog, sumatriptan selectively reduces carotid arterial blood flow with little or no effect on arterial blood pressure or total peripheral resistance.
Naproxen sodium is an NSAID with analgesic and antipyretic properties. The sodium salt of naproxen has been developed as a more rapidly absorbed formulation of naproxen for use as an analgesic. The mechanism of action of the naproxen anion, like that of other NSAIDs, is not completely understood but may be related to prostaglandin synthetase inhibition.
Pharmacokinetics
TREXIMET is a formulation of 85 mg of sumatriptan (as sumatriptan succinate) and 500 mg of naproxen sodium with a distinct pharmacokinetic profile. Cmax (median, range) for sumatriptan following administration of TREXIMET occurs at approximately 1 hour (0.3 to 4.0 hours). Cmax (median, range) for naproxen following administration of TREXIMET occurs at approximately 5 hours (0.3 to 12 hours). The sumatriptan half-life is approximately 2 hours (15% to 43% CV) and the naproxen half-life is approximately 19 hours (13% to 15% CV). The mean Cmax for sumatriptan when given as TREXIMET is similar to that of sumatriptan when given as IMITREX® (sumatriptan succinate) Tablets 100 mg alone. The median sumatriptan Tmax is only slightly different (1 hour for TREXIMET and 1.5 hours for IMITREX). The Cmax for naproxen is approximately 36% lower, and the Tmax occurs approximately 4 hours later from TREXIMET than from ANAPROX® DS (naproxen sodium tablets) 550 mg. AUC values for sumatriptan and for naproxen are similar for TREXIMET compared to IMITREX or ANAPROX DS, respectively. In a crossover study in 16 patients, the pharmacokinetics of both components administered as TREXIMET were similar during a migraine attack and during a migraine-free period.
Absorption and Bioavailability: Bioavailability of sumatriptan is approximately 15%, primarily due to presystemic (first-pass) metabolism and partly due to incomplete absorption.
Naproxen is rapidly and completely absorbed from the gastrointestinal tract with an in vivo bioavailability of 95%.
Food Effects: Food had no significant effect on the bioavailability of sumatriptan or naproxen administered as TREXIMET, but slightly delayed the Tmax of sumatriptan by about 0.6 hour. These data indicate that TREXIMET may be administered without regard to food.
Distribution: The volume of distribution of sumatriptan is 2.4 L/kg. Plasma protein binding is 14% to 21%. The effect of sumatriptan on the protein binding of other drugs has not been evaluated, but would be expected to be minor, given the low protein binding.
The volume of distribution of naproxen is 0.16 L/kg. At therapeutic levels naproxen is greater than 99% albumin bound. At doses of naproxen greater than 500 mg/day, there is a less than proportional increase in plasma levels due to an increase in clearance caused by saturation of plasma protein binding at higher doses (average trough Css = 36.5, 49.2, and 56.4 mg/L with 500, 1,000, and 1,500 mg daily doses of naproxen, respectively). However, the concentration of unbound naproxen continues to increase proportionally to dose.
Metabolism: Most of a radiolabeled dose of sumatriptan excreted in the urine is the major metabolite indole acetic acid (IAA) or the IAA glucuronide, both of which are inactive. Three percent of the dose can be recovered as unchanged sumatriptan. In vitro studies with human microsomes suggest that sumatriptan is metabolized by monoamine oxidase (MAO), predominantly the A isoenzyme, and inhibitors of that enzyme may alter sumatriptan pharmacokinetics to increase systemic exposure (see CONTRAINDICATIONS and PRECAUTIONS: Drug Interactions: Monoamine Oxidase-A Inhibitors). No significant effect was seen with an MAO-B inhibitor.
Naproxen is extensively metabolized to 6-0-desmethyl naproxen, and both parent and metabolites do not induce metabolizing enzymes.
Elimination: Radiolabeled 14C-sumatriptan administered orally is largely renally excreted (about 60%), with about 40% found in the feces. The elimination half-life of sumatriptan is approximately 2 hours.
The clearance of naproxen is 0.13 mL/min/kg. Approximately 95% of the naproxen from any dose is excreted in the urine, primarily as naproxen (less than 1%), 6-0-desmethyl naproxen (less than 1%), or their conjugates (66% to 92%). The plasma half-life of the naproxen anion in humans is approximately 19 hours. The corresponding half-lives of both metabolites and conjugates of naproxen are shorter than 12 hours, and their rates of excretion have been found to coincide closely with the rate of naproxen disappearance from the plasma. In patients with renal failure, metabolites may accumulate (see PRECAUTIONS: Renal Effects).
Special Populations
Renal Impairment: TREXIMET is not recommended for use in patients with creatinine clearance less than 30 mL/min (see PRECAUTIONS: Renal Effects). The effect of renal impairment on the pharmacokinetics of TREXIMET has not been studied.
Minimal change in clinical effect would be expected with regard to sumatriptan as it is largely metabolized to an inactive substance.
Since naproxen and its metabolites and conjugates are primarily excreted by the kidney, the potential exists for naproxen metabolites to accumulate in the presence of renal insufficiency. Elimination of naproxen is decreased in patients with severe renal impairment.
Hepatic Impairment: Because TREXIMET is a fixed-dose combination that cannot be adjusted for this patient population, it is contraindicated in patients with hepatic impairment (see CONTRAINDICATIONS and PRECAUTIONS: Hepatic Effects). The effect of hepatic impairment on the pharmacokinetics of TREXIMET has not been studied. Sumatriptan is contraindicated in patients with severe hepatic impairment and the dose is limited to 50 mg in patients with liver disease.
Age: The effect of age (elderly or pediatric patients) on the pharmacokinetics of TREXIMET has not been studied. Elderly patients are more likely to have decreased hepatic function and decreased renal function (see PRECAUTIONS: Geriatric Use).
The pharmacokinetics of oral sumatriptan in the elderly (mean age: 72 years, 2 males and 4 females) and in patients with migraine (mean age: 38 years, 25 males and 155 females) were similar to that in healthy male subjects (mean age: 30 years).
Gender: In a pooled analysis of 5 pharmacokinetic studies, there was no effect of gender on the systemic exposure of TREXIMET. In a study comparing the pharmacokinetics of sumatriptan in females and males, no differences were observed between genders for AUC, Cmax, Tmax, and T½.
Race: The effect of race on the pharmacokinetics of TREXIMET has not been studied. The systemic clearance and Cmax of sumatriptan were similar in black (n = 34) and Caucasian (n = 38) healthy male subjects.
Drug Interactions
No formal drug interaction studies have been conducted with TREXIMET.
Monoamine Oxidase Inhibitors: TREXIMET is contraindicated in patients taking MAO-A inhibitors (see CONTRAINDICATIONS and PRECAUTIONS: Drug Interactions). Treatment with MAO-A inhibitors generally leads to an increase of sumatriptan plasma levels. This interaction has not been seen with an MAO-B inhibitor.
Alcohol: The effect of alcohol consumption on the pharmacokinetics of TREXIMET has not been studied. Alcohol consumed 30 minutes prior to sumatriptan ingestion had no effect on the pharmacokinetics of sumatriptan.
-
CLINICAL TRIALS
The efficacy of TREXIMET in providing relief from migraine was demonstrated in 2 randomized, double-blind, multicenter, parallel-group trials utilizing placebo and each individual active component of TREXIMET (sumatriptan and naproxen sodium) as comparison treatments. Patients enrolled in these 2 trials were predominately female (87%) and Caucasian (88%), with a mean age of 40 years (range 18 to 65 years). Patients were instructed to treat a migraine of moderate to severe pain with 1 tablet. No rescue medication was allowed within 2 hours postdose. Patients evaluated their headache pain 2 hours after taking 1 dose of study medication; headache relief was defined as a reduction in headache severity from moderate or severe pain to mild or no pain. Associated symptoms of nausea, photophobia, and phonophobia were also evaluated. Sustained pain free was defined as a reduction in headache severity from moderate or severe pain to no pain at 2 hours postdose without a return of mild, moderate, or severe pain and no use of rescue medication for 24 hours postdose. The results from the 2 controlled clinical trials are summarized in Table 1. In both trials, the percentage of patients achieving headache pain relief 2 hours after treatment was significantly greater among patients receiving TREXIMET (65% and 57%) compared with those who received placebo (28% and 29%).
Further, the percentage of patients who remained pain free without use of other medications through 24 hours postdose was significantly greater among patients receiving a single dose of TREXIMET (25% and 23%) compared with those who received placebo (8% and 7%) or either sumatriptan (16% and 14%) or naproxen sodium (10%) alone.
Table 1. Percentage of Patients With 2-Hour Pain Relief and Sustained Pain Free Following Treatmenta ap values provided only for prespecified comparisons.
bp<0.05 versus placebo and sumatriptan.
cp<0.01 versus placebo, sumatriptan, and naproxen sodium.
TREXIMET
Sumatriptan
85 mg
Naproxen Sodium
500 mg
Placebo
2-Hour Pain Relief
Study 1 (all patients)
65%b
n = 364
55%
n = 361
44%
n = 356
28%
n = 360
Study 2 (all patients)
57%b
n = 362
50%
n = 362
43%
n = 364
29%
n = 382
Sustained Pain Free (2-24 Hours)
Study 1
25%c
n = 364
16%
n = 361
10%
n = 356
8%
n = 360
Study 2
23%c
n = 362
14%
n = 362
10%
n = 364
7%
n = 382
Note that comparisons of the performance of different drugs based upon results obtained in different clinical trials are never reliable. Because studies are generally conducted at different times, with different samples of patients, by different investigators, employing different criteria and/or different interpretations of the same criteria, under different conditions (dose, dosing regimen, etc.), quantitative estimates of treatment response and the timing of response may be expected to vary considerably from study to study.
The percentage of patients achieving initial headache pain relief within 2 hours following treatment with TREXIMET is shown in Figure 1.
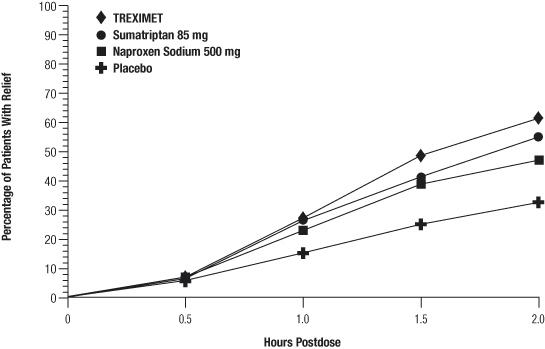
Figure 1. Percentage of Patients With Initial Headache Pain Relief Within 2 Hours
Compared with placebo, there was a decreased incidence of photophobia, phonophobia, and nausea 2 hours after the administration of TREXIMET. The estimated probability of taking a rescue medication over the first 24 hours is shown in Figure 2.
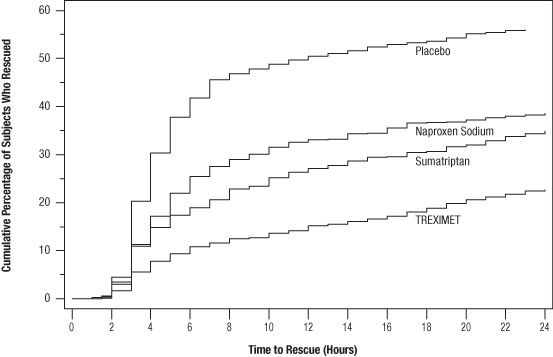
Figure 2. Cumulative Percentage of Subjects Taking a Rescue Medication Over the 24 Hours Following the First Dosea
aPlot also includes patients who had no response to the initial dose.
TREXIMET was more effective than placebo regardless of the presence of aura; duration of headache prior to treatment; gender, age, or weight of the patient; or concomitant use of oral contraceptives or common migraine prophylactic drugs (e.g., beta-blockers, anti-epileptic drugs, tricyclic antidepressants).
-
INDICATIONS AND USAGE
TREXIMET is indicated for the acute treatment of migraine attacks with or without aura in adults. Carefully consider the potential benefits and risks of TREXIMET and other treatment options when deciding to use TREXIMET.
TREXIMET is not intended for the prophylactic therapy of migraine or for use in the management of hemiplegic or basilar migraine (see CONTRAINDICATIONS). Safety and effectiveness of TREXIMET have not been established for cluster headache.
-
CONTRAINDICATIONS
Cardiac, Cerebrovascular, or Peripheral Vascular Disease
TREXIMET should not be given to patients with history, symptoms, or signs of ischemic cardiac, cerebrovascular, or peripheral vascular syndromes. In addition, patients with other significant underlying cardiovascular diseases should not receive TREXIMET, nor should patients who have had coronary artery bypass graft (CABG) surgery. Ischemic cardiac syndromes include, but are not limited to, angina pectoris of any type (e.g., stable angina of effort and vasospastic forms of angina, such as the Prinzmetal variant), all forms of myocardial infarction, and silent myocardial ischemia. Cerebrovascular syndromes include, but are not limited to, strokes of any type as well as transient ischemic attacks. Peripheral vascular disease includes, but is not limited to, ischemic bowel disease (see WARNINGS: Cardiovascular Effects).
Uncontrolled Hypertension
TREXIMET should not be given to patients with uncontrolled hypertension because the components have been shown to increase blood pressure.
Monoamine Oxidase-A Inhibitors
Concurrent administration of MAO-A inhibitors or use of TREXIMET within 2 weeks of discontinuation of MAO-A inhibitor therapy is contraindicated (see CLINICAL PHARMACOLOGY: Drug Interactions and PRECAUTIONS: Drug Interactions).
Ergotamine-Containing or Ergot-Type Medications
TREXIMET and any ergotamine-containing or ergot-type medication (like dihydroergotamine or methysergide) should not be used within 24 hours of each other (see PRECAUTIONS: Drug Interactions).
Other 5-HT1 Agonists
Since TREXIMET contains sumatriptan, it should not be administered within 24 hours of another 5-HT1 agonist.
Hemiplegic or Basilar Migraine
TREXIMET should not be administered to patients with hemiplegic or basilar migraine.
Hepatic Impairment
TREXIMET is contraindicated in patients with hepatic impairment (see CLINICAL PHARMACOLOGY: Special Populations, PRECAUTIONS: Hepatic Effects, and PRECAUTIONS: Geriatric Use).
Allergy to Naproxen/Asthma, Nasal Polyps, Urticaria, and Hypotension Associated With Nonsteroidal Anti-inflammatory Drugs
TREXIMET is contraindicated in patients who have had allergic reactions to prescription as well as to over-the-counter products containing naproxen. It is also contraindicated in patients in whom aspirin or other nonsteroidal anti-inflammatory/analgesic drugs induce the syndrome of asthma, rhinitis, and nasal polyps. Anaphylactic/anaphylactoid reactions to naproxen, whether of the true allergic type or the pharmacologic idiosyncratic type (e.g., aspirin hypersensitivity syndrome), usually but not always occur in patients with a known history of such reactions. Both types of reactions have the potential of being fatal. Therefore, careful questioning of patients for medical conditions such as asthma, nasal polyps, urticaria, and hypotension associated with NSAIDs before starting therapy is important. In addition, if such symptoms occur during therapy, treatment should be discontinued (see WARNINGS: Anaphylactic/Anaphylactoid Reactions and PRECAUTIONS: Preexisting Asthma).
-
WARNINGS
TREXIMET should only be used where a clear diagnosis of migraine headache has been established.
Cardiovascular Effects
Risk of Myocardial Ischemia and/or Infarction and Other Adverse Cardiac Events: TREXIMET should not be given to patients with documented ischemic or vasospastic coronary artery disease (CAD) or to patients with a history of CABG surgery (see CONTRAINDICATIONS). It is strongly recommended that sumatriptan-containing products not be given to patients in whom unrecognized CAD is predicted by the presence of risk factors (e.g., hypertension, hypercholesterolemia, smoker, obesity, diabetes, strong family history of CAD, female with surgical or physiological menopause, male over 40 years of age) unless a cardiovascular evaluation provides satisfactory clinical evidence that the patient is reasonably free of CAD and ischemic myocardial disease or other significant underlying cardiovascular disease. The sensitivity of cardiac diagnostic procedures to detect cardiovascular disease or predisposition to coronary artery vasospasm is modest, at best. If, during the cardiovascular evaluation, the patient’s medical history or electrocardiographic investigations reveal findings indicative of, or consistent with, coronary artery vasospasm or myocardial ischemia, TREXIMET should not be administered (see CONTRAINDICATIONS).
For patients with risk factors predictive of CAD who are determined to have a satisfactory cardiovascular evaluation, it is strongly recommended that administration of the first dose of TREXIMET take place in the setting of a physician’s office or similar medically staffed and equipped facility unless the patient has previously received sumatriptan. Because cardiac ischemia can occur in the absence of clinical symptoms, consideration should be given to obtaining an electrocardiogram (ECG) immediately following first-time use of TREXIMET in patients with risk factors.
It is recommended that patients who are intermittent long-term users of TREXIMET and who have or acquire risk factors predictive of CAD as described above undergo periodic cardiovascular evaluation as they continue to use TREXIMET.
The systematic approach described above is intended to reduce the likelihood that patients with unrecognized cardiovascular disease will be inadvertently exposed to sumatriptan-containing products.
Cardiac Events and Fatalities Associated With 5-HT1 Agonists: Serious adverse cardiac events, including acute myocardial infarction, life-threatening disturbances of cardiac rhythm, and death have been reported within a few hours following the administration of sumatriptan. Considering the extent of use of 5-HT1 agonists in patients with migraine, the incidence of these events is extremely low.
The fact that sumatriptan can cause coronary vasospasm, that some of these events have occurred in patients with no prior cardiac disease history and with documented absence of CAD, and the close proximity of the events to sumatriptan use support the conclusion that some of these cases were caused by the drug. In cases, however, where there has been known underlying coronary artery disease, the relationship is uncertain.
Cardiovascular Thrombotic Events and Fatalities Associated With Nonsteroidal Anti-inflammatory Drugs: Clinical trials of several COX-2 selective and nonselective NSAIDs of up to 3 years' duration have shown an increased risk of serious cardiovascular thrombotic events, myocardial infarction, and stroke, which can be fatal. All NSAIDs, both COX-2 selective and nonselective, may have a similar risk. Patients with known cardiovascular disease or risk factors for cardiovascular disease may be at greater risk. To minimize the potential risk for an adverse cardiovascular event in patients treated with an NSAID, the lowest effective dose should be used for the shortest duration possible. Physicians and patients should remain alert for the development of such events, even in the absence of previous cardiovascular symptoms. Patients should be informed about the signs and/or symptoms of serious cardiovascular events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious cardiovascular thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID does increase the risk of serious gastrointestinal events (see WARNINGS: Risk of Gastrointestinal Ulceration, Bleeding, and Perforation With Nonsteroidal Anti-inflammatory Drug Therapy).
Premarketing Experience With TREXIMET: Among 3,302 patients with migraine who received TREXIMET in premarketing controlled and uncontrolled clinical trials, a 47-year-old female with cardiac risk factors in an open-label 12-month safety study experienced signs and symptoms of acute coronary syndrome approximately 2 hours after receiving TREXIMET.
Drug-Associated Cerebrovascular Events and Fatalities: Cerebral hemorrhage, subarachnoid hemorrhage, stroke, and other cerebrovascular events have been reported in patients treated with oral or subcutaneous sumatriptan, and some have resulted in fatalities. The relationship of sumatriptan to these events is uncertain. In a number of cases, it appears possible that the cerebrovascular events were primary, sumatriptan having been administered in the incorrect belief that the symptoms experienced were a consequence of migraine when they were not. As with other acute migraine therapies, before treating headaches in patients not previously diagnosed as migraineurs, and in migraineurs who present with atypical symptoms, care should be taken to exclude other potentially serious neurological conditions. It should also be noted that patients with migraine may be at increased risk of certain cerebrovascular events (e.g., cerebrovascular accident, transient ischemic attack).
Other Vasospasm-Related Events: Sumatriptan may cause vasospastic reactions other than coronary artery vasospasm. Both peripheral vascular ischemia and colonic ischemia with abdominal pain and bloody diarrhea have been reported. Transient and permanent blindness and significant partial vision loss have been reported with the use of sumatriptan. Visual disorders may also be part of a migraine attack.
Increase in Blood Pressure: TREXIMET is contraindicated in patients with uncontrolled hypertension (see CONTRAINDICATIONS). TREXIMET should be used with caution in patients with controlled hypertension.
Significant elevation in blood pressure, including hypertensive crisis, has been reported in patients with and without a history of hypertension receiving sumatriptan. Sumatriptan-containing products should be administered with caution to patients with controlled hypertension as transient increases in blood pressure and peripheral vascular resistance have been observed.
NSAID-containing products can lead to onset of new hypertension or worsening of preexisting hypertension, either of which may contribute to the increased incidence of cardiovascular events. Patients taking thiazides or loop diuretics may have impaired response to these therapies when taking NSAIDs. The potential effect on blood pressure associated with long-term use of TREXIMET has not been studied. Blood pressure should be monitored closely during the initiation of NSAID treatment and throughout the course of therapy.
Congestive Heart Failure and Edema: TREXIMET should be used with caution in patients with fluid retention or heart failure. Fluid retention and edema have been observed in some patients taking NSAIDs. Since each TREXIMET tablet contains 61.2 mg of sodium (about 2.7 mEq/500 mg of naproxen sodium), this should be considered in patients whose overall intake of sodium must be severely restricted.
Serotonin Syndrome
The development of a potentially life-threatening serotonin syndrome may occur with triptans, including treatment with TREXIMET, particularly during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs). If concomitant treatment with TREXIMET and an SSRI (e.g., fluoxetine, paroxetine, sertraline, fluvoxamine, citalopram, escitalopram) or SNRI (e.g., venlafaxine, duloxetine) is clinically warranted, careful observation of the patient is advised, particularly during treatment initiation and dose increases. Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination), and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea) (see PRECAUTIONS: Drug Interactions).
Risk of Gastrointestinal Ulceration, Bleeding, and Perforation With Nonsteroidal Anti-inflammatory Drug Therapy
TREXIMET contains an NSAID. NSAID-containing products can cause serious gastrointestinal adverse events including inflammation, bleeding, ulceration, and perforation of the stomach, small intestine, or large intestine, which can be fatal.
These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with NSAIDs. Only 1 in 5 patients who develop a serious upper gastrointestinal adverse event on NSAID therapy is symptomatic. Upper gastrointestinal ulcers, gross bleeding, or perforation caused by NSAIDs appear to occur in approximately 1% of patients treated daily for 3 to 6 months and in about 2% to 4% of patients treated for 1 year. These trends continue with longer duration of use, increasing the likelihood of developing a serious gastrointestinal event at some time during the course of therapy. However, even short-term therapy is not without risk. Among 3,302 patients with migraine who received TREXIMET in premarketing controlled and uncontrolled clinical trials, 1 patient experienced a recurrence of gastric ulcer after taking 8 doses over 3 weeks, and 1 patient developed a gastric ulcer after treating an average of 8 attacks per month over 7 months.
NSAID-containing products, including TREXIMET, should be prescribed with extreme caution in those with a prior history of ulcer disease or gastrointestinal bleeding. Patients with a prior history of peptic ulcer disease and/or gastrointestinal bleeding who use NSAIDs have a greater than 10-fold increased risk for developing gastrointestinal bleeding compared to patients with neither of these risk factors. Other factors that increase the risk for gastrointestinal bleeding in patients treated with NSAIDs include concomitant use of oral corticosteroids or anticoagulants, longer duration of NSAID therapy, smoking, use of alcohol, older age, and poor general health status. Most spontaneous reports of fatal gastrointestinal events are in elderly or debilitated patients, and therefore special care should be taken in treating this population.
To minimize the potential risk for an adverse gastrointestinal event in patients treated with an NSAID-containing product, the lowest effective dose should be used for the shortest possible duration. Patients and physicians should remain alert for signs and symptoms of gastrointestinal ulceration and bleeding during NSAID therapy and promptly initiate additional evaluation and treatment if a serious gastrointestinal adverse event is suspected. This should include discontinuation of the NSAID until a serious gastrointestinal adverse event is ruled out. For high-risk patients, alternate therapies that do not involve NSAIDs should be considered.
NSAIDs should be given with care to patients with a history of inflammatory bowel disease (ulcerative colitis, Crohn's disease) as their condition may be exacerbated.
Renal Effects
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients administration of an NSAID may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and angiotensin-converting enzyme (ACE) inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
Advanced Renal Disease: Treatment with TREXIMET is not recommended in patients with advanced renal disease. If therapy with TREXIMET must be initiated, close monitoring of the patient’s renal function is advisable (see CLINICAL PHARMACOLOGY: Pharmacokinetics and PRECAUTIONS: Renal Effects). No information is available from controlled clinical studies regarding the use of TREXIMET in patients with advanced renal disease.
Anaphylactic/Anaphylactoid Reactions
As with other NSAID-containing products, anaphylactic/anaphylactoid reactions may occur in patients without known prior exposure to naproxen. TREXIMET should not be given to patients with the aspirin triad. This symptom complex typically occurs in patients with asthma who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs (see CONTRAINDICATIONS, PRECAUTIONS: Preexisting Asthma, and PRECAUTIONS: Drug Interactions).
Anaphylactic/anaphylactoid reactions have occurred in patients receiving sumatriptan. Such reactions can be life-threatening or fatal. In general, anaphylactic reactions to drugs are more likely to occur in individuals with a history of sensitivity to multiple allergens (see CONTRAINDICATIONS). Emergency help should be sought in cases where an anaphylactoid reaction occurs. Anaphylactoid reactions, like anaphylaxis, may have a fatal outcome.
Skin Reactions
NSAID-containing products, including TREXIMET, can cause serious adverse events such as exfoliative dermatitis, Stevens-Johnson syndrome, and toxic epidermal necrolysis, which can be fatal. These serious events may occur without warning. Patients should be informed about the signs and symptoms of serious skin manifestations and use of the drug should be discontinued at the first appearance of skin rash or any other sign of hypersensitivity.
Pregnancy
TREXIMET should not be used in late pregnancy because NSAID-containing products have been shown to cause premature closure of the ductus arteriosus. TREXIMET should not be used during early pregnancy unless the potential benefit justifies the potential risk to the fetus (see PRECAUTIONS: Pregnancy).
-
PRECAUTIONS
Naproxen-Containing Products
TREXIMET and other naproxen-containing products should not be used concomitantly since they all circulate in the plasma as the naproxen anion.
Chest, Jaw, or Neck Pain/Discomfort
Chest discomfort and jaw or neck tightness have been reported following use of sumatriptan. Only rarely have these symptoms been associated with ischemic ECG changes. However, because sumatriptan may cause coronary artery vasospasm, patients who experience signs or symptoms suggestive of angina following TREXIMET should be evaluated for the presence of CAD or a predisposition to Prinzmetal variant angina before receiving additional doses of TREXIMET and should be monitored electrocardiographically if dosing is resumed and similar symptoms recur. Similarly, patients who experience other symptoms or signs suggestive of decreased arterial flow, such as ischemic bowel syndrome or Raynaud syndrome, following TREXIMET should be evaluated for atherosclerosis or predisposition to vasospasm (see WARNINGS: Cardiovascular Effects).
Diseases That May Alter the Absorption, Metabolism, or Excretion of Drugs
TREXIMET should also be administered with caution to patients with diseases that may alter the absorption, metabolism, or excretion of drugs, such as impaired renal function.
Seizures
TREXIMET should be used with caution in patients with a history of epilepsy or conditions associated with a lowered seizure threshold. There have been reports of seizure following administration of sumatriptan.
Other Potentially Serious Neurologic Conditions
Care should be taken to exclude other potentially serious neurologic conditions before treating headache in patients not previously diagnosed with migraine headache or who experience a headache that is atypical for them. There have been reports where patients received sumatriptan for severe headaches that were subsequently shown to have been secondary to an evolving neurologic lesion (see WARNINGS: Drug-Associated Cerebrovascular Events and Fatalities). For a given attack, if a patient does not respond to the first dose of TREXIMET, the diagnosis of migraine should be reconsidered before administration of a second dose.
Hepatic Effects
TREXIMET is contraindicated in patients with hepatic impairment (see CONTRAINDICATIONS and CLINICAL PHARMACOLOGY).A patient with symptoms and/or signs suggesting liver dysfunction or in whom an abnormal liver test has occurred should be evaluated for evidence of the development of a more severe hepatic reaction while on therapy with TREXIMET. Borderline elevations of 1 or more liver tests may occur in up to 15% of patients who take NSAID-containing products. These abnormalities may progress, may remain essentially unchanged, or may be transient with continued therapy. Notable (3 times the upper limit of normal) elevations of SGPT (ALT) or SGOT (AST) have been reported in approximately 1% of patients in clinical trials with NSAIDs. In addition, cases of severe hepatic reactions, including jaundice and fatal fulminant hepatitis, liver necrosis, and hepatic failure, some of them with fatal outcomes, have been reported with NSAIDs. A patient with symptoms and/or signs suggesting liver dysfunction, or in whom an abnormal liver test has occurred, should be evaluated for evidence of the development of a more severe hepatic reaction while on therapy with TREXIMET. If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash), TREXIMET should be discontinued.
Overuse
Overuse of acute migraine treatments has been associated with the exacerbation of headache (medication overuse headache) in susceptible patients. Withdrawal of the treatment may be necessary.
Binding to Melanin-Containing Tissues
In rats treated with a single subcutaneous dose (0.5 mg/kg) or oral dose (2 mg/kg) of radiolabeled sumatriptan, the elimination half-life of radioactivity from the eye was 15 and 23 days, respectively, suggesting that sumatriptan and/or its metabolites bind to the melanin of the eye. Because there could be an accumulation in melanin-rich tissues over time, sumatriptan could possibly cause toxicity in these tissues after extended use. However, no effects on the retina related to treatment with sumatriptan were noted in any of the oral or subcutaneous toxicity studies. Although no systematic monitoring of ophthalmologic function was undertaken in clinical trials and no specific recommendations for ophthalmologic monitoring are offered, prescribers should be aware of the possibility of long-term ophthalmologic effects.
Corneal Opacities
Sumatriptan causes corneal opacities and defects in the corneal epithelium in dogs (see ANIMAL TOXICOLOGY). Adverse eye findings have also been observed in animal studies with some NSAIDs. Patients were not systematically evaluated for these changes in clinical trials. However, since the animal findings raise the possibility that adverse effects on the eye may occur in humans, it is recommended that ophthalmic studies be carried out if any change or disturbance in vision occurs.
Renal Effects
Caution is recommended in patients with preexisting kidney disease or dehydration (see WARNINGS: Renal Effects). Naproxen and its metabolites are eliminated primarily by the kidneys; therefore, TREXIMET should be used with caution in patients with significantly impaired renal function, and monitoring of serum creatinine and/or creatinine clearance is advised in these patients. TREXIMET is not recommended for use in patients with creatinine clearance less than 30 mL/min (see CLINICAL PHARMACOLOGY: Special Populations).
Hematological Effects
Patients on long-term treatment with NSAIDs, including TREXIMET, should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia. Anemia is sometimes seen in patients receiving NSAIDs. This may be due to fluid retention, occult or gross gastrointestinal blood loss, or an incompletely described effect upon erythropoiesis. Patients receiving TREXIMET who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants, should be carefully monitored. NSAID-containing products inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Unlike aspirin, their effect on platelet function is quantitatively less, of shorter duration, and reversible.
Preexisting Asthma
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm that can be fatal. Since cross reactivity, including bronchospasm, between aspirin and other NSAIDs has been reported in such aspirin-sensitive patients, TREXIMET should not be administered to patients with this form of aspirin sensitivity and should be used with caution in patients with preexisting asthma.
Information for Patients
Patients should be informed of the following information before initiating therapy with TREXIMET and periodically during the course of ongoing therapy. Patients should also be encouraged to read the Medication Guide that accompanies each prescription dispensed.
- TREXIMET may cause serious cardiovascular side effects such as myocardial infarction or stroke, which may result in hospitalization and even death. Although serious cardiovascular events can occur without warning symptoms, patients should be alert for the signs and symptoms of chest pain, shortness of breath, weakness, slurring of speech, and should ask for medical advice when observing any indicative sign or symptoms. Patients should be apprised of the importance of this follow-up (see WARNINGS: Cardiovascular Effects).
- TREXIMET, like other NSAID-containing products, may cause gastrointestinal discomfort and, rarely, serious gastrointestinal side effects such as ulcers and bleeding, which may result in hospitalization and even death. Although serious gastrointestinal tract ulcerations and bleeding can occur without warning symptoms, patients should be alert for the signs and symptoms of ulcerations and bleeding and should ask for medical advice when observing any indicative sign or symptoms, including epigastric pain, dyspepsia, melena, and hematemesis. Patients should be apprised of the importance of this follow-up (see WARNINGS: Risk of Gastrointestinal Ulceration, Bleeding, and Perforation With Nonsteroidal Anti-inflammatory Drug Therapy).
- TREXIMET, like other NSAID-containing products, may increase the risk of serious skin side effects such as exfoliative dermatitis, Stevens-Johnson syndrome, and toxic epidermal necrolysis, which may result in hospitalizations and even death. Although serious skin reactions may occur without warning, patients should be alert for the signs and symptoms of skin rash and blisters, fever, or other signs of hypersensitivity such as itching and should ask for medical advice when observing any indicative signs or symptoms. Patients should be advised to stop the drug immediately if they develop any type of rash and contact their physicians as soon as possible.
- Patients should promptly report signs or symptoms of unexplained weight gain or edema to their physicians.
- Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness, flu-like symptoms). If these occur, patients should be instructed to stop therapy and seek immediate medical therapy.
- Patients should be informed of the signs of an anaphylactic/anaphylactoid reaction (e.g., difficulty breathing, swelling of the face or throat). If these occur, patients should be instructed to seek immediate emergency help (see WARNINGS: Anaphylactic/Anaphylactoid Reactions).
- TREXIMET should not be used in late pregnancy because NSAID-containing products have been shown to cause premature closure of the ductus arteriosus. TREXIMET should not be used during early pregnancy unless the potential benefit justifies the potential risk to the fetus.
- Patients should be cautioned about the risk of serotonin syndrome, particularly during concomitant use with SSRIs or SNRIs.
- Caution should be exercised by patients whose activities require alertness if they experience drowsiness, dizziness, vertigo, or depression during therapy with TREXIMET.
Laboratory Tests
Because serious gastrointestinal tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs or symptoms of gastrointestinal bleeding. If clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash), or abnormal liver tests persist or worsen, TREXIMET should be discontinued.
Drug Interactions
Monoamine Oxidase-A Inhibitors: The use of TREXIMET in patients receiving MAO-A inhibitors is contraindicated (see CLINICAL PHARMACOLOGY: Drug Interactions and CONTRAINDICATIONS). MAO-A inhibitors reduce sumatriptan clearance, significantly increasing systemic exposure. In patients taking MAO-A inhibitors, sumatriptan plasma levels attained after treatment with recommended doses are 7-fold higher following oral administration than those obtained under other conditions.
Ergot-Containing Drugs: Ergot-containing drugs have been reported to cause prolonged vasospastic reactions. Because there is a theoretical basis that these effects may be additive, use of ergotamine-containing or ergot-type medications (e.g., dihydroergotamine, methysergide) and TREXIMET within 24 hours of each other should be avoided (see CONTRAINDICATIONS).
Methotrexate: Caution should be used if TREXIMET is administered concomitantly with methotrexate. Naproxen sodium and other NSAIDs have been reported to reduce the tubular secretion of methotrexate in an animal model, possibly increasing the toxicity of methotrexate. Concomitant administration of some NSAIDs with high-dose methotrexate therapy has been reported to elevate and prolong serum methotrexate levels, resulting in deaths from severe hematologic and gastrointestinal toxicity.
Aspirin: When naproxen is administered with aspirin, its protein binding is reduced, although the clearance of free naproxen is not altered. The clinical significance of this interaction is not known; however, as with other NSAID-containing products, concomitant administration of TREXIMET and aspirin is not generally recommended because of the potential of increased adverse effects.
Selective Serotonin Reuptake Inhibitors/Serotonin Norepinephrine Reuptake Inhibitors and Serotonin Syndrome: Cases of life-threatening serotonin syndrome have been reported during combined use of SSRIs or SNRIs and triptans (see WARNINGS: Serotonin Syndrome).
Angiotensin-Converting Enzyme Inhibitors: Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE inhibitors. The use of TREXIMET in patients who are receiving ACE inhibitors may potentiate renal disease states (see WARNINGS: Renal Effects).
Furosemide: Clinical studies, as well as postmarketing observations, have shown that NSAIDs can reduce the natriuretic effect of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis. During concomitant therapy with NSAIDs, the patient should be observed closely for signs of renal failure (see WARNINGS: Renal Effects), as well as to assure diuretic efficacy.
Lithium: NSAIDs have produced an elevation of plasma lithium levels and a reduction in renal lithium clearance. The mean minimum lithium concentration increased 15%, and the renal clearance was decreased by approximately 20%. These effects have been attributed to inhibition of renal prostaglandin synthesis by the NSAID. Thus, when TREXIMET and lithium are administered concurrently, patients should be observed carefully for signs of lithium toxicity.
Probenecid: Probenecid given concurrently increases naproxen anion plasma levels and extends its plasma half-life significantly.
Propranolol and Other Beta-Blockers: Propranolol 80 mg given twice daily had no significant effect on sumatriptan pharmacokinetics. Naproxen and other NSAIDs can reduce the antihypertensive effect of propranolol and other beta-blockers.
Warfarin: The effects of warfarin and NSAIDs on gastrointestinal bleeding are synergistic, such that patients taking both drugs have a higher risk of serious gastrointestinal bleeding than patients taking either drug alone.
Drug/Laboratory Test Interactions
The ability of TREXIMET to interfere with commonly employed clinical laboratory tests has not been investigated.
Sumatriptan is not known to interfere with commonly employed clinical laboratory tests. Naproxen may decrease platelet aggregation and prolong bleeding time. This effect should be kept in mind when bleeding times are determined.
The administration of naproxen sodium may result in increased urinary values for 17-ketogenic steroids because of an interaction between the drug and/or its metabolites with m-di-nitrobenzene used in this assay. Although 17-hydroxy-corticosteroid measurements (Porter-Silber test) do not appear to be artifactually altered, it is suggested that therapy with naproxen be temporarily discontinued 72 hours before adrenal function tests are performed if the Porter-Silber test is to be used.
Naproxen may interfere with some urinary assays of 5-hydroxy indoleacetic acid (5HIAA).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: The carcinogenic potential of TREXIMET has not been studied.
The carcinogenic potential of sumatriptan was evaluated in oral carcinogenicity studies in mice (78 weeks) and rats (104 weeks). The highest dose administered to mice and rats (160 mg/kg/day) is approximately 9 and 18 times, respectively, the recommended human oral daily dose of 85 mg sumatriptan on a mg/m2 basis. There was no evidence of an increase in tumors in either species related to sumatriptan administration.
The carcinogenic potential of naproxen sodium was evaluated in a 2-year oral carcinogenicity study in rats at doses of 8, 16, and 24 mg/kg/day and in another 2-year oral carcinogenicity study in rats at a dose of 8 mg/kg/day. No evidence of tumorigenicity was found in either study, at doses up to approximately 0.5 times the recommended human oral daily dose of 500 mg/day naproxen sodium on a mg/m2 basis.
Mutagenesis: Sumatriptan and naproxen sodium tested alone and in combination were negative in an in vitro bacterial reverse mutation assay, and in an in vivo micronucleus assay in mice.
The combination of sumatriptan and naproxen sodium was negative in an in vitro mouse lymphoma tk assay in the presence and absence of metabolic activation. However, in separate in vitro mouse lymphoma tk assays, naproxen sodium alone was reproducibly positive in the presence of metabolic activation.
Naproxen sodium alone and in combination with sumatriptan was positive in an in vitro clastogenicity assay in mammalian cells in the presence and absence of metabolic activation. The clastogenic effect for the combination was reproducible within this assay and was greater than observed with naproxen sodium alone. Sumatriptan alone was negative in these assays.
Chromosomal aberrations were not induced in peripheral blood lymphocytes following 7 days of twice-daily dosing with TREXIMET in human volunteers.
In previous studies, sumatriptan alone was not mutagenic in 2 gene mutation assays (the Ames test and the in vitro Chinese Hamster V79/HGPRT assay) and was not clastogenic in 2 cytogenetics assays (the in vitro human lymphocyte assay and the in vivo rat micronucleus assay).
Impairment of Fertility: The effect of TREXIMET on fertility in animals has not been studied.
In a study in which male and female rats were dosed daily with oral sumatriptan prior to and throughout the mating period, there was a treatment-related decrease in fertility secondary to a decrease in mating in animals treated with 50 and 500 mg/kg/day. The highest no-effect dose for this finding was 5 mg/kg/day, or approximately 0.5 times the recommended human oral daily dose of 85 mg sumatriptan on a mg/m2 basis. It is not clear whether the problem is associated with treatment of the males or females or both combined. In a similar study of sumatriptan by the subcutaneous route there was no evidence of impaired fertility at doses up to 60 mg/kg/day.
Pregnancy
Pregnancy Category C. In developmental toxicity studies in rabbits, oral treatment with sumatriptan combined with naproxen sodium (5/9, 25/45, or 50/90 mg/kg/day sumatriptan/naproxen sodium) or each drug alone (50/0 or 0/90 mg/kg/day sumatriptan/naproxen sodium) resulted in decreased fetal body weight in all treated groups and in increased embryofetal death at the highest dose of naproxen, alone and in combination with sumatriptan. Naproxen sodium, alone and in combination with sumatriptan, increased the total incidences of fetal abnormalities at all doses and increased the incidences of specific malformations (cardiac interventricular septal defect in the 50/90-mg/kg/day group, fused caudal vertebrae in the 50/0- and 0/90-mg/kg/day groups) and variations (absent intermediate lobe of the lung, irregular ossification of the skull, incompletely ossified sternal centra) in the 50/0- and 0/90-mg/kg/day groups. A no-effect dose for development toxicity in rabbits was not established. The lowest effect dose was 5/9 mg/kg/day sumatriptan/naproxen sodium, which was associated with plasma exposures (AUC) to sumatriptan and naproxen that were 1.4 and 0.14 times, respectively, those attained at the maximum recommended human oral daily dose of 85 mg sumatriptan and 500 mg naproxen sodium.
In previous developmental toxicity studies in rats and rabbits, oral treatment with sumatriptan was associated with embryolethality, fetal abnormalities, and pup mortality. Oral treatment of pregnant rats with sumatriptan during the period of organogenesis resulted in an increased incidence of fetal blood vessel (cervicothoracic and umbilical) abnormalities and decreased pup survival at doses of 250 mg/kg/day or higher. The highest no-effect dose was approximately 60 mg/kg/day, which is approximately 7 times the recommended human oral daily dose of 85 mg sumatriptan on a mg/m2 basis. Oral treatment of pregnant rabbits with sumatriptan during the period of organogenesis resulted in an increased incidence of cervicothoracic vascular and skeletal abnormalities at a dose of 50 mg/kg/day and embryolethality at 100 mg/kg/day. The highest no-effect dose for embryotoxicity in rabbits was 15 mg/kg/day, or approximately 3 times the recommended human oral daily dose of 85 mg sumatriptan on a mg/m2 basis.
Inhibitors of prostaglandin synthesis (including naproxen) are known to delay parturition. Because of this and the known effects of drugs of this class on the human fetal cardiovascular system (closure of the ductus arteriosus), use during third trimester should be avoided.
There are no adequate and well-controlled studies in pregnant women.
TREXIMET should not be used during pregnancy unless the potential benefit justifies the potential risk to the fetus.
Pregnancy Registry: To monitor fetal outcomes of pregnant women exposed to TREXIMET, GlaxoSmithKline maintains a TREXIMET Pregnancy Registry. Physicians are encouraged to register patients as soon as possible after they become pregnant and (if possible) before the outcome of the pregnancy is known by calling (800) 336-2176.
Labor and Delivery
In rat studies with NSAIDs, as with other drugs known to inhibit prostaglandin synthesis, an increased incidence of dystocia, delayed parturition, and decreased pup survival occurred. Naproxen-containing products are not recommended in labor and delivery because, through its prostaglandin synthesis inhibitory effect, naproxen may adversely affect fetal circulation and inhibit uterine contractions, thus increasing the risk of uterine hemorrhage.
Nursing Mothers
Both active components of TREXIMET, sumatriptan and naproxen sodium, have been reported to be excreted in human breast milk. Because of the possible adverse effects of these drugs on neonates, use of TREXIMET in nursing mothers should be avoided.
Geriatric Use
TREXIMET is contraindicated for use in elderly patients who have abnormal hepatic function, and is not recommended for use in elderly patients who have decreased renal function, higher risk for unrecognized CAD, and increases in blood pressure that may be more pronounced in the elderly (see CONTRAINDICATIONS: Hepatic Impairment, WARNINGS: Cardiovascular Effects, and CLINICAL PHARMACOLOGY: Pharmacokinetics).
-
ADVERSE REACTIONS
The adverse reactions reported below are specific to the clinical trials with TREXIMET. See also the full prescribing information for naproxen and sumatriptan products.
Incidence in Controlled Clinical Trials
Table 2 lists adverse events that occurred in 2 placebo-controlled clinical trials evaluating patients who took at least 1 dose of study drug. Only events that occurred at a frequency of 2% or more with TREXIMET and were more frequent than in the placebo group are included in Table 2. The events cited reflect experience gained under closely monitored conditions of clinical trials in a highly selected patient population. In actual clinical practice or in other clinical trials, these frequency estimates may not apply, as the conditions of use, reporting behavior, and the kinds of patients treated may differ.
Table 2. Treatment-Emergent Adverse Events Reported by at Least 2% of Patients in 2 Controlled Migraine Trialsa aEvents that occurred at a frequency of 2% or more in the group treated with TREXIMET and that occurred more frequently in the group treated with TREXIMET than in the placebo group.
Adverse Event Percent of Patients Reporting TREXIMET
(n = 737)Placebo
(n = 752)Sumatriptan
85 mg
(n = 735)Naproxen Sodium
500 mg
(n = 732)Nervous system disorders Dizziness 4 2 2 2 Somnolence 3 2 2 2 Paresthesia 2 <1 2 <1 Gastrointestinal disorders Nausea 3 1 3 <1 Dyspepsia 2 1 2 1 Dry mouth 2 1 2 <1 Pain and other pressure sensations Chest discomfort/chest pain 3 <1 2 1 Neck/throat/jaw pain/tightness/pressure 3 1 3 1 Other events that occurred in more than 1% of patients receiving TREXIMET and occurred at a frequency greater than the placebo group included asthenia, feeling hot, muscle tightness, and palpitations.
TREXIMET was generally well tolerated. Most adverse reactions were mild and transient. The incidence of adverse events in controlled clinical trials was not affected by gender or age of the patients. There were insufficient data to assess the impact of race on the incidence of adverse events.
Other Events Observed in Migraine Clinical Trials Associated With the Administration of TREXIMET
The occurrence of less commonly reported adverse clinical events is presented in this section. Because the reports include events observed in an open-label, long-term safety study in which TREXIMET was used as needed for up to 12 months, the role of TREXIMET cannot be reliably determined. Furthermore, variability associated with adverse event reporting, the terminology used to describe adverse events, etc., limit the value of quantitative frequency estimates provided. Event frequencies are calculated as the number of patients who used TREXIMET and reported an event divided by the total number of patients (N = 3,302) exposed to TREXIMET. Events listed in the previous table and text are not included below. Those events described too generally to be informative or those unlikely to be associated with the use of TREXIMET are excluded. Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are those occurring in at least 1/100 patients, infrequent adverse events are those occurring in 1/100 to 1/1,000 patients, and rare adverse events are those occurring in fewer than 1/1,000 patients.
Blood and Lymphatic Disorders: Infrequent was lymphadenopathy. Rare were anemia, ecchymosis, leukopenia.
Cardiac Disorders: Infrequent was tachycardia. Rare were acute coronary syndrome, cardiac flutter, congestive cardiac failure, right ventricular failure, ventricular extrasystoles.
Ear and Labyrinth Disorders: Infrequent were ear pain, tinnitus. Rare were motion sickness, vertigo.
Endocrine, Metabolic, and Nutrition Disorders: Rare were diabetes mellitus, goiter, hypoglycemia, hypothyroidism.
Eye Disorders: Infrequent was conjunctivitis. Rare were cataract, conjunctival hemorrhage, visual disturbance.
Gastrointestinal Disorders: Frequent was abdominal pain. Infrequent were abdominal distention, constipation, diarrhea, dysgeusia, dysphagia, flatulence, gastritis, gastroesophageal reflux disease, vomiting. Rare were colitis, diverticulitis, gastric ulcer, irritable bowel syndrome, oral mucosal blistering, swollen tongue.
General Disorders: Frequent was fatigue. Infrequent were feeling jittery, lethargy, malaise, peripheral edema, pyrexia, temperature intolerance, thirst. Rare was difficulty in walking.
Hepatobiliary Disorders: Rare was biliary colic.
Infections and Infestations: Rare were kidney infection, pneumonia, sepsis, staphylococcal infection, viral myocarditis.
Musculoskeletal and Connective Tissue: Infrequent were arthralgia, back pain, muscular weakness, myalgia, sensation of heaviness.
Nervous System Disorders: Infrequent were burning sensation, disturbance of attention, insomnia, mental impairment, tremor. Rare were aphasia, facial palsy, impairment of psychomotor skills, sedation.
Psychiatric Disorders: Infrequent were anxiety, depression, irritability, nervousness. Rare were disorientation, panic attack.
Renal and Urinary Disorders: Infrequent was nephrolithiasis. Rare was renal insufficiency.
Respiratory, Thoracic, and Mediastinal: Infrequent were asthma, cough, dyspnea, oropharyngeal swelling. Rare was pleurisy.
Skin and Subcutaneous Disorders: Infrequent were facial swelling, hyperhydrosis, pruritus, rash, urticaria. Rare was systemic lupus erythematosus.
Vascular Disorders: Infrequent were flushing, hot flush, hypertension. Rare were epistaxis, peripheral coldness.
-
DRUG ABUSE AND DEPENDENCE
The potential for abuse with TREXIMET has not been studied.
One clinical study with sumatriptan succinate injection enrolling 12 patients with a history of substance abuse failed to induce subjective behavior and/or physiologic response ordinarily associated with drugs that have an established potential for abuse.
-
OVERDOSAGE
Because strategies for the management of overdose are continually evolving, it is advisable to contact a Poison Control Center to determine the latest recommendations for the management of an overdose of any drug.
There have been no reports of overdosage with TREXIMET. Since sumatriptan and naproxen have pharmacologically different actions, it is difficult to predict how an individual will respond to an overdosage with TREXIMET.
Patients (N = 670) have received single oral doses of 140 to 300 mg of sumatriptan without significant adverse effects. Volunteers (N = 174) have received single oral doses of 140 to 400 mg without serious adverse events. Overdose of sumatriptan in animals has been fatal and has been heralded by convulsions, tremor, paralysis, inactivity, ptosis, erythema of the extremities, abnormal respiration, cyanosis, ataxia, mydriasis, salivation, and lacrimation.
Significant naproxen overdosage may be characterized by lethargy, dizziness, drowsiness, epigastric pain, abdominal discomfort, heartburn, indigestion, nausea, transient alterations in liver function, hypoprothrombinemia, renal dysfunction, metabolic acidosis, apnea, disorientation, or vomiting. Gastrointestinal bleeding can occur. Hypertension, acute renal failure, respiratory depression, and coma may occur, but are rare. Anaphylactoid reactions have been reported with therapeutic ingestion of NSAIDs, and may occur following an overdose. Because naproxen sodium may be rapidly absorbed, high and early blood levels should be anticipated. A few patients have experienced seizures, but it is not clear whether or not these were drug related. It is not known what dose of the drug would be life threatening.
In animals 0.5 g/kg of activated charcoal was effective in reducing plasma levels of naproxen. Patients should be managed by symptomatic and supportive care. There are no specific antidotes. Hemodialysis does not decrease the plasma concentration of naproxen because of the high degree of its protein binding. It is unknown what effect hemodialysis or peritoneal dialysis has on the serum concentrations of sumatriptan. Emesis and/or activated charcoal (60 to 100 g in adults, 1 to 2 g/kg in children) and/or osmotic cathartic may be indicated in patients seen within 4 hours of ingestion with symptoms or following a large overdose. Forced diuresis, alkalinization of urine, or hemoperfusion may not be useful due to high protein binding.
-
DOSAGE AND ADMINISTRATION
TREXIMET is a fixed combination containing doses of sumatriptan (85 mg) and naproxen sodium (500 mg) within the approved dosage ranges of the individual components (25 to 100 mg of sumatriptan and 220 to 825 mg of naproxen sodium). TREXIMET contains a dose of sumatriptan higher than the lowest effective dose. Individuals may vary in response to doses of sumatriptan. The choice of the dose of sumatriptan, and of the use of a fixed combination such as in TREXIMET should therefore be made on an individual basis, weighing the possible benefit of a higher dose of sumatriptan with the potential for a greater risk of adverse events. Carefully consider the potential benefits and risks of TREXIMET and other treatment options when deciding to use TREXIMET.
The recommended dose is 1 tablet. In controlled clinical trials, single doses of TREXIMET were effective for the acute treatment of migraine in adults (see CLINICAL TRIALS).
The efficacy of taking a second dose has not been established. Do not take more than 2 TREXIMET tablets in 24 hours. Dosing of tablets should be at least 2 hours apart. The safety of treating an average of more than 5 migraine headaches in a 30-day period has not been established.
TREXIMET may be administered with or without food. Tablets should not be split, crushed, or chewed.
The combined use of TREXIMET with MAO-A inhibitors or use of TREXIMET within 2 weeks of discontinuation of MAO-A inhibitor therapy is contraindicated (see CONTRAINDICATIONS, CLINICAL PHARMACOLOGY: Drug Interactions, PRECAUTIONS: Drug Interactions).
TREXIMET and any ergotamine-containing or ergot-type medication (like dihydroergotamine or methysergide) should not be used within 24 hours of each other. TREXIMET and other 5-HT1 agonists should not be administered within 24 hours of each other (see CONTRAINDICATIONS and PRECAUTIONS: Drug Interactions).
TREXIMET is contraindicated in patients with hepatic impairment (see CONTRAINDICATIONS and CLINICAL PHARMACOLOGY: Special Populations).
TREXIMET is not recommended for use in patients with creatinine clearance less than 30 mL/min (see CLINICAL PHARMACOLOGY: Special Populations and PRECAUTIONS: Renal Effects).
-
HOW SUPPLIED
TREXIMET contains 119 mg of sumatriptan succinate equivalent to 85 mg of sumatriptan and 500 mg of naproxen sodium and is supplied as blue film-coated tablets debossed on one side with TREXIMET in compact containers of 9 tablets with a specially formulated, non-removable desiccant (NDC 21695-954-09).
Store at 25°C (77°F); excursions permitted to 15°-30°C (59°-86°F) [see USP Controlled Room Temperature]. Do not repackage; dispense and store in original container.
-
ANIMAL TOXICOLOGY
Corneal Opacities
Dogs receiving oral sumatriptan developed corneal opacities and defects in the corneal epithelium. Corneal opacities were seen at the lowest dosage tested, 2 mg/kg/day, and were present after 1 month of treatment. Defects in the corneal epithelium were noted in a 60-week study. Earlier examinations for these toxicities were not conducted and no-effect doses were not established; the lowest dose tested is approximately 0.8 times the recommended human oral daily dose of 85 mg sumatriptan on a mg/m2 basis. There was evidence of alterations in corneal appearance on the first day of intranasal dosing to dogs at all doses tested.
-
MEDICATION GUIDE
MEDICATION GUIDE
TREXIMET®[trex' i-met] Tablets
(sumatriptan and naproxen sodium)
What is the most important information I should know about TREXIMET?
TREXIMET, which contains sumatriptan and naproxen sodium [a nonsteroidal anti-inflammatory drug (NSAID)], may increase the chance of a heart attack or stroke that can lead to death. This chance increases:
- with longer use of NSAID medicines
- in people who have heart disease.
NSAID-containing medicines, such as TREXIMET, should never be used right before or after a heart surgery called a coronary artery bypass graft (CABG).
NSAID-containing medicines, such as TREXIMET, can cause ulcers and bleeding in the stomach and intestines at any time during treatment. Ulcers and bleeding:
- can happen without warning symptoms
- may cause death.
The chance of a person getting an ulcer or bleeding increases with:
- the use of medicines called steroid hormones (corticosteroids) and blood thinners (anticoagulants)
- longer use
- more frequent use
- smoking
- drinking alcohol
- older age
- having poor health.
TREXIMET is not recommended for people with risk factors for heart disease unless a heart exam is done and shows no problems.
The risk factors for heart disease include:
- high blood pressure
- high cholesterol levels
- smoking
- obesity
- diabetes
- family history of heart disease
- female who has gone through menopause
- male over age 40.
“Serotonin syndrome” is a serious and life-threatening problem that may occur with TREXIMET, especially if used with antidepressant medicines called selective serotonin reuptake inhibitors (SSRIs) or selective norepinephrine reuptake inhibitors (SNRIs).
Commonly used SSRIs are:
- CELEXA® (citalopram HBr)
- LEXAPRO® (escitalopram oxalate)
- PAXIL® (paroxetine)
- PROZAC®/SARAFEM® (fluoxetine)
- SYMBYAX® (olanzapine/fluoxetine)
- ZOLOFT® (sertraline)
- LUVOX® (fluvoxamine).
Commonly used SNRIs are:
- CYMBALTA® (duloxetine)
- EFFEXOR® (venlafaxine).
Call your healthcare provider if you have symptoms of serotonin syndrome, which include:
- mental changes (hallucinations, agitation, coma)
- fast heartbeat
- changes in blood pressure
- high body temperature or sweating
- tight muscles
- trouble walking
- nausea, vomiting, diarrhea.
TREXIMET should only be used:
- exactly as prescribed
- at the lowest dose possible for your treatment
- for the shortest time needed.
TREXIMET already contains an NSAID (naproxen). Do not use TREXIMET with other medicines to lessen pain or fever without talking to your healthcare provider first, because they may contain an NSAID also.
What is TREXIMET?
TREXIMET is a prescription medicine used to treat migraine attacks in adults. It does not prevent or lessen the number of migraines you have, and it is not for other types of headaches. TREXIMET contains 2 medicines: sumatriptan and naproxen sodium (an NSAID). This Medication Guide provides important information you need to know before taking TREXIMET. It does not take the place of talking with your healthcare provider about your medical condition or your treatment.
How should I take TREXIMET?
- Take 1 TREXIMET tablet to treat your migraine headache. Do not take more than 2 TREXIMET tablets in 24 hours. Doses should be separated by at least 2 hours.
- TREXIMET can be taken with or without food.
- Do not split, crush, or chew TREXIMET tablets.
- If you take too much TREXIMET, call the Poison Control Center at 1-800-222-1222.
Who should not take TREXIMET?
Do not take TREXIMET right before or after heart bypass surgery.
Do not take TREXIMET if you have or have had:
- uncontrolled high blood pressure
- hemiplegic or basilar migraine. (Ask your doctor if you are not sure what type of migraine you have.)
- liver problems
- an asthma attack, hives, or other allergic reaction with aspirin or any other NSAID medicine
- a heart attack or a history or symptoms of heart disease (such as chest pain or angina)
- a stroke, mini-stroke (transient ischemic attack or TIA), or other stroke-like syndrome
- problems with blood circulation to parts of your body, such as less blood flow to your intestines (ischemic bowel disease)
- allergic reactions to sumatriptan, naproxen, or other ingredients in TREXIMET.
Do not take TREXIMET if you take or have taken an antidepressant medicine called a monoamine oxidase (MAO) inhibitor within the last 2 weeks. Common MAO inhibitors are isocarboxazid (MARPLAN®), phenelzine (NARDIL®), tranylcypromine (PARNATE®), and selegiline (ELDEPRYL®, EMSAM®). Ask your healthcare provider if you are not sure if your medicine is an MAO inhibitor.
Do not take TREXIMET if you have taken other migraine medicines in the last 24 hours such as:
- ergotamine-containing medicine or
- another triptan medicine.
Before starting TREXIMET, tell your healthcare provider about:
- all of your medical conditions including kidney or liver problems
- all allergies to any medicines
- chest pain, shortness of breath, irregular heartbeats
- medicines you may take for migraines, depression, or other health problems such as MAO inhibitors, SSRIs, or SNRIs
- all the prescription and non-prescription medicines you take, including vitamins and herbal supplements. Some medicines can interact with TREXIMET and cause serious side effects.
Keep a list of your medicines to show to your healthcare provider.
Before starting TREXIMET, tell your healthcare provider if you:
- are pregnant, think you might be pregnant, or are trying to become pregnant. TREXIMET should not be used by pregnant women late in their pregnancy.
- are breastfeeding
- have a headache that is different from your usual migraine
- have or have had epilepsy or seizures.
What are the possible side effects of TREXIMET?
Serious side effects include: - heart attack
- heartbeat problems
- stroke
- high blood pressure
- heart failure from body swelling (fluid retention)
- kidney problems including kidney failure
- bleeding and ulcers in the stomach and intestine
- low red blood cells (anemia)
- life-threatening skin reactions
- life-threatening allergic reactions
- liver problems including liver failure
- asthma attacks in people who have asthma
- loss of blood circulation to areas of your body
- serotonin syndrome (See list of symptoms in “What is the most important information I should know about TREXIMET?”)
Other side effects include:
- pain, tightness, or pressure in the chest, neck, and throat
- stomach pain
- constipation
- diarrhea
- gas
- heartburn
- nausea
- vomiting
- dizziness
- drowsiness
- tiredness
- weakness
- tingling and numbness
- unusual body sensations
- redness of face (flushed)
Get emergency help right away if you have any of the following symptoms:
- shortness of breath or trouble breathing
- chest pain
- swelling of the face or throat
- weakness in one part or on one side of your body
- slurred speech.
Stop TREXIMET and call your healthcare provider right away if you have any of the following symptoms:
- nausea that seems out of proportion to your migraine
- stomach pain
- sudden/severe pain in your belly
- vomit blood
- blood in your bowel movement or it is black and sticky like tar
- itching
- skin rash or blisters with fever
- yellow skin or eyes
- swelling of the arms and legs, hands, feet, face, lips, or tongue
- unusual weight gain
- more tired or weaker than usual
- flu-like symptoms
- serotonin syndrome. See list of symptoms in “What is the most important information I should know about TREXIMET?”
Tell your healthcare provider if you have any side effects that bother you or do not go away. These are not all of the side effects of TREXIMET. For more information ask your healthcare provider.
Call your healthcare provider for medical advice about side effects. You may report side effects at FDA at 1-800-FDA-1088.
How should I store TREXIMET?
- Store TREXIMET at room temperature, 59° to 86°F (15° to 30°C).
- Keep TREXIMET and all medicines out of the reach of children.
General information about TREXIMET
- Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TREXIMET for a condition for which it was not prescribed.
- Do not give TREXIMET to other people, even if they have the same problem you have. It may harm them.
- This Medication Guide contains the most important information about TREXIMET. If you would like more information, talk with your healthcare provider.
- You can ask your healthcare provider for information written for healthcare professionals.
- For more information call 1-888-825-5249 (toll-free), or visit www.TREXIMET.com.
What are the ingredients in TREXIMET?
Active ingredients: sumatriptan succinate and naproxen sodium
Inactive ingredients: croscarmellose sodium, dextrose monohydrate, dibasic calcium phosphate, FD&C Blue No. 2, lecithin, magnesium stearate, maltodextrin, microcrystalline cellulose, povidone, sodium bicarbonate, sodium carboxymethylcellulose, talc, and titanium dioxide.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
June 2009 TRX:3MG
TREXIMET is a trademark of GlaxoSmithKline. IMITREX, PARNATE, and PAXIL are registered trademarks of GlaxoSmithKline.
The other brands listed are trademarks of their respective owners and are not trademarks of GlaxoSmithKline. The makers of these brands are not affiliated with and do not endorse GlaxoSmithKline or its products.
GlaxoSmithKline
Research Triangle Park, NC 27709
©2009, GlaxoSmithKline. All rights reserved.
December 2009 TRX:5PI
Repackaged by:
Rebel Distributors Corp
Thousand Oaks, CA 91320
- Principal Display Panel
-
INGREDIENTS AND APPEARANCE
TREXIMET
sumatriptan succinate and naproxen sodium tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:21695-954(NDC:0173-0750) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SUMATRIPTAN SUCCINATE (UNII: J8BDZ68989) (SUMATRIPTAN - UNII:8R78F6L9VO) SUMATRIPTAN SUCCINATE 85 mg NAPROXEN SODIUM (UNII: 9TN87S3A3C) (NAPROXEN - UNII:57Y76R9ATQ) NAPROXEN SODIUM 500 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) DEXTROSE MONOHYDRATE (UNII: LX22YL083G) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) LECITHIN, SOYBEAN (UNII: 1DI56QDM62) MAGNESIUM STEARATE (UNII: 70097M6I30) MALTODEXTRIN (UNII: 7CVR7L4A2D) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POVIDONE (UNII: FZ989GH94E) SODIUM BICARBONATE (UNII: 8MDF5V39QO) CARBOXYMETHYLCELLULOSE SODIUM (UNII: K679OBS311) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color BLUE Score no score Shape CAPSULE (capsule-shaped) Size 19mm Flavor Imprint Code TREXIMET Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:21695-954-09 9 in 1 CONTAINER Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021926 04/25/2008 Labeler - Rebel Distributors Corp (118802834) Establishment Name Address ID/FEI Business Operations Rebel Distributors Corp 118802834 RELABEL, REPACK