Label: DOXORUBICIN HYDROCHLORIDE- doxorubicin hydrochloride injection, solution




- NDC Code(s): 63323-101-61, 63323-883-05, 63323-883-10, 63323-883-30
- Packager: Fresenius Kabi USA, LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated December 31, 2019
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
WARNING
- Severe local tissue necrosis will occur if there is extravasation during administration (see DOSAGE AND ADMINISTRATION). Doxorubicin must not be given by the intramuscular or subcutaneous route.
- Myocardial toxicity manifested in its most severe form by potentially fatal congestive heart failure (CHF) may occur either during therapy or months to years after termination of therapy. The probability of developing impaired myocardial function based on a combined index of signs, symptoms and decline in left ventricular ejection fraction (LVEF) is estimated to be 1 to 2% at a total cumulative dose of 300 mg/m 2 of doxorubicin, 3 to 5% at a dose of 400 mg/m 2, 5 to 8% at 450 mg/m 2 and 6 to 20% at 500 mg/m 2. The risk of developing CHF increases rapidly with increasing total cumulative doses of doxorubicin in excess of 400 mg/m 2. Risk factors (active or dormant cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, concomitant use of other cardiotoxic drugs) may increase the risk of cardiac toxicity. Cardiac toxicity with doxorubicin may occur at lower cumulative doses whether or not cardiac risk factors are present. Pediatric patients are at increased risk for developing delayed cardiotoxicity.
- Secondary acute myelogenous leukemia (AML) or myelodysplastic syndrome (MDS) has been reported in patients treated with anthracyclines, including doxorubicin (see ADVERSE REACTIONS). The occurrence of refractory secondary AML or MDS is more common when anthracyclines are given in combination with DNA-damaging anti-neoplastic agents or radiotherapy, when patients have been heavily pretreated with cytotoxic drugs, or when doses of anthracyclines have been escalated. The rate of developing secondary AML or MDS has been estimated in an analysis of 8,563 patients with early breast cancer treated in 6 studies conducted by the National Surgical Adjuvant Breast and Bowel Project (NSABP), including NSABP B-15. Patients in these studies received standard doses of doxorubicin and standard or escalated doses of cyclophosphamide (AC) adjuvant chemotherapy and were followed for 61,810 patient years. Among 4,483 such patients who received conventional doses of AC, 11 cases of AML or MDS were identified, for an incidence of 0.32 cases per 1,000 patient years (95% Cl, 0.16 to 0.57) and a cumulative incidence at 5 years of 0.21% (95% Cl, 0.11 to 0.41%). In another analysis of 1,474 patients with breast cancer who received adjuvant treatment with doxorubicin-containing regimens in clinical trials conducted at University of Texas M.D. Anderson Cancer Center, the incidence was estimated at 1.5% at 10 years. In both experiences, patients who received regimens with higher cyclophosphamide dosages, who received radiotherapy, or who were aged 50 or older had an increased risk of secondary AML or MDS. Pediatric patients are also at risk of developing secondary AML.
- Dosage should be reduced in patients with impaired hepatic function.
- Severe myelosuppression may occur.
- Doxorubicin should be administered only under the supervision of a physician who is experienced in the use of cancer chemotherapeutic agents.
-
DESCRIPTION:
Doxorubicin is a cytotoxic anthracycline antibiotic isolated from cultures of Streptomyces peucetius var .caesius. Doxorubicin consists of a naphthacenequinone nucleus linked through a glycosidic bond at ring atom 7 to an amino sugar, daunosamine. Chemically, doxorubicin hydrochloride is: 5,12-Naphthacenedione, 10-[(3-amino-2,3,6-trideoxy-α-L- lyxo-hexopyranosyl)oxy]-7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(hydroxylacetyl)-1-methoxy-, hydrochloride (8 S- cis)-. (8 S, 10 S)-10-[(3-Amino-2,3,6-trideoxy-α-L- lyxo-hexopyranosyl)-oxy]-8-glycoloyl-7,8,9,10-tetrahydro-6,8,11-trihydroxy-1-methoxy-5,12-naphthacenedione hydrochloride [ 25316-40-9].
The structural formula is as follows:
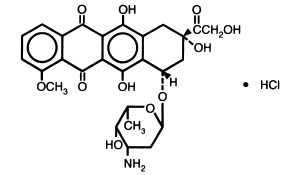
C 27H 29NO 11• HCl M.W.579.99
Doxorubicin binds to nucleic acids, presumably by specific intercalation of the planar anthracycline nucleus with the DNA double helix. The anthracycline ring is lipophilic, but the saturated end of the ring system contains abundant hydroxyl groups adjacent to the amino sugar, producing a hydrophilic center. The molecule is amphoteric, containing acidic functions in the ring phenolic groups and a basic function in the sugar amino group. It binds to cell membranes as well as plasma proteins.Doxorubicin Hydrochloride Injection, USP is a sterile, isotonic, preservative-free solution for intravenous use. It is available in 5 mL (10 mg), 10 mL (20 mg) and 25 mL (50 mg) single dose vials and 100 mL (200 mg) multiple dose vials.
Each mL contains: Doxorubicin hydrochloride 2 mg; sodium chloride 9 mg for Isotonicity: Water for Injection q.s. Hydrochloric acid and/or sodium hydroxide may have been added for pH adjustment (2.5 to 4.5).
-
CLINICAL PHARMACOLOGY:
The cytotoxic effect of doxorubicin on malignant cells and its toxic effects on various organs are thought to be related to nucleotide base intercalation and cell membrane lipid binding activities of doxorubicin. Intercalation inhibits nucleotide replication and action of DNA and RNA polymerases. The interaction of doxorubicin with topoisomerase II to form DNA-cleavable complexes appears to be an important mechanism of doxorubicin cytocidal activity.
Doxorubicin cellular membrane binding may affect a variety of cellular functions. Enzymatic electron reduction of doxorubicin by a variety of oxidases, reductases and dehydrogenases generates highly reactive species including the hydroxyl free radical OH•. Free radical formation has been implicated in doxorubicin cardiotoxicity by means of Cu (II) and Fe (III) reduction at the cellular level.
Cells treated with doxorubicin have been shown to manifest the characteristic morphologic changes associated with apoptosis or programmed cell death. Doxorubicin-induced apoptosis may be an integral component of the cellular mechanism of action relating to therapeutic effects, toxicities, or both.
Animal studies have shown activity in a spectrum of experimental tumors, immunosuppression, carcinogenic properties in rodents, induction of a variety of toxic effects, including delayed and progressive cardiac toxicity, myelosuppression in all species and atrophy to testes in rats and dogs.
Pharmacokinetics
Pharmacokinetic studies, determined in patients with various types of tumors undergoing either single or multi-agent therapy have shown that doxorubicin follows a multiphasic disposition after intravenous injection. In four patients, doxorubicin has demonstrated dose-independent pharmacokinetics in the dose range of 30 to 70 mg/m 2.
Distribution
The initial distribution half-life of approximately 5 minutes suggests rapid tissue uptake of doxorubicin, while its slow elimination from tissues is reflected by a terminal half-life of 20 to 48 hours. Steady-state distribution volume ranges from 809 to 1,214 L/m 2 and is indicative of extensive drug uptake into tissues. Binding of doxorubicin and its major metabolite, doxorubicinol, to plasma proteins is about 74 to 76% and is independent of plasma concentration of doxorubicin up to 1.1 mcg/mL.
Doxorubicin was excreted in the milk of one lactating patient, with peak milk concentration at 24 hours after treatment being approximately 4.4-fold greater than the corresponding plasma concentration. Doxorubicin was detectable in the milk up to 72 hours after therapy with 70 mg/m 2 of doxorubicin given as a 15-minute intravenous infusion and 100 mg/m 2 of cisplatin as a 26-hour intravenous infusion. The peak concentration of doxorubicinol in milk at 24 hours was 0.11 mcg/mL and AUC up to 24 hours was 9 mcg•h/mL while the AUC for doxorubicin was 5.4 mcg•h/mL.
Doxorubicin does not cross the blood brain barrier.
Metabolism
Enzymatic reduction at the 7 position and cleavage of the daunosamine sugar yields aglycones which are accompanied by free radical formation, the local production of which may contribute to the cardiotoxic activity of doxorubicin. Disposition of doxorubicinol (DOX-OL) in patients is formation rate limited, with the terminal half-life of DOX-OL being similar to doxorubicin. The relative exposure of DOX-OL, i.e., the ratio between the AUC of DOX-OL and the AUC of doxorubicin, compared to doxorubicin ranges between 0.4 and 0.6.
Excretion
Plasma clearance is in the range 324 to 809 mL/min/m 2 and is predominately by metabolism and biliary excretion. Approximately 40% of the dose appears in the bile in 5 days, while only 5 to 12% of the drug and its metabolites appear in the urine during the same time period. In urine, < 3% of the dose was recovered as DOX-OL over 7 days.
Systemic clearance of doxorubicin is significantly reduced in obese women with ideal body weight greater than 130%. There was a significant reduction in clearance without any change in volume of distribution in obese patients when compared with normal patients with less than 115% ideal body weight.
Pharmacokinetics in Special Populations
Pediatric
Following administration of 10 to 75 mg/m 2 doses of doxorubicin to 60 children and adolescents ranging from 2 months to 20 years of age, doxorubicin clearance averaged 1,443 ± 114 mL/min/m 2. Further analysis demonstrated that clearance in 52 children greater than 2 years of age (1,540 mL/min/m 2) was increased compared with adults. However, clearance in infants younger than 2 years of age (813 mL/min/m 2) was decreased compared with older children and approached the range of clearance values determined in adults.
Geriatric
While the pharmacokinetics of elderly subjects (≥65 years of age) have been evaluated, no dosage adjustment is recommended based on age (see PRECAUTIONS, Geriatric Use).
Gender
A published clinical study involving 6 men and 21 women with no prior anthracycline therapy reported a significantly higher median doxorubicin clearance in the men compared to the women (1,088 mL/min/m 2 versus 433 mL/min/m 2). However, the terminal half-life of doxorubicin was longer in men compared to the women (54 versus 35 hours).
Race
The influence of race on the pharmacokinetics of doxorubicin has not been evaluated.
Hepatic Impairment
The clearance of doxorubicin and doxorubicinol was reduced in patients with impaired hepatic function (see DOSAGE AND ADMINISTRATION).
Renal Impairment
The influence of renal function on the pharmacokinetics of doxorubicin has not been evaluated.
-
CLINICAL STUDIES:
The effectiveness of doxorubicin-containing regimens in the adjuvant therapy of early breast cancer has primarily been established based on data collected in a meta-analysis published in 1998 by the Early Breast Cancer Trialists Collaborative Group (EBCTCG). The EBCTCG obtains primary data on all relevant studies, both published and unpublished, for early stage breast cancer and regularly updates these analyses. The principal endpoints for the adjuvant chemotherapy trials were disease-free survival (DFS) and overall survival (OS). The meta-analyses allowed comparisons of cyclophosphamide, methotrexate, and 5-fluorouracil (CMF) to no chemotherapy (19 trials including 7,523 patients) and comparisons of doxorubicin-containing regimens with CMF as an active control (6 trials including 3,510 patients). The pooled estimates of DFS and OS from these trials were used to calculate the effect of CMF relative to no therapy. The hazard ratio for DFS for CMF compared to no chemotherapy was 0.76 (95% Cl, 0.71 to 0.82) and for OS was 0.86 (95% Cl, 0.8 to 0.93). Based on a conservative estimate of CMF effect (lower 2-sided 95% confidence limit of hazard ratio) and 75% retention of CMF effect on DFS, it was determined that the doxorubicin-containing regimens would be considered as non-inferior to CMF if the upper 2-sided 95% confidence limit of the hazard ratio was less than 1.06, i.e., not more than 6% worse than CMF. A similar calculation for OS would require a non-inferiority margin of 1.02.
Six randomized trials in the EBCTCG meta-analysis compared doxorubicin-containing regimens to CMF. A total of 3,510 women with early breast cancer involving axillary lymph nodes were evaluated; approximately 70% were premenopausal and 30% were postmenopausal. At the time of the meta-analysis, 1,745 first recurrences and 1,348 deaths had occurred. Analyses demonstrated that doxorubicin-containing regimens retained at least 75% of the historical CMF adjuvant effect on DFS and are effective. The hazard ratio for DFS (dox:CMF) was 0.91 (95% Cl, 0.82 to 1.01) and for OS was 0.91 (95% Cl, 0.81 to 1.03). Results of these analyses for both DFS and OS are provided in Table 1 and Figures 1 and 2.
Table 1. Summary of Randomized Trials Comparing Doxorubicin-Containing Regimens Versus CMF in EBCTCG Meta-Analysis
Study
(starting year)
Regimens
No. of Cycles
No. of Patients
Doxorubicin-Containing Regimens vs CMF
HR (95% CI)
DFS
OS
NSABP B-15
(1984)
AC
4
1,562*
0.93 (0.82 to 1.06)
0.97 (0.83 to 1.12)
CMF
6
776
SECSG 2
(1976)
FAC
6
260
0.86 (0.66 to 1.13)
0.93 (0.69 to 1.26)
CMF
6
268
ONCOFRANCE
(1978)
FACV
12
138
0.71 (0.49 to 1.03)
0.65 (0.44 to 0.96)
CMF
12
113
SE Sweden BCG A (1980)
AC
6
21
0.59 (0.22 to 1.61)
0.53 (0.21 to 1.37)
CMF
6
22
NSABC Israel Br0283 (1983)
AVbCMF †
CMF
4
6
6
55
50
0.91 (0.53 to 1.57)
0.88 (0.47 to 1.63)
Austrian BCSG 3 (1984)
CMFVA
6
121
1.07 (0.73 to 1.55)
0.93 (0.64 to 1.35)
CMF
8
124
Combined Studies
Doxorubicin-Containing Regimens
2,157
0.91 (0.82 to 1.01)
0.91 (0.81 to 1.03)
CMF
1,353
Abbreviations: DFS = disease free survival; OS = overall survival; AC = doxorubicin, cyclophosphamide; AVbCMF = doxorubicin, vinblastine, cyclophosphamide, methotrexate, 5-fluorouracil; CMF = cyclophosphamide, methotrexate, 5-fluorouracil; CMFVA = cyclophosphamide, methotrexate, 5-fluorouracil, vincristine, doxorubicin; FAC = 5-fluorouracil, doxorubicin, cyclophosphamide; FACV = 5-fluorouracil, doxorubicin, cyclophosphamide, vincristine; HR = hazard ratio; CI = confidence interval
_________________________________________________________________________________________________________________________
* Includes pooled data from patients who received either AC alone for 4 cycles, or who were treated with AC for 4 cycles followed by 3 cycles of CMF.
† Patients received alternating cycles of AVb and CMF.
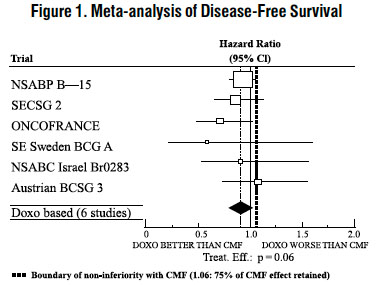
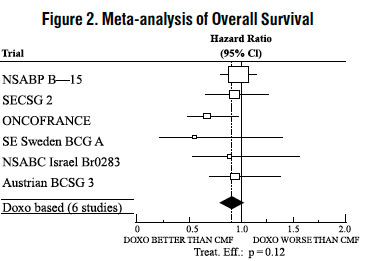
With respect to DFS, 2 of 6 studies (NSABP B-15 and ONCOFRANCE) met the non-inferiority standard individually and with respect to OS, 1 study met the non-inferiority margin individually (ONCOFRANCE). The largest of the 6 studies in the EBCTCG meta-analysis, a randomized, open-label, multicenter trial (NSABP B-15) was conducted in approximately 2,300 women (80% premenopausal; 20% postmenopausal) with early breast cancer involving axillary lymph nodes. In this trial, 6 cycles of conventional CMF was compared to 4 cycles of doxorubicin and cyclophosphamide (AC) and 4 cycles of AC followed by 3 cycles of CMF. No statistically significant differences in terms of DFS or OS were observed (see Table 1).
-
INDICATIONS AND USAGE:
Doxorubicin Hydrochloride Injection, USP has been used successfully to produce regression in disseminated neoplastic conditions such as acute lymphoblastic leukemia, acute myeloblastic leukemia, Wilms' tumor, neuroblastoma, soft tissue and bone sarcomas, breast carcinoma, ovarian carcinoma, transitional cell bladder carcinoma, thyroid carcinoma, gastric carcinoma, Hodgkin's disease, malignant lymphoma and bronchogenic carcinoma in which the small cell histologic type is the most responsive compared to other cell types.
Doxorubicin is also indicated for use as a component of adjuvant therapy in women with evidence of axillary lymph node involvement following resection of primary breast cancer.
-
CONTRAINDICATIONS:
Patients should not be treated with doxorubicin if they have any of the following conditions: baseline neutrophil count <1,500 cells/mm 3; severe hepatic impairment; recent myocardial infarction; severe myocardial insufficiency; severe arrhythmias; previous treatment with complete cumulative doses of doxorubicin, daunorubicin, idarubicin, and/or other anthracyclines and anthracenediones; or hypersensitivity to doxorubicin, any of its excipients, or other anthracyclines or anthracenediones (see WARNINGS and DOSAGE AND ADMINISTRATION).
-
WARNINGS:
General
Doxorubicin should be administered only under the supervision of qualified physicians experienced in the use of cytotoxic therapy. Patients should recover from acute toxicities of prior cytotoxic treatment (such as stomatitis, neutropenia, thrombocytopenia, and generalized infections) before beginning treatment with doxorubicin. Also, initial treatment with doxorubicin should be preceded by a careful baseline assessment of blood counts; serum levels of total bilirubin, AST, and creatinine; and cardiac function as measured by left ventricular ejection function (LVEF). Patients should be carefully monitored during treatment for possible clinical complications due to myelosuppression. Supportive care may be necessary for the treatment of severe neutropenia and severe infectious complications. Monitoring for potential cardiotoxicity is also important, especially with greater cumulative exposure to doxorubicin. Doxorubicin may potentiate the toxicity of other anticancer therapies (see PRECAUTIONS, Drug Interactions).
Cardiac Function
Cardiotoxicity is a known risk of anthracycline treatment. Anthracycline-induced cardiotoxicity may be manifested by early (or acute) or late (delayed) events. Early cardiotoxicity of doxorubicin consists mainly of sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes. Tachyarrhythmias, including premature ventricular contractions and ventricular tachycardia, bradycardia, as well as atrioventricular and bundle-branch block have also been reported. These effects do not usually predict subsequent development of delayed cardiotoxicity, are rarely of clinical importance, and are generally not considered an indication for the suspension of doxorubicin treatment.
Delayed cardiotoxicity usually develops late in the course of therapy with doxorubicin or within 2 to 3 months after treatment termination, but later events, several months to years after completion of treatment, have also been reported. Delayed cardiomyopathy is manifested by a reduction in LVEF and/or signs and symptoms of congestive heart failure (CHF) such as tachycardia, dyspnea, pulmonary edema, dependent edema, cardiomegaly and hepatomegaly, oliguria, ascites, pleural effusion, and gallop rhythm. Subacute effects such as pericarditis/myocarditis have also been reported. Life-threatening CHF is the most severe form of anthracycline-induced cardiomyopathy and represents the cumulative dose-limiting toxicity of the drug.
The probability of developing impaired myocardial function, based on a combined index of signs, symptoms and decline in left ventricular ejection fraction (LVEF) is estimated to be 1 to 2% at a total cumulative dose of 300 mg/m 2 of doxorubicin, 3 to 5% at a dose of 400 mg/m 2, 5 to 8% at a dose of 450 mg/m 2 and 6 to 20% at a dose of 500 mg/m 2 given in a schedule of a bolus injection once every 3 weeks. In a retrospective review, the probability of developing congestive heart failure was reported to be 5/168 (3%) at a cumulative dose of 430 mg/m 2 of doxorubicin, 8/110 (7%) at 575 mg/m 2, and 3/14 (21%) at 728 mg/m 2. In a prospective study of doxorubicin in combination with cyclophosphamide, fluorouracil and/or vincristine in patients with breast cancer or small cell lung cancer, the probability of CHF at various cumulative doses of doxorubicin was 1.5% at 300 mg/m 2, 4.9% at 400 mg/m 2, 7.7% at 450 mg/m 2 and 20.5% at 500 mg/m 2. The risk of developing CHF increases rapidly with increasing total cumulative doses of doxorubicin in excess of 400 mg/m 2.
Cardiotoxicity may occur at lower doses in patients with prior mediastinal/pericardial irradiation, concomitant use of other cardiotoxic drugs, doxorubicin exposure at an early age, and advanced age. Data also suggest that pre-existing heart disease is a co-factor for increased risk of doxorubicin cardiotoxicity. In such cases, cardiac toxicity may occur at doses lower than the recommended cumulative dose of doxorubicin. Studies have suggested that concomitant administration of doxorubicin and calcium channel entry blockers or cardiotoxic drugs, especially those with long half-lives, e.g. trastuzumab, may increase the risk of doxorubicin cardiotoxicity (see PRECAUTIONS, General, DOSAGE AND ADMINISTRATION). The total dose of doxorubicin administered to the individual patient should also take into account previous or concomitant therapy with related compounds such as daunorubicin, idarubicin and mitoxantrone. Although not formally tested, it is probable that the toxicity of doxorubicin and other anthracyclines or anthracenediones is additive. Cardiomyopathy and/or congestive heart failure may be encountered several months or years after discontinuation of doxorubicin therapy.
The risk of acute manifestations of doxorubicin cardiotoxicity in pediatric patients may be as much or lower than in adults. Pediatric patients appear to be at particular risk for developing delayed cardiac toxicity in that doxorubicin-induced cardiomyopathy impairs myocardial growth as pediatric patients mature, subsequently leading to possible development of congestive heart failure during early adulthood. As many as 40% of pediatric patients may have subclinical cardiac dysfunction and 5 to 10% of pediatric patients may develop congestive heart failure on long term follow-up. This late cardiac toxicity may be related to the dose of doxorubicin. The longer the length of follow-up, the greater the increase in the detection rate. Treatment of doxorubicin-induced congestive heart failure includes the use of digitalis, diuretics, after load reducers such as angiotensin I converting enzyme (ACE) inhibitors, low salt diet, and bed rest. Such intervention may relieve symptoms and improve the functional status of the patient.
Monitoring Cardiac Function
The risk of serious cardiac impairment may be decreased through regular monitoring of LVEF during the course of treatment with prompt discontinuation of doxorubicin at the first sign of impaired function. The preferred method for assessment of cardiac function is evaluation of LVEF measured by multi-gated radionuclide angiography (MUGA) or echocardiography (ECHO). An ECG may also be done. A baseline cardiac evaluation with a MUGA scan or an ECHO is recommended, especially in patients with risk factors for increased cardiac toxicity. Repeated MUGA or ECHO determinations of LVEF should be performed, particularly with higher, cumulative anthracycline doses. The technique used for assessment should be consistent through follow-up. In patients with risk factors, particularly prior anthracycline or anthracenedione use, the monitoring of cardiac function must be particularly strict and the risk-benefit of continuing treatment with doxorubicin in patients with impaired cardiac function must be carefully evaluated.
Endomyocardial biopsy is recognized as the most sensitive diagnostic tool to detect anthracycline-induced cardiomyopathy; however, this invasive examination is not practically performed on a routine basis. ECG changes such as dysrhythmias, a reduction of the QRS voltage, or a prolongation beyond normal limits of the systolic time interval may be indicative of anthracycline-induced cardiomyopathy, but ECG is not a sensitive or specific method for following anthracycline-related cardiotoxicity.
Pediatric patients are at increased risk for developing delayed cardiotoxicity following doxorubicin administration and therefore a follow-up cardiac evaluation is recommended periodically to monitor for this delayed cardiotoxicity.
In adults, a 10% decline in LVEF to below the lower limit of normal or an absolute LVEF of 45%, or a 20% decline in LVEF at any level is indicative of deterioration in cardiac function. In pediatric patients, deterioration in cardiac function during or after the completion of therapy with doxorubicin is indicated by a drop in fractional shortening (FS) by an absolute value of ≥ 10 percentile units or below 29%, and a decline in LVEF of 10 percentile units or an LVEF below 55%. In general, if test results indicate deterioration in cardiac function associated with doxorubicin, the benefit of continued therapy should be carefully evaluated against the risk of producing irreversible cardiac damage. Acute life-threatening arrhythmias have been reported to occur during or within a few hours after doxorubicin administration.
Hematologic Toxicity
As with other cytotoxic agents, doxorubicin may produce myelosuppression. Myelosuppression requires careful monitoring. Total and differential WBC, red blood cell (RBC), and platelet counts should be assessed before and during each cycle of therapy with doxorubicin. A dose-dependent, reversible leukopenia and/or granulocytopenia (neutropenia) are the predominant manifestations of doxorubicin hematologic toxicity and is the most common acute dose-limiting toxicities of this drug. With the recommended dose schedule, leukopenia is usually transient, reaching its nadir 10 to 14 days after treatment with recovery usually occurring by the 21st day. Thrombocytopenia and anemia may also occur. Clinical consequences of severe myelosuppression include fever, infections, sepsis/septicemia, septic shock, hemorrhage, tissue hypoxia, or death.
Secondary Leukemia
The occurrence of secondary AML or MDS has been reported most commonly in patients treated with chemotherapy regimens containing anthracyclines (including doxorubicin) and DNA-damaging antineoplastic agents, in combination with radiotherapy, when patients have been heavily pretreated with cytotoxic drugs, or when doses of anthracyclines have been escalated. Such cases generally have a 1 to 3 year latency period. The rate of developing secondary AML or MDS has been estimated in an analysis of 8,563 patients with early breast cancer treated in 6 studies conducted by the National Surgical Adjuvant Breast and Bowel Project (NSABP), including NSABP B-15. Patients in these studies received standard doses of doxorubicin and standard or escalated doses of cyclophosphamide (AC) adjuvant chemotherapy and were followed for 61,810 patient years. Among 4,483 such patients who received conventional doses of AC, 11 cases of AML or MDS were identified, for an incidence of 0.32 cases per 1,000 patient years (95% Cl, 0.16 to 0.57) and a cumulative incidence at 5 years of 0.21% (95% Cl, 0.11 to 0.41%). In another analysis of 1,474 patients with breast cancer who received adjuvant treatment with doxorubicin-containing regimens in clinical trials conducted at University of Texas M.D. Anderson Cancer Center, the incidence was estimated at 1.5% at 10 years. In both experiences, patients who received regimens with higher cyclophosphamide dosages, who received radiotherapy, or who were aged 50 or older had an increased risk of secondary AML or MDS.
Pediatric patients are also at risk of developing secondary AML.
Effects at Site of Injection
Phlebosclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Following the recommended administration procedures may minimize the risk of phlebitis/thrombophlebitis at the injection site (see DOSAGE AND ADMINISTRATION, Instruction for Use/Handling).
Extravasation
On intravenous administration of doxorubicin, extravasation may occur with or without an accompanying stinging or burning sensation, even if blood returns well on aspiration of the infusion needle. If any signs or symptoms of extravasation have occurred, the injection or infusion should be immediately terminated and restarted in another vein (see DOSAGE AND ADMINISTRATION).
Hepatic Impairment
Since metabolism and excretion of doxorubicin occurs predominantly by the hepatobiliary route, toxicity of recommended doses of doxorubicin can be enhanced by hepatic impairment; therefore, prior to individual dosing, evaluation of hepatic function is recommended using conventional laboratory tests such as SGOT, SGPT, alkaline phosphatase, and bilirubin (see DOSAGE AND ADMINISTRATION).
Immunosuppressant Effects/Increased Susceptibility to Infections
Administration of live or live attenuated vaccines in patients immunocompromised by chemotherapeutic agents including doxorubicin, may result in serious or fatal infections. Vaccination with a live vaccine should be avoided in patients receiving doxorubicin. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
Pregnancy Category D
Doxorubicin can cause fetal harm when administered to a pregnant woman. Doxorubicin was teratogenic and embryotoxic at doses of 0.8 mg/kg/day (about 1/13 the recommended human dose on a body surface area basis) when administered during the period of organogenesis in rats. Teratogenicity and embryotoxicity were also seen using discrete periods of treatment. The most susceptible was the 6- to 9-day gestation period at doses of 1.25 mg/kg/day and greater. Characteristic malformations included esophageal and intestinal atresia, tracheoesophageal fistula, hypoplasia of the urinary bladder and cardiovascular anomalies. Doxorubicin was embryotoxic (increase in embryofetal deaths) and abortifacient at 0.4 mg/kg/day (about 1/14 the recommended human dose on a body surface area basis) in rabbits when administered during the period of organogenesis.
There are no adequate and well-controlled studies in pregnant women. If doxorubicin is to be used during pregnancy, or if the patient becomes pregnant during therapy, the patient should be apprised of the potential hazard to the fetus. Women of childbearing age should be advised to avoid becoming pregnant.
-
PRECAUTIONS:
General
Doxorubicin is not an anti-microbial agent. Doxorubicin is emetigenic. Antiemetics may reduce nausea and vomiting; prophylactic use of antiemetics should be considered before administration of doxorubicin, particularly when given in conjunction with other emetigenic drugs. Doxorubicin should not be administered in combination with other cardiotoxic agents unless the patient's cardiac function is closely monitored. Patients receiving doxorubicin after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, may also be at an increased risk of developing cardiotoxicity. Physicians should avoid doxorubicin-based therapy for up to 24 weeks after stopping trastuzumab when possible. If doxorubicin used before this time, careful monitoring of cardiac function is recommended (see WARNINGS, DOSAGE AND ADMINISTRATION).
Information for Patients
Patients should be informed of the expected adverse effects of doxorubicin, including gastrointestinal symptoms (nausea, vomiting, diarrhea, and stomatitis) and potential neutropenic complications. Patients should consult their physician if vomiting, dehydration, fever, evidence of infection, symptoms of CHF, or injection-site pain occurs following therapy with doxorubicin. Patients should be informed that they will almost certainly develop alopecia. Patients should be advised that their urine may appear red for 1 to 2 days after administration of doxorubicin and that they should not be alarmed. Patients should understand that there is a risk of irreversible myocardial damage associated with treatment with doxorubicin, as well as a risk of treatment-related leukemia. Because doxorubicin may induce chromosomal damage in sperm, men undergoing treatment with doxorubicin should use effective contraceptive methods. Women treated with doxorubicin may develop irreversible amenorrhea, or premature menopause.
Drug Interactions
Doxorubicin is extensively metabolized by the liver. Changes in hepatic function induced by concomitant therapies may affect doxorubicin metabolism, pharmacokinetics, therapeutic efficacy, and/or toxicity. Toxicities associated with doxorubicin, especially hematologic and gastrointestinal events, may be increased when doxorubicin is used in combination with other cytotoxic drugs.
Paclitaxel
There have been a number of reports in the literature that describe an increase in cardiotoxicity when doxorubicin is co-administered with paclitaxel. Two published studies report that initial administration of paclitaxel infused over 24 hours followed by doxorubicin administered over 48 hours resulted in a significant decrease in doxorubicin clearance with more profound neutropenic and stomatitis episodes than the reverse sequence of administration.
Progesterone
In a published study, progesterone was given intravenously to patients with advanced malignancies (ECOG PS< 2) at high doses (up to 10 g over 24 hours) concomitantly with a fixed doxorubicin dose (60 mg/m 2) via bolus injection. Enhanced doxorubicin-induced neutropenia and thrombocytopenia were observed.
Verapamil
A study of the effects of verapamil on the acute toxicity of doxorubicin in mice revealed higher initial peak concentrations of doxorubicin in the heart with a higher incidence and severity of degenerative changes in cardiac tissue resulting in a shorter survival.
Cyclosporine
The addition of cyclosporine to doxorubicin may result in increases in AUC for both doxorubicin and doxorubicinol possibly due to a decrease in clearance of parent drug and a decrease in metabolism of doxorubicinol. Literature reports suggest that adding cyclosporine to doxorubicin results in more profound and prolonged hematologic toxicity than doxorubicin alone. Coma and/or seizures have also been described.
Dexrazoxane
In a clinical study of women with metastatic breast cancer, the concurrent use of the cardioprotectant, dexrazoxane, with the initiation of a regimen of fluorouracil, doxorubicin, and cyclophosphamide (FAC) was associated with a lower tumor response rate. Later initiation of dexrazoxane (after administration of a cumulative doxorubicin dose of 300 mg/m 2 of doxorubicin had been given as a component of FAC) was not associated with a reduction in chemotherapy activity. Dexrazoxane is only indicated for use in women with metastatic breast cancer who have received a cumulative doxorubicin dose of 300 mg/m 2 and are continuing with doxorubicin therapy.
Cytarabine
Necrotizing colitis manifested by typhlitis (cecal inflammation), bloody stools and severe and sometimes fatal infections have been associated with a combination of doxorubicin given by intravenous push daily for 3 days and cytarabine given by continuous infusion daily for 7 or more days.
Sorafenib
In clinical studies, both an increase of 21% and 47% and no change in the AUC of doxorubicin were observed with concomitant treatment with sorafenib 400 mg twice daily. The clinical significance of these findings is unknown.
CyclophosphamideThe addition of cyclophosphamide to doxorubicin treatment does not affect exposure to doxorubicin, but may result in an increase in exposure to doxorubicinol, a metabolite. Doxorubicinol only has 5% of the cytotoxic activity of doxorubicin. Concurrent treatment with doxorubicin has been reported to exacerbate cyclophosphamide-induced hemorrhagic cystitis. Acute myeloid leukemia has been reported as a second malignancy after treatment with doxorubicin and cyclophosphamide.
Literature reports have also described the following drug interactions
Phenobarbital increases the elimination of doxorubicin; phenytoin levels may be decreased by doxorubicin; streptozocin (Zanosar ®) may inhibit hepatic metabolism of doxorubicin; saquinavir in combination with cyclophosphamide, doxorubicin, and etoposide increased mucosal toxicity in patients with HIV-associated non-Hodgkin's lymphoma; and administration of live vaccines to immunosuppressed patients including those undergoing cytotoxic chemotherapy may be hazardous (see WARNINGS).
Laboratory Tests
Initial treatment with doxorubicin requires observation of the patient and periodic monitoring of complete blood counts, hepatic function tests, and left ventricular ejection fraction (see WARNINGS). Abnormalities of hepatic function tests may occur. Like other cytotoxic drugs, doxorubicin may induce "tumor-lysis syndrome" and hyperuricemia in patients with rapidly growing tumors. Blood uric acid levels, potassium, calcium, phosphate, and creatinine should be evaluated after initial treatment. Hydration, urine alkalinization, and prophylaxis with allopurinol to prevent hyperuricemia may minimize potential complications of tumor-lysis syndrome.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with doxorubicin. Secondary acute myelogenous leukemia (AML) or myelodysplastic syndrome (MDS) have been reported in patients treated with doxorubicin-containing combination chemotherapy regimens (see WARNINGS). Pediatric patients treated with doxorubicin or other topoisomerase II inhibitors are at risk for developing acute myelogenous leukemia and other neoplasms. Doxorubicin was mutagenic in the in vitro Ames assay, and clastogenic in multiple in vitro assays (CHO cell, V79 hamster cell, human lymphoblast, and SCE assays) and the in vivo mouse micronucleus assay.
Doxorubicin decreased fertility in female rats at the doses of 0.05 and 0.2 mg/kg/day (about 1/200 and 1/50 the recommended human dose on a body surface area basis) when administered from 14 days before mating through late gestation period. A single IV dose of doxorubicin at 0.1 mg/kg (about 1/100 the recommended human dose on a body surface area basis) was toxic to male reproductive organs producing testicular atrophy and oligospermia in rats. Doxorubicin is mutagenic as it induced DNA damage in rabbit spermatozoa and dominant lethal mutations in mice. Therefore, doxorubicin may potentially induce chromosomal damage in human spermatozoa. Oligospermia or azoospermia were evidenced in men treated with doxorubicin, mainly in combination therapies. Men undergoing doxorubicin treatment should use effective contraceptive methods.
Doxorubicin was toxic to male reproductive organs in animal studies, producing testicular atrophy, diffuse degeneration of the seminiferous tubules, and hypospermia. Doxorubicin is mutagenic as it induces DNA damage in rabbit spermatozoa and dominant lethal mutations in mice. Therefore, doxorubicin can potentially induce chromosomal damage in human spermatozoa. Oligospermia or azoospermia were evidenced in men treated with doxorubicin, mainly in combination therapies. This effect may be permanent. However, sperm counts have been reported to return to normal levels in some instances. This may occur several years after the end of the therapy. Men undergoing doxorubicin treatment should use effective contraceptive methods.
In women, doxorubicin may cause infertility during the time of drug administration. Doxorubicin may cause amenorrhea. Ovulation and menstruation may return after termination of therapy, although premature menopause can occur. Recovery of menses is related to age at treatment.
Secondary acute myelogenous leukemia (AML) or myelodysplastic syndrome (MDS) have been reported in patients treated with anthracycline-containing adjuvant combination chemotherapy regimens (see WARNINGS, Hematologic Toxicity).
Nursing Mothers
Doxorubicin and its major metabolite, doxorubicinol have been detected in the milk of at least one lactating patient (see CLINICAL PHARMACOLOGY, Pharmacokinetics). Because of the potential for serious adverse reactions in nursing infants from doxorubicin, mothers should be advised to discontinue nursing during doxorubicin therapy.
Pediatric Use
Pediatric patients are at increased risk for developing delayed cardiotoxicity. Follow-up cardiac evaluations are recommended periodically to monitor for this delayed cardiotoxicity (see WARNINGS). Doxorubicin, as a component of intensive chemotherapy regimens administered to pediatric patients, may contribute to prepubertal growth failure. It may also contribute to gonadal impairment, which is usually temporary. Pediatric patients treated with doxorubicin or other topoisomerase II inhibitors are at a risk for developing acute myelogenous leukemia and other neoplasms. Pediatric patients receiving concomitant doxorubicin and actinomycin-D have manifested acute "recall" pneumonitis at variable times after local radiation therapy.
Geriatric Use
An estimated 4,600 patients who were 65 and over were included in the reported clinical experience of doxorubicin use for various indications. No overall differences in safety and effectiveness were observed between these patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out. The decision to use doxorubicin in the treatment of older patients should be based upon a consideration of overall performance status and concurrent illnesses, in addition to age of the individual patient.
-
ADVERSE REACTIONS:
Dose limiting toxicities of therapy are myelosuppression and cardiotoxicity. Other reactions reported are:
Cutaneous
Reversible complete alopecia occurs in most cases. Hyperpigmentation of nailbeds and dermal creases, primarily in pediatric patients, and onycholysis have been reported in a few cases. Radiation recall reaction has occurred with doxorubicin administration. Rash, itching, or photosensitivity may occur.
Gastrointestinal
Acute nausea and vomiting occurs frequently and may be severe. This may be alleviated by antiemetic therapy. Mucositis (stomatitis and esophagitis) may occur within 5 to 10 days of beginning therapy, and most patients recover from this adverse event within another 5 to 10 days. The effect may be severe leading to ulceration and represents a site of origin for severe infections. The dosage regimen consisting of administration of doxorubicin on three successive days results in greater incidence and severity of mucositis. Ulceration and necrosis of the colon, especially the cecum, may occur leading to bleeding or severe infections which can be fatal. This reaction has been reported in patients with acute non-lymphocytic leukemia treated with a 3-day course of doxorubicin combined with cytarabine. Anorexia, abdominal pain, dehydration, diarrhea, and hyperpigmentation of the oral mucosa have been occasionally reported.
Hypersensitivity
Fever, chills and urticaria have been reported occasionally. Anaphylaxis may occur. A case of apparent cross sensitivity to lincomycin has been reported.
Neurological
Peripheral neurotoxicity in the form of local-regional sensory and/or motor disturbances have been reported in patients treated intra-arterially with doxorubicin, mostly in combination with cisplatin. Animal studies have demonstrated seizures and coma in rodents and dogs treated with intra-carotid doxorubicin. Seizures and coma have been reported in patients treated with doxorubicin in combination with cisplatin or vincristine.
Adverse Reactions in Patients with Early Breast Cancer Receiving Doxorubicin-Containing Adjuvant Therapy
Safety data were collected from approximately 2,300 women who participated in a randomized, open-label trial (NSABP B-15) evaluating the use of AC versus CMF in the treatment of early breast cancer involving axillary lymph nodes. In the safety analysis, the follow-up data from all patients receiving AC were combined (N=1,492 evaluable patients) and compared with data from patients receiving conventional CMF (i.e., oral cyclophosphamide; N=739 evaluable patients). The most relevant adverse events reported in this study are provided in Table 2.
Table 2. Relevant Adverse Events in Patients with Early Breast Cancer Involving Axillary Lymph Nodes
AC*
Conventional
CMF
N=1,492
N=739
Treatment administration
Mean number of cycles
3.8
5.5
Total cycles
5,676
4,068
Adverse events, % of patients
Leukopenia
Grade 3 (1,000 to 1,999 /mm 3)
3.4
9.4
Grade 4 (<1000/mm 3)
0.3
0.3
Thrombocytopenia
Grade 3 (25,000 to 49,999 /mm 3)
0
0.3
Grade 4 (<25,000 /mm 3)
0.1
0
Shock, sepsis
1.5
0.9
Systemic infection
2.4
1.2
Nausea and vomiting
Nausea only
15.5
42.8
Vomiting ≤ 12 hours
34.4
25.2
Vomiting >12 hours
36.8
12
Intractable
4.7
1.6
Alopecia
92.4
71.4
Partial
22.9
56.3
Complete
69.5
15.1
Weight loss
5 to 10%
6.2
5.7
>10%
2.4
2.8
Weight gain
5 to 10%
10.6
27.9
>10%
3.8
14.3
Cardiac function
Asymptomatic
0.2
0.1
Transient
0.1
0
Symptomatic
0.1
0
Treatment-related death
0
0
*Includes pooled data from patients who received either AC alone for 4 cycles, or who were treated with AC for 4 cycles followed by 3 cycles of CMF
-
OVERDOSAGE:
Acute overdosage with doxorubicin enhances the toxic effects of mucositis, leukopenia and thrombocytopenia. Treatment of acute overdosage consists of treatment of the severely myelosuppressed patient with hospitalization, antimicrobials, platelet transfusions and symptomatic treatment of mucositis. Use of hemopoietic growth factor (G-CSF, GM-CSF) may be considered.
The 200 mg doxorubicin hydrochloride injection vial is packaged as a multiple dose vial and caution should be exercised to prevent inadvertent overdosage.
Cumulative dosage with doxorubicin increases the risk of cardiomyopathy and resultant congestive heart failure (see WARNINGS). Treatment consists of vigorous management of congestive heart failure with digitalis preparations, diuretics, and after-load reducers such as ACE inhibitors.
-
DOSAGE AND ADMINISTRATION:
When possible, to reduce the risk of developing cardiotoxicity in patients receiving doxorubicin after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, doxorubicin-based therapy should be delayed until the other agents have cleared from the circulation (see WARNINGS and PRECAUTIONS, General).
Care in the administration of doxorubicin will reduce the chance of perivenous infiltration (see WARNINGS). It may also decrease the chance of local reactions such as urticaria and erythematous streaking. On intravenous administration of doxorubicin, extravasation may occur with or without an accompanying burning or stinging sensation, even if blood returns well on aspiration of the infusion needle. If any signs or symptoms of extravasation have occurred, the injection or infusion should be immediately terminated and restarted in another vein. If extravasation is suspected, intermittent application of ice to the site for 15 min. q.i.d. x 3 days may be useful. The benefit of local administration of drugs has not been clearly established. Because of the progressive nature of extravasation reactions, close observation and plastic surgery consultation is recommended. Blistering, ulceration and/or persistent pain are indications for wide excision surgery, followed by split-thickness skin grafting.
The most commonly used dose schedule when used as a single agent is 60 to 75 mg/m 2 as a single intravenous injection administered at 21-day intervals. The lower dosage should be given to patients with inadequate marrow reserves due to old age, or prior therapy, or neoplastic marrow infiltration.
Doxorubicin has been used concurrently with other approved chemotherapeutic agents. Evidence is available that in some types of neoplastic disease, combination chemotherapy is superior to single agents. The benefits and risks of such therapy continue to be elucidated. When used in combination with other chemotherapy drugs, the most commonly used dosage of doxorubicin is 40 to 60 mg/m 2 given as a single intravenous injection every 21 to 28 days.
In a large randomized study (NSABP B-15) of patients with early breast cancer involving axillary lymph nodes (see CLINICAL PHARMACOLOGY, CLINICAL STUDIES and ADVERSE REACTIONS, Adverse Reactions in Patients with Early Breast Cancer Receiving Doxorubicin-Containing Adjuvant Therapy), the combination dosage regimen of AC (doxorubicin 60 mg/m 2 and cyclophosphamide 600 mg/m 2) was administered intravenously on day 1 of each 21-day treatment cycle. Four cycles of treatment were administered.
Dose Modifications
Patients in the NSABP B-15 study could have dose modifications of AC to 75% of the starting doses for neutropenic fever/infection. When necessary, the next cycle of treatment cycle was delayed until the absolute neutrophil count (ANC) was ≥ 1,000 cells/mm 3 and the platelet count was ≥ 100,000 cells/mm 3 and nonhematologic toxicities had resolved.
Doxorubicin dosage must be reduced in case of hyperbilirubinemia as follows:
Plasma bilirubin concentration (mg/dL)
Dosage reduction (%)
1.2 to 3
50
3.1 to 5
75
Reconstitution Directions
It is recommended that doxorubicin be slowly administered into the tubing of a freely running intravenous infusion of Sodium Chloride Injection, USP, or 5% Dextrose Injection, USP. The tubing should be attached to a Butterfly ® needle inserted preferably into a large vein. If possible, avoid veins over joints or in extremities with compromised venous or lymphatic drainage. The rate of administration is dependent on the size of the vein and the dosage. However, the dose should be administered in not less than 3 to 5 minutes. Local erythematous streaking along the vein as well as facial flushing may be indicative of too rapid an administration. A burning or stinging sensation may be indicative of perivenous infiltration and, if this occurs, the infusion should be immediately terminated and restarted in another vein. Perivenous infiltration may occur painlessly.
Doxorubicin should not be mixed with heparin or fluorouracil since it has been reported that these drugs are incompatible to the extent that a precipitate may form. Contact with alkaline solutions should be avoided since this can lead to hydrolysis of doxorubicin. Until specific compatibility data are available, it is not recommended that doxorubicin be mixed with other drugs.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Handling and Disposal
Procedures for proper handling and disposal of anti-cancer drugs should be considered. Several guidelines on this subject have been published. 1-4 There is no general agreement that all the procedures recommended in the guidelines are necessary or appropriate. However, given the toxic nature of this substance, the following protective recommendations are provided:
- Personnel should be trained in good technique for reconstitution and handling.
- Pregnant staff should be excluded from working with this drug.
- Personnel handling doxorubicin should wear protective clothing: goggles, gowns and disposable gloves and masks.
- A designated area should be defined for reconstitution (preferably under a laminar flow system). The work surface should be protected by disposable, plastic-backed, absorbent paper.
- All items used for reconstitution, administration or cleaning, including gloves, should be placed in high-risk waste-disposal bags for high-temperature incineration.
- Spillage or leakage should be treated with dilute sodium hypochlorite (1% available chlorine) solution, preferably by soaking, and then water.
- All cleaning materials should be disposed of as indicated previously.
- In case of skin contact, thoroughly wash the affected area with soap and water or sodium bicarbonate solution. However, do not abrade the skin by using a scrub brush.
- In case of contact with the eye(s), hold back the eyelid(s) and flush the affected eye(s) with copious amounts of water for at least 15 minutes. Then seek medical evaluation by a physician.
- Always wash hands after removing gloves.
Caregivers of pediatric patients receiving doxorubicin should be counseled to take precautions (such as wearing latex gloves) to prevent contact with the patient’s urine and other body fluids for at least 5 days after each treatment.
-
HOW SUPPLIED:
Doxorubicin Hydrochloride Injection, USP, 2 mg per mL, a sterile product which contains no preservatives, is available as follows:
Product No.
NDC
No.
88305
63323-883-05
Doxorubicin hydrochloride 10 mg in a 5 mL single dose flip-top vial, packaged individually.
88310
63323-883-10
Doxorubicin hydrochloride 20 mg in a 10 mL single dose flip-top vial, packaged individually.
88330
63323-883-30
Doxorubicin hydrochloride 50 mg in a 25 mL single dose flip-top vial, packaged individually.
100161
63323-101-61
Doxorubicin hydrochloride 200 mg in a 100 mL multiple dose vial, packaged individually.
REFRIGERATE AT: 2° to 8°C ( 36° to 46°F).
Protect from light (keep in outer carton). Preservative Free. Discard unused portion.
The container closure is not made with natural rubber latex.
-
REFERENCES:
- NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://osha.gov/dts/osta/otm/otm_vi/otm _vi_2.html
- American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. 2006; 63:1172-1193.
- Polovich, M., White, J. M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for human practice (2nd. ed.) Pittsburgh, PA: Oncology Nursing Society.
The brand names mentioned in this document are the trademarks of their respective owners.
-
PATIENT INFORMATION
Doxorubicin
(dakse-RU-besen)
Hydrochloride Injection, USPRead this Patient information before you start receiving doxorubicin and before each infusion. This information leaflet does not take the place of talking with your doctor about your medical condition or your treatment.
What is the most important information I should know about doxorubicin?
Doxorubicin may cause serious side effects including:
• Heart problems. Doxorubicin may cause heart problems that may lead to death. These problems can happen during your treatment or months to years after stopping treatment. In some cases heart problems are irreversible. Your chance of heart problems is higher if you:
• already have heart problems
• have a history of radiation therapy or are currently receiving radiation therapy to your chest
• have had treatment with certain other anti-cancer medicines
• take other medicines that can affect your heart
Tell your doctor if you get any of these symptoms of heart problems:
• shortness of breath
• cough
• swelling of your feet and ankles
• fast heartbeat
Your doctor should do tests to check your heart before, during, and after your treatment with doxorubicin.
• Secondary cancers. Some people who have received doxorubicin have developed acute myelogenous leukemia (AML) or myelodysplastic syndrome (MDS). Your chance of developing a secondary cancer is higher if you receive doxorubicin along with other anti-cancer medicines or with radiation therapy.
• Decreased blood cell counts. Doxorubicin can cause a severe decrease in neutrophils (a type of white blood cells important in fighting in bacterial infections), red blood cells (blood cells that carry oxygen to the tissues), and platelets (important for clotting and to control bleeding).Your doctor will check your blood cell count during your treatment with doxorubicin and after you have stopped your treatment.
What is doxorubicin?
Doxorubicin is a prescription anti-cancer medicine used to treat certain types of cancers. Doxorubicin may be used alone or along with other anti-cancer medicines.
Who should not receive doxorubicin?
Do not receive doxorubicin if:
• your blood cell counts are too low: platelets (which help your blood to clot), red blood cells (which help to carry iron and oxygen throughout your body), and white blood cells (which help to fight infection)
• you have a severe liver problem
• you have had a recent heart attack or have severe heart problems
• you have had previous treatment with doxorubicin or certain other anticancer medicines and received the maximum dose allowed
• you are allergic to certain other anti-cancer medicines, doxorubicin hydrochloride, or any other ingredient in Doxorubicin Hydrochloride Injection, USP. See the end of this leaflet for a complete list of ingredients in Doxorubicin Hydrochloride Injection, USP.
Talk to your doctor before receiving doxorubicin if you have any of the conditions listed above.
What should I tell my doctor before receiving doxorubicin?
Before you receive doxorubicin, tell your doctor if you:
• have heart problems
• have had radiation treatment or currently receiving radiation therapy
• are over the age of 50
• have liver problems
• plan to receive any vaccines. Talk to your doctor about which vaccines are safe for you to receive during your treatment with doxorubicin. See “What should I avoid while receiving doxorubicin?”
• have any other medical conditions
• are pregnant or plan to become pregnant. Doxorubicin can harm your unborn baby. Women who may become pregnant should use effective birth control (contraception). Talk to your doctor about the best way to prevent pregnancy while receiving doxorubicin.
• are breastfeeding or plan to breast feed. Doxorubicin can pass into your breast milk and harm your baby. You and your doctor should decide if you will receive doxorubicin or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Doxorubicin can interact with other medicines. Do not start any new medicine before you talk with the doctor that prescribed doxorubicin.
Know the medicines you take. Keep a list to show your doctor and pharmacist each time you get a new medicine.
How will I receive doxorubicin?
• Your doctor will prescribe doxorubicin in an amount that is right for you.
• Doxorubicin will be given to you by intravenous (IV) infusion into your vein.
• Your doctor will do regular blood tests to check for side effects of doxorubicin.
• Before receiving doxorubicin you may receive other medicines to prevent or treat side effects.
• Caregivers of children receiving doxorubicin should take precautions (such as wearing latex gloves) to prevent contact with the patient’s urine and other body fluids for at least 5 days after each treatment.
What should I avoid while taking doxorubicin?
• Avoid receiving live vaccines during treatment with doxorubicin. Talk to your doctor to find out which vaccines are safe for you while receiving doxorubicin. See “What should I tell my doctor before receiving doxorubicin?”
What are the possible side effects of doxorubicin?
Doxorubicin can cause serious side effects including:
• See "What is the most important information I should know about doxorubicin?"
Infusion site reactions. Serious infusion site reactions can happen with doxorubicin. Symptoms of infusion reaction may include:
• pain at injection site
• skin redness or swelling
• burning or stinging
• open skin sores at injection site
Your doctor will watch you closely while you are receiving doxorubicin and after your infusion for signs of a reaction. You may experience these reactions immediately or within 2 hours of infusion.
Change in the color of your urine. You may have red colored urine for 1 to 2 days after your infusion of doxorubicin. This is normal. Tell your doctor if it does not stop in a few days, or if you see what looks like blood or blood clots in your urine.
Infection. Call your doctor right away if you get any of the following signs of infection:
• fever (temperature of 100.4 F or greater) chills or shivering
• cough that brings up mucus
• burning or pain with urination
Doxorubicin may cause lower sperm counts and sperm problems in men.
This could affect your ability to father a child and cause birth defects. Men should use effective birth control (contraception) while receiving doxorubicin. Do not have unprotected sexual contact with a female who could become pregnant. Tell your doctor if you do have unprotected sexual contact with a female who could become pregnant. Talk to your doctor if this is a concern for you.
Irreversible amenorrhea or early menopause. Your periods (menstrual cycle) may completely stop when you receive doxorubicin. Your periods may or may not return after you complete your treatment of doxorubicin.
The most common side effects of doxorubicin include:
• hair loss (alopecia). Your hair may re-grow after your treatment.
• darkening of your nails or separation of your nails from your nailbed
• nausea
• vomiting
• lack of appetite or increased thirst
• bruise or bleed more easily
• abnormal heart beat
• a secondary cancer may occur when doxorubicin is combined with other chemotherapy agents.
• mouth sores
• weight changes
• stomach (abdominal) pain
• diarrhea
• eye problems
• allergic reactions. Call your doctor right away if you have any of the following symptoms of an allergic reaction:
• rash
• flushed face
• fever
• hives
• dizziness or feel faint
• itching
• shortness of breath or trouble breathing
• swelling of your lips or tongue
Tell your doctor or nurse if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects of doxorubicin. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of doxorubicin.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet.
This leaflet summarizes the most important information about doxorubicin. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about doxorubicin that is written for healthcare professionals.
For more information, call 1-800-551-7176.
What are the ingredients of Doxorubicin Hydrochloride Injection, USP?
Active ingredient: Doxorubicin hydrochloride
Inactive ingredient: sodium chloride 0.9%, water for injection, hydrochloric acid and/or sodium hydroxide.
This Patient Information has been approved by the U.S. Food and Drug Administration.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL
PACKAGE LABEL - PRINCIPAL DISPLAY - Doxorubicin 5 mL Single Dose Vial Label
DOXOrubicin Hydrochloride Injection, USP
10 mg per 5 mL
(2 mg per mL)For intravenous use only.
Preservative free.5 mL Single Dose Vial Rx only

PACKAGE LABEL - PRINCIPAL DISPLAY - Doxorubicin 5 mL Single Dose Vial Carton Panel
DOXOrubicin Hydrochloride Injection, USP
10 mg per 5 mL
(2 mg per mL)For intravenous use only.
Preservative free.
Rx only
5 mL
Single Dose Vial

PACKAGE LABEL - PRINCIPAL DISPLAY - Doxorubicin 100 mL Multiple Dose Vial Label
DOXOrubicin Hydrochloride Injection, USP
200 mg per 100 mL
(2 mg per mL)For intravenous use only.
Preservative free.
100 mL
Multiple Dose Vial Rx only
PACKAGE LABEL - PRINCIPAL DISPLAY - Doxorubicin 100 mL Multiple Dose Vial Carton Panel
DOXOrubicin Hydrochloride Injection, USP
200 mg per 100 mL
(2 mg per mL)For intravenous use only.
Preservative free.Rx only
100 mL
Multiple Dose Vial
-
INGREDIENTS AND APPEARANCE
DOXORUBICIN HYDROCHLORIDE
doxorubicin hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63323-883 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DOXORUBICIN HYDROCHLORIDE (UNII: 82F2G7BL4E) (DOXORUBICIN - UNII:80168379AG) DOXORUBICIN HYDROCHLORIDE 2 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 9 mg in 1 mL HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63323-883-05 1 in 1 CARTON 04/14/2000 1 5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:63323-883-10 1 in 1 CARTON 04/14/2000 2 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 3 NDC:63323-883-30 1 in 1 CARTON 04/14/2000 3 25 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA063277 04/14/2000 DOXORUBICIN HYDROCHLORIDE
doxorubicin hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63323-101 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DOXORUBICIN HYDROCHLORIDE (UNII: 82F2G7BL4E) (DOXORUBICIN - UNII:80168379AG) DOXORUBICIN HYDROCHLORIDE 2 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 9 mg in 1 mL HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63323-101-61 1 in 1 CARTON 06/14/2000 1 100 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA063277 06/14/2000 Labeler - Fresenius Kabi USA, LLC (608775388) Establishment Name Address ID/FEI Business Operations Fresenius Kabi USA, LLC 023648251 manufacture(63323-101, 63323-883)
