Label: trovan- trovafloxacin mesylate tablet, film coated
trovan- trovafloxacin mesylate injection, solution, concentrate
-
Contains inactivated NDC Code(s)
NDC Code(s): 0049-3780-30, 0049-3780-43, 0049-3790-30, 0049-3790-43, view more0049-3890-28, 0049-3900-28 - Packager: Roerig
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
Drug Label Information
Updated February 6, 2006
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- N/A - Section Title Not Found In Database
-
BOXED WARNING
(What is this?)
TROVAN® HAS BEEN ASSOCIATED WITH SERIOUS LIVER INJURY LEADING TO LIVER TRANSPLANTATION AND/OR DEATH. TROVAN-ASSOCIATED LIVER INJURY HAS BEEN REPORTED WITH BOTH SHORT-TERM AND LONG-TERM DRUG EXPOSURE. TROVAN USE EXCEEDING 2 WEEKS IN DURATION IS ASSOCIATED WITH A SIGNIFICANTLY INCREASED RISK OF SERIOUS LIVER INJURY. LIVER INJURY HAS ALSO BEEN REPORTED FOLLOWING TROVAN RE-EXPOSURE. TROVAN SHOULD BE RESERVED FOR USE IN PATIENTS WITH SERIOUS, LIFE- OR LIMB-THREATENING INFECTIONS WHO RECEIVE THEIR INITIAL THERAPY IN AN IN-PATIENT HEALTH CARE FACILITY (I.E., HOSPITAL OR LONG-TERM NURSING CARE FACILITY). TROVAN SHOULD NOT BE USED WHEN SAFER, ALTERNATIVE ANTIMICROBIAL THERAPY WILL BE EFFECTIVE. (SEE WARNINGS.)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
TROVAN Tablets
TROVAN Tablets contain trovafloxacin mesylate, a synthetic broad-spectrum antibacterial agent for oral administration. Chemically, trovafloxacin mesylate, a fluoronaphthyridone related to the fluoroquinolone antibacterials, is (1α, 5α, 6α)-7-(6-amino-3-azabicyclo[3.1.0]hex-3-yl)-1-(2,4-difluorophenyl)-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid, monomethanesulfonate. Trovafloxacin mesylate differs from other quinolone derivatives by having a 1,8-naphthyridine nucleus.
The chemical structure is:
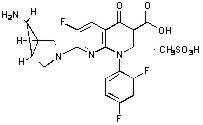
Its empirical formula is C20H15F3N4O3•CH3SO3H and its molecular weight is 512.46.
Trovafloxacin mesylate is a white to off-white powder.
Trovafloxacin mesylate is available in 100 mg and 200 mg (trovafloxacin equivalent) blue, film-coated tablets. TROVAN Tablets contain microcrystalline cellulose, cross-linked sodium carboxymethylcellulose and magnesium stearate. The tablet coating is a mixture of hydroxypropylcellulose, hydroxypropylmethylcellulose, titanium dioxide, polyethylene glycol and FD&C blue #2 aluminum lake.
TROVAN I.V.
TROVAN I.V. contains alatrofloxacin mesylate, the L-alanyl-L-alanyl prodrug of trovafloxacin mesylate. Chemically, alatrofloxacin mesylate is (1α, 5α, 6α)-L-alanyl-N-[3-[6-carboxy-8-(2,4-difluorophenyl)-3-fluoro-5,8-dihydro-5-oxo-1,8-naphthyridine-2-yl]-3-azabicyclo[3.1.0]hex-6-yl]-L-alaninamide, monomethanesulfonate. It is intended for administration by intravenous infusion.
Following intravenous administration, the alanine substituents in alatrofloxacin are rapidly hydrolyzed in vivo to yield trovafloxacin. (See CLINICAL PHARMACOLOGY.)
The chemical structure is:
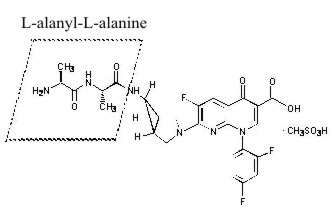
Its empirical formula is C26H25F3N6O5•CH3SO3H and its molecular weight is 654.62.
Alatrofloxacin mesylate is a white to light yellow powder.
TROVAN I.V. is available in 40 mL and 60 mL single use vials as a sterile, preservative-free aqueous concentrate of 5 mg trovafloxacin/mL as alatrofloxacin mesylate intended for dilution prior to intravenous administration of doses of 200 mg or 300 mg of trovafloxacin, respectively. (See HOW SUPPLIED.)
The formulation contains Water for Injection, and may contain sodium hydroxide or hydrochloric acid for pH adjustment. The pH range for the 5 mg/mL aqueous concentrate is 3.5 to 4.3.
-
CLINICAL PHARMACOLOGY
After intravenous administration, alatrofloxacin is rapidly converted to trovafloxacin. Plasma concentrations of alatrofloxacin are below quantifiable levels within 5 to 10 minutes of completion of a 1 hour infusion.
Absorption
Trovafloxacin is well-absorbed from the gastrointestinal tract after oral administration. The absolute bioavailability is approximately 88%. For comparable dosages, no dosage adjustment is necessary when switching from parenteral to oral administration (Figure 1). (See DOSAGE AND ADMINISTRATION.)
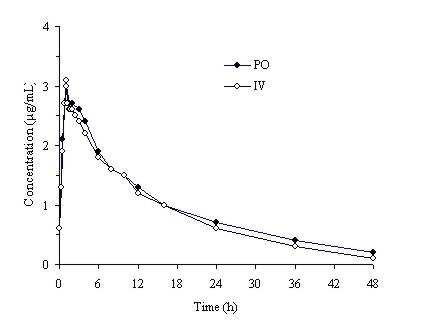
Figure 1. Mean trovafloxacin serum concentrations determined following 1 hour intravenous infusions of alatrofloxacin at daily doses of 200 mg (trovafloxacin equivalents) to healthy male volunteers and following daily oral administration of 200 mg trovafloxacin for 7 days to six male and six female healthy young volunteers.
Pharmacokinetics
The mean pharmacokinetic parameters (±SD) of trovafloxacin after single and multiple 100 mg and 200 mg oral doses and 1 hour intravenous infusions of alatrofloxacin in doses of 200 and 300 mg (trovafloxacin equivalents) appear in the chart below.
TROVAFLOXACIN PHARMACOKINETIC PARAMETERS Cmax
(µg/mL)Tmax
(hrs)AUC*
(µg•h/mL)T½
(hrs)Vdss
(L/Kg)CL
(mL/hr/Kg)CLr
(mL/hr/Kg)Cmax=Maximum serum concentration; Tmax=Time to Cmax; AUC=Area under concentration vs. time curve; T1/2=serum half-life; Vdss=Volume of distribution; Cl=Total clearance; Clr=Renal clearance Trovafloxacin 100 mg Single dose 1.0±0.3 0.9±0.4 11.2±2.2 9.1 — — — Multiple dose 1.1±0.2 1.0±0.5 11.8±1.8 10.5 — — — Trovafloxacin 200 mg Single dose 2.1±0.5 1.8±0.9 26.7±7.5 9.6 — — — Multiple dose 3.1±1.0 1.2±0.5 34.4±5.7 12.2 — — — Alatrofloxacin 200 mg† Single dose 2.7±0.4 1.0±0.0 28.1±5.1 9.4 1.2±0.2 93.0±17.4 6.5±3.5 Multiple dose 3.1±0.6 1.0±0.0 32.2±7.3 11.7 1.3±0.1 81.7±17.8 8.6±2.4 Alatrofloxacin 300 mg† Single dose 3.6±0.6 1.3±0.4 46.1±5.2 11.2 1.2±0.1 84.6±6.0 6.9±0.5 Multiple dose 4.4±0.6 1.2±0.2 46.3±3.9 12.7 1.4±0.1 84.5±11.1 8.4±1.8 Serum concentrations of trovafloxacin are dose proportional after oral administration of trovafloxacin in the dose range of 30 to 1000 mg or after intravenous administration of alatrofloxacin in the dose range of 30 to 400 mg (trovafloxacin equivalents). Steady state concentrations are achieved by the third daily oral or intravenous dose of trovafloxacin with an accumulation factor of approximately 1.3 times the single dose concentrations.
Oral absorption of trovafloxacin is not altered by concomitant food intake; therefore, it can be administered without regard to food.
The systemic exposure to trovafloxacin (AUC0–∞) administered as crushed tablets via nasogastric tube into the stomach was identical to that of orally administered intact tablets. Administration of concurrent enteral feeding solutions had no effect on the absorption of trovafloxacin given via nasogastric tube into the stomach. When trovafloxacin was administered as crushed tablets into the duodenum via nasogastric tube, the AUC0–∞ and peak serum concentration (Cmax) were reduced by 30% relative to the orally administered intact tablets. Time to peak serum level (Tmax) was also decreased from 1.7 hrs to 1.1 hrs.
Distribution
The mean plasma protein bound fraction is approximately 76%, and is concentration-independent. Trovafloxacin is widely distributed throughout the body. Rapid distribution of trovafloxacin into tissues results in significantly higher trovafloxacin concentrations in most target tissues than in plasma or serum.
Fluid or Tissue Tissue-Fluid/Serum Ratio* (Range) - *
- Mean values in adults over 2–29 hours following drug administration, except individual lung tissues, which were single time points of 6 hours following drug administration
- †
- Ratio of composite AUC(0–24) in CSF/composite AUC(0–24) in serum in 22 pediatric patients aged 1 to 12 years after 1 hour I.V. infusion of single dose alatrofloxacin (equivalent trovafloxacin dose range: 4.5–9.9 mg/kg)
Respiratory bronchial macrophages (multiple dose) 24.1 (9.6–41.8) lung mucosa 1.1 (0.7–1.5) lung epithelial lining fluid (multiple dose) 5.8 (1.1–17.5) whole lung 2.1 (0.42–5.03) Skin, Musculoskeletal skin 1.0 (0.20–1.88) subcutaneous tissue 0.4 (0.15–0.68) skin blister fluid 0.7–0.9 (blister/plasma) skeletal muscle 1.5 (0.50–2.90) bone 1.0 (0.55–1.67) Gastrointestinal colonic tissue 0.7 (0.0–1.47) peritoneal fluid 0.4 (0.0–1.25) bile 15.4 (11.9–21.0) Central Nervous System cerebrospinal fluid (CSF), adults 0.25 (0.03–0.33) cerebrospinal fluid (CSF), children 0.28† Reproductive prostatic tissue 1.0 (0.5–1.6) cervix (multiple dose) 0.6 (0.5–0.7) ovary 1.6 (0.3–2.2) fallopian tube 0.7 (0.2–1.1) myometrium (multiple dose) 0.6 (0.4–0.8) uterus 0.6 (0.3–0.8) vaginal fluid (multiple dose) 4.7 (0.8–20.8) Presence in Breast Milk
Trovafloxacin was found in measurable concentrations in the breast milk of three lactating subjects. The average measurable breast milk concentration was 0.8 µg/mL (range: 0.3–2.1 µg/mL) after single I.V. alatrofloxacin (300 mg trovafloxacin equivalents) and repeated oral trovafloxacin (200 mg) doses.
Metabolism
Trovafloxacin is metabolized by conjugation (the role of cytochrome P450 oxidative metabolism of trovafloxacin is minimal). Thirteen percent of the administered dose appears in the urine in the form of the ester glucuronide and 9% appears in the feces as the N-acetyl metabolite (2.5% of the dose is found in the serum as the active N-acetyl metabolite). Other minor metabolites (diacid, sulfamate, hydroxycarboxylic acid) have been identified in both urine and feces in small amounts (<4% of the administered dose).
Excretion
Approximately 50% of an oral dose is excreted unchanged (43% in the feces and 6% in the urine).
After multiple 200 mg doses, to healthy subjects, mean (±SD) cumulative urinary trovafloxacin concentrations were 12.1±3.4 µg/mL. With these levels of trovafloxacin in urine, crystals of trovafloxacin have not been observed in the urine of human subjects.
Special Populations
Geriatric
In adult subjects, the pharmacokinetics of trovafloxacin are not affected by age (range 19–78 years).
Pediatric
Limited information is available in the pediatric population (see Distribution). The pharmacokinetics of trovafloxacin have not been fully characterized in pediatric populations less than 18 years of age.
Gender
There are no significant differences in trovafloxacin pharmacokinetics between males and females when differences in body weight are taken into account. After single 200 mg doses, trovafloxacin Cmax and AUC(0–∞) were 60% and 32% higher, respectively, in healthy females compared to healthy males. Following repeated daily administration of 200 mg for 7 days, the Cmax for trovafloxacin was 38% higher and AUC(0–24) was 16% higher in healthy females compared to healthy males. The clinical importance of the increases in serum levels of trovafloxacin in females has not been established. (See PRECAUTIONS: Information for Patients.)
Chronic Hepatic Disease
Following repeated administration of 100 mg for 7 days to patients with mild cirrhosis (Child-Pugh Class A), the AUC(0–24) for trovafloxacin was increased ~45% compared to matched controls. Repeated administration of 200 mg for 7 days to patients with moderate cirrhosis (Child-Pugh Class B) resulted in an increase of ~50% in AUC(0–24) compared to matched controls. There appeared to be no significant effect on trovafloxacin Cmax for either group. The oral clearance of trovafloxacin was reduced ~30% in both cirrhosis groups, which corresponded to prolongation of half life by 2–2.5 hours (25–30% increase) compared to controls. There are no data in patients with severe cirrhosis (Child-Pugh Class C). Dosage adjustment is recommended in patients with mild to moderate cirrhosis. (See DOSAGE AND ADMINISTRATION.)
Renal Insufficiency
The pharmacokinetics of trovafloxacin are not affected by renal impairment. Trovafloxacin serum concentrations are not significantly altered in subjects with severe renal insufficiency (creatinine clearance <20 mL/min), including patients on hemodialysis.
Photosensitivity Potential
In a study of the skin response to ultraviolet and visible radiation conducted in 48 healthy volunteers (12 per group), the minimum erythematous dose (MED) was measured for ciprofloxacin, lomefloxacin, trovafloxacin and placebo before and after drug administration for 5 days. In this study, trovafloxacin (200 mg q.d.) was shown to have a lower potential for producing delayed photosensitivity skin reactions than ciprofloxacin (500 mg b.i.d.) or lomefloxacin (400 mg q.d.), although greater than placebo. (See PRECAUTIONS: Information for Patients.)
Drug-drug Interactions
The systemic availability of trovafloxacin following oral tablet administration is significantly reduced by the concomitant administration of antacids containing aluminum and magnesium salts, sucralfate, vitamins or minerals containing iron, and concomitant intravenous morphine administration.
Administration of trovafloxacin (300 mg p.o.) 30 minutes after administration of an antacid containing magnesium hydroxide and aluminum hydroxide resulted in reductions in systemic exposure to trovafloxacin (AUC) of 66% and peak serum concentration (Cmax) of 60%. (See PRECAUTIONS: Drug Interactions, DOSAGE AND ADMINISTRATION.)
Concomitant sucralfate administration (1g) with trovafloxacin 200 mg p.o. resulted in a 70% decrease in trovafloxacin systemic exposure (AUC) and a 77% reduction in peak serum concentration (Cmax). (See PRECAUTIONS: Drug Interactions, DOSAGE AND ADMINISTRATION.)
Concomitant administration of ferrous sulfate (120 mg elemental iron) with trovafloxacin 200 mg p.o. resulted in a 40% reduction in trovafloxacin systemic exposure (AUC) and a 48% decrease in trovafloxacin Cmax. (See PRECAUTIONS: Drug Interactions, DOSAGE AND ADMINISTRATION.)
Concomitant administration of intravenous morphine (0.15 mg/kg) with oral trovafloxacin (200 mg) resulted in a 36% reduction in trovafloxacin AUC and a 46% decrease in trovafloxacin Cmax. Trovafloxacin administration had no effect on the pharmacokinetics of morphine or its pharmacologically active metabolite, morphine-6-β-glucuronide. (See PRECAUTIONS: Drug Interactions, DOSAGE AND ADMINISTRATION.)
Minor pharmacokinetic interactions that are most likely without clinical significance include calcium carbonate, omeprazole and caffeine.
Concomitant administration of calcium carbonate (1000 mg) with trovafloxacin 200 mg p.o. resulted in a 20% reduction in trovafloxacin AUC and a 17% reduction in peak serum trovafloxacin concentration (Cmax).
A 40 mg dose of omeprazole given 2 hours prior to trovafloxacin (300 mg p.o.) resulted in a 17% reduction in trovafloxacin AUC and a 17% reduction in trovafloxacin peak serum concentration (Cmax).
Administration of trovafloxacin (200 mg) concomitantly with caffeine (200 mg) resulted in a 17% increase in caffeine AUC and a 15% increase in caffeine Cmax. These changes in caffeine exposure are not considered clinically significant.
No significant pharmacokinetic interactions were seen when TROVAN was co-administered with cimetidine, theophylline, digoxin, warfarin and cyclosporine.
Cimetidine co-administration (400 mg twice daily for 5 days) with trovafloxacin (200 mg p.o. daily for 3 days) resulted in changes in trovafloxacin AUC and Cmax of less than 5%.
Trovafloxacin (200 mg p.o. daily for 7 days) co-administration with theophylline (300 mg twice daily for 14 days) resulted in no change in theophylline AUC and Cmax.
Trovafloxacin (200 mg p.o. daily for 10 days) co-administration with digoxin (0.25 mg daily for 20 days) did not significantly alter systemic exposure (AUC) to digoxin or the renal clearance of digoxin.
Trovafloxacin (200 mg p.o. daily for 7 days) did not interfere with either the pharmacokinetics or the pharmacodynamics of warfarin (daily for 21 days).
Concomitant oral administration of trovafloxacin did not affect the systemic exposure (AUC) or peak plasma concentrations (Cmax) of the S or R isomers of warfarin, nor did it influence prothrombin times. (See PRECAUTIONS: Drug Interactions.)
Trovafloxacin (200 mg p.o. daily for 7 days) co-administration with cyclosporine (daily doses from 150–450 mg for 7 days) resulted in decreases of 10% or less in systemic exposure to cyclosporine (AUC) and in the peak blood concentrations of cyclosporine.
Microbiology
Trovafloxacin is a fluoronaphthyridone related to the fluoroquinolones with in vitro activity against a wide range of gram-negative and gram-positive aerobic, and anaerobic microorganisms. The bactericidal action of trovafloxacin results from inhibition of DNA gyrase and topoisomerase IV. DNA gyrase is an essential enzyme that is involved in the replication, transcription and repair of bacterial DNA. Topoisomerase IV is an enzyme known to play a key role in the partitioning of the chromosomal DNA during bacterial cell division. Mechanism of action of fluoroquinolones including trovafloxacin is different from that of penicillins, cephalosporins, aminoglycosides, macrolides, and tetracyclines. Therefore, fluoroquinolones may be active against pathogens that are resistant to these antibiotics. There is no cross-resistance between trovafloxacin and the mentioned classes of antibiotics. The overall results obtained from in vitro synergy studies, testing combinations of trovafloxacin with beta-lactams and aminoglycosides, indicate that synergy is strain specific and not commonly encountered. This agrees with results obtained previously with other fluoroquinolones. Resistance to trovafloxacin in vitro develops slowly via multiple-step mutation in a manner similar to other fluoroquinolones. Resistance to trovafloxacin in vitro occurs at a general frequency of between 1×10-7 to 10-10. Although cross-resistance has been observed between trovafloxacin and some other fluoroquinolones, some microorganisms resistant to other fluoroquinolones may be susceptible to trovafloxacin.
Trovafloxacin has been shown to be active against most strains of the following microorganisms, both in vitro and in clinical infections as described in the INDICATIONS AND USAGE section:
Aerobic gram-positive microorganisms
Enterococcus faecalis (many strains are only moderately susceptible)
Staphylococcus aureus (methicillin-susceptible strains)
Streptococcus agalactiae
Streptococcus pneumoniae (penicillin-susceptible strains)
Viridans group streptococciAerobic gram-negative microorganisms
Escherichia coli
Gardnerella vaginalis
Haemophilus influenzae
Klebsiella pneumoniae
Moraxella catarrhalis
Proteus mirabilis
Pseudomonas aeruginosaAnaerobic microorganisms
Bacteroides fragilis
Peptostreptococcus species
Prevotella speciesOther microorganisms
Chlamydia pneumoniae
Legionella pneumophila
Mycoplasma pneumoniaeThe following in vitro data are available, but their clinical significance is unknown.
Trovafloxacin exhibits in vitro minimum inhibitory concentrations (MICs) of ≤2 µg/mL against most (90%) strains of the following microorganisms; however, the safety and effectiveness of trovafloxacin in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled clinical trials.
Aerobic gram-positive microorganisms
Streptococcus pneumoniae (penicillin resistant strains)Aerobic gram-negative microorganisms
Citrobacter freundii
Enterobacter aerogenes
Morganella morganii
Proteus vulgarisAnaerobic microorganisms
Bacteroides distasonis
Bacteroides ovatus
Clostridium perfringensOther microorganisms
Mycoplasma hominis
Ureaplasma urealyticumNOTE: Mycobacterium tuberculosis and Mycobacterium avium-intracellulare complex organisms are commonly resistant to trovafloxacin.
NOTE: The activity of trovafloxacin against Treponema pallidum has not been evaluated; however, other quinolones are not active against Treponema pallidum. (See WARNINGS.)
Susceptibility Tests
Dilution Techniques: Quantitative methods are used to determine antimicrobial minimum inhibitory concentrations (MICs). These MICs provide estimates of the susceptibility of bacteria to antimicrobial compounds. The MICs should be determined using a standardized procedure. Standardized procedures are based on dilution methods1 (broth or agar) or equivalent with standardized inoculum concentrations and standardized concentrations of trovafloxacin mesylate powder. The MIC values should be interpreted according to the following criteria:
For testing non-fastidious aerobic organisms:
MIC (µg/mL) Interpretation ≤2.0 Susceptible (S) 4.0 Intermediate (I) ≥8.0 Resistant (R) For testing Haemophilus spp.1
MIC (µg/mL) Interpretation* - *
- The current absence of data on resistant strains precludes defining any results other than "Susceptible". Strains yielding MIC results suggestive of a "nonsusceptible" category should be submitted to a reference laboratory for further testing.
≤1.0 Susceptible (S) For testing Streptococcus spp. including Streptococcus pneumoniae2:
MIC (µg/mL) Interpretation ≤1.0 Susceptible (S) 2.0 Intermediate (I) ≥4.0 Resistant (R) A report of "Susceptible" indicates that the pathogen is likely to be inhibited if the antimicrobial compound in the blood reaches the concentration usually achievable. A report of "Intermediate" indicates that the result should be considered equivocal, and, if the microorganism is not fully susceptible to alternative, clinically feasible drugs, the test should be repeated. This category implies possible clinical applicability in body sites where the drug is physiologically concentrated or in situations where high dosage of drug can be used. This category also provides a buffer zone which prevents small uncontrolled technical factors from causing major discrepancies in interpretation. A report of "Resistant" indicates that the pathogen is not likely to be inhibited if the antimicrobial compound in the blood reaches the concentration usually achievable; other therapy should be selected.
Standardized susceptibility test procedures require the use of laboratory control microorganisms to control the technical aspects of the laboratory procedures. Standard trovafloxacin mesylate powder should provide the following MIC values:
Microorganism MIC Range (µg/mL) - *
- This quality control range is applicable to only H. influenzae ATCC 49247 tested by a microdilution procedure using HTM1.
- †
- This quality control range is applicable to only S. pneumoniae ATCC 49619 tested by a microdilution procedure using cation-adjusted Mueller-Hinton broth with 2–5% lysed horse blood.
Escherichia coli ATCC 25922 0.004–0.016 Staphylococcus aureus ATCC 29213 0.008–0.03 Pseudomonas aeruginosa ATCC 27853 0.25–2.0 Enterococcus faecalis ATCC 29212 0.06–0.25 Haemophilus influenzae* ATCC 49247 0.004–0.016 Streptococcus pneumoniae† ATCC 49619 0.06–0.25 Diffusion Techniques: Quantitative methods that require measurement of zone diameters also provide reproducible estimates of the susceptibility of bacteria to antimicrobial compounds. One such standardized procedure2 requires the use of standardized inoculum concentrations. This procedure uses paper disks impregnated with trovafloxacin mesylate equivalent to 10 µg trovafloxacin to test the susceptibility of microorganisms to trovafloxacin.
Reports from the laboratory providing results of the standard single-disk susceptibility test with a trovafloxacin mesylate disk (equivalent to 10 µg trovafloxacin) should be interpreted according to the following criteria:
The following zone diameter interpretive criteria should be used for testing non-fastidious aerobic organisms:
Zone Diameter (mm) Interpretation ≥17 Susceptible (S) 14–16 Intermediate (I) ≤13 Resistant (R) For testing Haemophilus spp.3:
Zone Diameter (mm) Interpretation* - *
- The current absence of data on resistant strains precludes defining any results other than "Susceptible". Strains yielding MIC results suggestive of a "nonsusceptible" category should be submitted to a reference laboratory for further testing.
≥22 Susceptible (S) For testing Streptococcus spp. including Streptococcus pneumoniae4:
Zone Diameter (mm) Interpretation ≥19 Susceptible (S) 18–16 Intermediate (I) ≤15 Resistant (R) Interpretation should be as stated above for results using dilution techniques. Interpretation involves correlation of the diameter obtained in the disk test with the MIC for trovafloxacin.
As with standardized dilution techniques, diffusion methods require the use of laboratory control microorganisms that are used to control the technical aspects of the laboratory procedures. For the diffusion technique, the trovafloxacin mesylate equivalent to 10-µg trovafloxacin disk should provide the following zone diameters in these laboratory quality control strains:
Microorganism Zone Diameter Range (mm) Escherichia coli ATCC 25922 29–36 Staphylococcus aureus ATCC 25923 29–35 Pseudomonas aeruginosa ATCC 27853 21–27 Haemophilus influenzae* ATCC 49247 32–39 Streptococcus pneumoniae† ATCC 49619 25–32 Anaerobic Techniques: For anaerobic bacteria, the susceptibility to trovafloxacin as MICs can be determined by standardized test methods3. The MIC values obtained should be interpreted according to the following criteria:
MIC (µg/mL) Interpretation ≤2.0 Susceptible (S) 4.0 Intermediate (I) ≥8.0 Resistant (R) Interpretation is identical to that stated above for results using dilution techniques.
As with other susceptibility techniques, the use of laboratory control microorganisms is required to control the technical aspects of the laboratory standardized procedures. Standardized trovafloxacin mesylate powder should provide the following MIC values:
Microorganism MIC* (µg/mL) - *
- kThese quality control ranges were derived from tests performed in the broth formulation of Wilkins-Chalgren agar.
Bacteroides fragilis ATCC 25285 0.125–0.5 Bacteroides thetaiotamicron ATCC 29741 0.25–1.0 Eubacterium lentum ATCC 43055 0.25–1.0
- 1
- This interpretive standard is applicable only to broth microdilution susceptibility tests with Haemophilus spp. using Haemophilus Test Medium (HTM)1.
- 2
- These interpretive standards are applicable only to broth microdilution susceptibility tests using cation adjusted Mueller-Hinton broth with 2–5% lysed horse blood.
- 3
- This zone diameter standard is applicable only to tests with Haemophilus spp. using HTM2.
- 4
- These zone diameter standards only apply to tests performed using Mueller-Hinton agar supplemented with 5% sheep blood incubated in 5% CO2.
-
INDICATIONS AND USAGE
TROVAN is indicated for the treatment of patients initiating therapy in in-patient health care facilities (i.e., hospitals and long term nursing care facilities) with serious, life- or limb-threatening infections caused by susceptible strains of the designated microorganisms in the conditions listed below. (See DOSAGE AND ADMINISTRATION.)
Nosocomial pneumonia caused by Escherichia coli, Pseudomonas aeruginosa, Haemophilus influenzae, or Staphylococcus aureus. As with other antimicrobials, where Pseudomonas aeruginosa is a documented or presumptive pathogen, combination therapy with either an aminoglycoside or aztreonam may be clinically indicated.
Community acquired pneumonia caused by Streptococcus pneumoniae, Haemophilus influenzae, Klebsiella pneumoniae, Staphylococcus aureus, Mycoplasma pneumoniae, Moraxella catarrhalis, Legionella pneumophila, or Chlamydia pneumoniae.
Complicated intra-abdominal infections, including post-surgical infections caused by Escherichia coli, Bacteroides fragilis, viridans group streptococci, Pseudomonas aeruginosa, Klebsiella pneumoniae, Peptostreptococcus species, or Prevotella species.
Gynecologic and pelvic infections including endomyometritis, parametritis, septic abortion and post-partum infections caused by Escherichia coli, Bacteroides fragilis, viridans group streptococci, Enterococcus faecalis, Streptococcus agalactiae, Peptostreptococcus species, Prevotella species, or Gardnerella vaginalis.
Complicated skin and skin structure infections, including diabetic foot infections, caused by Staphylococcus aureus, Streptococcus agalactiae, Pseudomonas aeruginosa, Enterococcus faecalis, Escherichia coli, or Proteus mirabilis. NOTE: TROVAN has not been studied in the treatment of osteomyelitis. (See WARNINGS.)
- CONTRAINDICATIONS
-
WARNINGS
(See boxed WARNING.) TROVAN-ASSOCIATED LIVER ENZYME ABNORMALITIES, SYMPTOMATIC HEPATITIS, JAUNDICE, AND LIVER FAILURE (INCLUDING RARE REPORTS OF ACUTE HEPATIC NECROSIS WITH EOSINOPHILIC INFILTRATION, LIVER TRANSPLANTATION AND/OR DEATH) HAVE BEEN REPORTED WITH BOTH SHORT-TERM AND LONG-TERM DRUG EXPOSURE IN MEN AND WOMEN. TROVAN USE EXCEEDING 2 WEEKS IN DURATION IS ASSOCIATED WITH A SIGNIFICANTLY INCREASED RISK OF SERIOUS LIVER INJURY. LIVER INJURY HAS ALSO BEEN REPORTED FOLLOWING TROVAN RE-EXPOSURE. CLINICIANS SHOULD MONITOR LIVER FUNCTION TESTS (e.g., AST, ALT, BILIRUBIN) IN TROVAN RECIPIENTS WHO DEVELOP SIGNS OR SYMPTOMS CONSISTENT WITH HEPATITIS. CLINICIANS SHOULD CONSIDER DISCONTINUING TROVAN IN THOSE PATIENTS WHO DEVELOP LIVER FUNCTION TEST ABNORMALITIES.
THE SAFETY AND EFFECTIVENESS OF TROVAFLOXACIN IN PEDIATRIC PATIENTS AND ADOLESCENTS LESS THAN 18 YEARS OF AGE, PREGNANT WOMEN, AND NURSING WOMEN HAVE NOT BEEN ESTABLISHED. (See PRECAUTIONS: Pediatric Use, Pregnancy, and Nursing Mothers subsections.)
As with other members of the quinolone class, trovafloxacin has caused arthropathy and/or chondrodysplasia in immature rats and dogs. The significance of these findings to humans is unknown. (See ANIMAL PHARMACOLOGY.)
Convulsions, increased intracranial pressure and psychosis have been reported in patients receiving quinolones. Quinolones may also cause central nervous system stimulation which may lead to tremors, restlessness, lightheadedness, confusion, hallucinations, paranoia, depression, nightmares and insomnia. These reactions may occur following the first dose. If these reactions occur in patients receiving trovafloxacin or alatrofloxacin, the drug should be discontinued and appropriate measures instituted. (See PRECAUTIONS: General,Information for Patients, Drug Interactions and ADVERSE REACTIONS.)
As with other quinolones, TROVAN should be used with caution in patients with known or suspected CNS disorders, such as severe cerebral atherosclerosis, epilepsy, and other factors that predispose to seizures. (See ADVERSE REACTIONS.)
Serious and occasionally fatal hypersensitivity and/or anaphylactic reactions have been reported in patients receiving therapy with TROVAN. These reactions may occur following the first dose. Some reactions have been accompanied by cardiovascular collapse, hypotension/shock, seizure, loss of consciousness, tingling, angioedema (including tongue, laryngeal, throat or facial edema/swelling), airway obstruction (including bronchospasm, shortness of breath and acute respiratory distress), dyspnea, urticaria, itching and other serious skin reactions, including generalized erythema.
Life-threatening hypotension has been reported with alatrofloxacin administration. This has occurred in patients receiving alatrofloxacin at either the recommended rate of infusion or if given more rapidly. Hypotension may be potentiated with the concomitant administration of anesthetic agents. Alatrofloxacin should only be administered by slow intravenous infusion over a period of 60 minutes. Blood pressure should be monitored closely during infusion.
TROVAN should be discontinued at the first appearance of a skin rash or any other sign of hypersensitivity. Serious acute hypersensitivity reactions may require treatment with epinephrine and other resuscitative measures, including oxygen, intravenous fluids, antihistamines, corticosteroids, pressor amines and airway management, as clinically indicated. (See PRECAUTIONS and ADVERSE REACTIONS.)
Serious and sometimes fatal events, some due to hypersensitivity and some due to uncertain etiology, have been reported in patients receiving therapy with all antibiotics. These events may be severe and generally occur following the administration of multiple doses. Clinical manifestations may include one or more of the following: fever, rash or severe dermatologic reactions (e.g., toxic epidermal necrolysis, Stevens-Johnson Syndrome); vasculitis, arthralgia, myalgia, serum sickness; allergic pneumonitis, interstitial nephritis; acute renal insufficiency or failure; hepatitis, jaundice, acute hepatic necrosis or failure; anemia, including hemolytic and aplastic; thrombocytopenia, including thrombotic thrombocytopenic purpura; leukopenia; agranulocytosis; pancytopenia; and/or other hematologic abnormalities.
Pseudomembranous colitis has been reported with nearly all antibacterial agents, including TROVAN, and may range in severity from mild to life-threatening. Therefore, it is important to consider this diagnosis in patients who present with diarrhea subsequent to the administration of any antibacterial agent.
Treatment with antibacterial agents alters the flora of the colon and may permit overgrowth of clostridia. Studies indicate that a toxin produced by Clostridium difficile is the primary cause of "antibiotic-associated colitis."
After the diagnosis of pseudomembranous colitis has been established, therapeutic measures should be initiated. Mild cases of pseudomembranous colitis usually respond to drug discontinuation alone. In moderate to severe cases, consideration should be given to management with fluids and electrolytes, protein supplementation, and treatment with an antibacterial drug clinically effective against C. difficile colitis. (See ADVERSE REACTIONS.)
Although not seen in TROVAN clinical trials, ruptures of the shoulder, hand, and Achilles tendons that required surgical repair or resulted in prolonged disability have been reported in patients receiving quinolones. TROVAN should be discontinued if the patient experiences pain, inflammation or rupture of a tendon. Patients should rest and refrain from exercise until the diagnosis of tendinitis or tendon rupture has been confidently excluded. Tendon rupture can occur during or after therapy with quinolones.
Trovafloxacin has not been shown to be effective in the treatment of syphilis. Antimicrobial agents used in high doses for short periods of time to treat gonorrhea may mask or delay the symptoms of incubating syphilis. All patients with gonorrhea should have a serologic test for syphilis at the time of diagnosis.
-
PRECAUTIONS
General
Moderate to severe phototoxicity reactions have been observed in patients who are exposed to direct sunlight while receiving some drugs in this class. Therapy should be discontinued if phototoxicity (e.g., a skin eruption, etc.) occurs.
The safety and efficacy of TROVAN in patients with severe cirrhosis (Child-Pugh Class C) have not been studied.
Symptomatic pancreatitis has been reported on therapy. Clinicians should monitor pancreatic tests in patients who develop symptoms consistent with pancreatitis as clinically indicated.
Because a rapid or bolus intravenous injection may result in life-threatening hypotension, alatrofloxacin should only be administered by slow intravenous infusion over a period of 60 minutes. Profound hypotension has also been reported in patients receiving alatrofloxacin at the recommended rate of infusion. (See WARNINGS and DOSAGE AND ADMINISTRATION: Intravenous Administration.)
Information For Patients
Patients should be advised:
- to discontinue therapy and to inform their physician immediately if they develop symptoms suggestive of hepatic dysfunction including fatigue, anorexia, vomiting, abdominal pain, jaundice, dark urine or pale stool. (See WARNINGS.)
- to inform their physician if they develop symptoms suggestive of pancreatitis including abdominal pain and/or nausea and vomiting. (See PRECAUTIONS: General.)
- that TROVAN Tablets may be taken without regard to meals;
- that vitamins or minerals containing iron, aluminum- or magnesium-base antacids, antacids containing citric acid buffered with sodium citrate, or sucralfate or Videx®, (Didanosine), chewable/buffered tablets or the pediatric powder for oral solution, should be taken at least 2 hours before or 2 hours after taking TROVAN tablets. (See PRECAUTIONS: Drug Interactions.);
- that TROVAN may cause lightheadedness and/or dizziness. Dizziness and/or lightheadedness was the most common adverse reaction reported, and for females under 45 years, it was reported significantly more frequently than in other groups. The incidence of dizziness may be substantially reduced if TROVAN Tablets are taken at bedtime or with food. Patients should know how they react to trovafloxacin before they operate an automobile or machinery or engage in activities requiring mental alertness and coordination. (See WARNINGS and ADVERSE REACTIONS.);
- to discontinue treatment and inform their physician if they experience pain, inflammation or rupture of a tendon, and to rest and refrain from exercise until the diagnosis of tendinitis or tendon rupture has been confidently excluded;
- that TROVAN may be associated with hypersensitivity reactions, even following the first dose, and to discontinue the drug at the first sign of a skin rash, hives or other skin reactions, difficulty in swallowing or breathing, any swelling suggesting angioedema (e.g., swelling of the lips, tongue, face, tightness of the throat, hoarseness), or other symptoms of an allergic reaction. (See WARNINGS and ADVERSE REACTIONS.);
- to avoid excessive sunlight or artificial ultraviolet light (e.g., tanning beds) while taking TROVAN and to discontinue therapy if phototoxicity (e.g., sunburn-like reaction or skin eruption) occurs.
- that convulsions have been reported in patients taking quinolones, including trovafloxacin, and to notify their physician before taking this drug if there is a history of this condition
Drug Interactions
Antacids, Sucralfate, and Iron: The absorption of oral trovafloxacin is significantly reduced by the concomitant administration of some antacids containing magnesium or aluminum, citric acid/sodium citrate (Bicitra®), as well as sucralfate and iron (ferrous ions). These agents as well as formulations containing divalent and trivalent cations such as Videx®, (Didanosine), chewable/buffered tablets or the pediatric powder for oral solution, should be taken at least 2 hours before or 2 hours after oral trovafloxacin administration. (See CLINICAL PHARMACOLOGY.)
Morphine: Co-administration of intravenous morphine significantly reduces the absorption of oral trovafloxacin. Intravenous morphine should be administered at least 2 hours after oral TROVAN dosing in the fasted state and at least 4 hours after oral TROVAN is taken with food. Trovafloxacin administration had no effect on the pharmacokinetics of morphine or its metabolite, morphine-6-β-glucuronide. (See CLINICAL PHARMACOLOGY.)
Warfarin: There have been reports during the post-marketing experience that trovafloxacin/alatrofloxacin enhance the effects of warfarin, including cases of bleeding. The mechanism for this reaction is unknown. Prothrombin time, International Normalized Ratio (INR) or other suitable anticoagulation tests should be closely monitored if trovafloxacin/alatrofloxacin is administered concomitantly with warfarin. Patients should also be monitored for evidence of bleeding.
Minor pharmacokinetic interactions without clinical significance have been observed with co-administration of TROVAN Tablets with caffeine, omeprazole and calcium carbonate. (See CLINICAL PHARMACOLOGY.)
No significant pharmacokinetic interactions with theophylline, cimetidine, digoxin, warfarin, or cyclosporine have been observed with TROVAN Tablets. (See CLINICAL PHARMACOLOGY.)
Alatrofloxacin should not be co-administered with any solution containing multivalent cations, e.g., magnesium, through the same intravenous line. (See DOSAGE AND ADMINISTRATION.)
Carcinogenesis, Mutagenesis, Impairment Of Fertility
Long term studies in animals to determine the carcinogenic potential of trovafloxacin or alatrofloxacin have not been conducted.
TROVAN did not shorten the time to development of UV-induced skin tumors in hairless albino (Skh-1) mice; thus, it was not photo co-carcinogenic in this model. These mice received oral trovafloxacin and concurrent irradiation with simulated sunlight 5 days per week for 40 weeks followed by a 12-week treatment-free observation period. The daily dose of UV radiation used in this study was approximately 30% of the minimal dose of UV radiation that would induce erythema in Caucasian humans. The median time to the development of skin tumors in the hairless mice (42–43 weeks) was similar in the vehicle control group and those given 10 or 30 mg/kg of trovafloxacin daily. At a dose level of 30 mg/kg/day, the mice had skin trovafloxacin concentrations of approximately 7 µg/g. Following multiple 200 mg daily doses of trovafloxacin, the amount in human skin is estimated to be about 3 µg/g, based upon plasma concentrations measured at this dose level.
Trovafloxacin was not mutagenic in the Ames Salmonella reversion assay or CHO/HGPRT mammalian cell gene mutation assay and it was not clastogenic in mitogen-stimulated human lymphocytes or mouse bone marrow cells. A mouse micronucleus test conducted with alatrofloxacin was also negative. The positive response observed in the E. coli bacterial mutagenicity assay may be due to the inhibition of DNA gyrase by trovafloxacin.
Trovafloxacin and alatrofloxacin did not affect the fertility of male or female rats at oral and I.V. doses of 75 mg/kg/day and 50 mg/kg/day, respectively. These doses are 15 and 10 times the recommended maximum human dose based on mg/kg or approximately 2 times based on mg/m2. However, oral doses of trovafloxacin at 200 mg/kg/day (40 times the recommended maximum human dose based on mg/kg or about 6 times based on mg/m2) were associated with increased preimplantation loss in rats.
Pregnancy
Teratogenic Effects
Pregnancy Category C
An increase in skeletal variations was observed in rat fetuses after daily oral 75 mg/kg maternal doses of trovafloxacin (approximately 15 times the highest recommended human dose based on mg/kg or 2 times based upon body surface area) were administered during organogenesis. However, fetal skeletal variations were not observed in rats dosed orally with 15 mg/kg trovafloxacin. Evidence of fetotoxicity (increased perinatal mortality and decreased body weights) was also observed in rats at 75 mg/kg. Daily oral doses of trovafloxacin at 45 mg/kg (approximately 9 times the highest recommended human dose based on mg/kg or 2.7 times based upon body surface area) in the rabbit were not associated with an increased incidence of fetal skeletal variations or malformations.
An increase in skeletal variations and malformations was observed in rat fetuses after daily intravenous doses of alatrofloxacin at ≥20 mg/kg/day (approximately 4 times the highest recommended human dose based on mg/kg or 0.6 times based upon body surface area) were administered to dams during organogenesis. In the rabbit, an increase in fetal skeletal malformations was also observed when 20 mg/kg/day (approximately equal to the highest recommended human dose based upon body surface area) of alatrofloxacin was given intravenously during the period of organogenesis. Intravenous dosing of alatrofloxacin at 6.5 mg/kg in the rat or rabbit was not associated with an increased incidence of skeletal variations or malformations. Fetotoxicity and fetal skeletal malformations have been associated with other quinolones.
Oral doses of trovafloxacin >5mg/kg were associated with an increased gestation time in rats, and several dams at 75 mg/kg experienced uterine dystocia.
There are no adequate and well-controlled studies in pregnant women. TROVAN should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. (See WARNINGS.)
Nursing Mothers
Trovafloxacin is excreted in human milk and was found in measurable concentrations in the breast milk of lactating subjects. (See CLINICAL PHARMACOLOGY, Distribution.)
Because of the potential for unknown effects from trovafloxacin in nursing infants from mothers taking trovafloxacin, a decision should be made either to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
The safety and effectiveness of trovafloxacin in pediatric patients and adolescents less than 18 years of age have not been established. Quinolones, including trovafloxacin, cause arthropathy and osteochondrosis in juvenile animals of several species. (See WARNINGS.)
Geriatric Use
In multiple-dose clinical trials of trovafloxacin, 27% of patients were ≥65 years of age and 12% of patients were ≥75 years of age. The overall incidence of drug-related adverse reactions, including central nervous system and gastrointestinal side effects, was less in the ≥65 year group than the other age groups.
-
ADVERSE REACTIONS
Over 6000 patients have been treated with TROVAN in multidose clinical efficacy trials worldwide.
In TROVAN studies the majority of adverse reactions were described as mild in nature (over 90% were described as mild or moderate). TROVAN was discontinued for adverse events thought related to drug in 5% of patients (dizziness 2.4%, nausea 1.9%, headache 1.1%, and vomiting 1.0%).
TROVAN Drug-Related Adverse Reactions (frequency ≥1%) in Multiple-Dose Clinical Trials 200 mg oral
qd
(N=3259)200 mg I.V.→
200 mg oral qd
(N=634)300 mg I.V.→
200 mg oral qd
(N=623)Dizziness 11% 2% 2% Lightheadedness 4% 2% <1% Nausea 8% 5% 4% Headache 5% 5% 1% Vomiting 3% 1% 3% Diarrhea 2% 2% 2% Abdominal pain 1% 1% 0% Application/ injection/insertion site n/a 5% 2% reaction/ Vaginitis 2% 2% <1% Pruritus <1% 2% 2% Rash <1% 2% 2% Dizziness/lightheadedness on TROVAN is generally mild, lasts for a few hours following a dose, and in most cases, resolves with continued dosing. The incidence of dizziness and lightheadedness in TROVAN patients over 65 years is 3.1% and 0.6%, respectively. (See PRECAUTIONS: Information for Patients.)
TROVAN appears to have a low potential for phototoxicity. In clinical trials with TROVAN, only mild, treatment-related phototoxicity was observed in less than 0.03% (2/7096) of patients.
Additional reported drug-related events in clinical trials (remotely, possibly, probably or unknown) that occurred in <1% of TROVAN-treated patients are:
APPLICATION/INJECTION/INSERTION SITE: Application/injection/insertion site device complications, inflammation, pain, edema
AUTONOMIC NERVOUS: flushing, increased sweating, dry mouth, cold clammy skin, increased saliva
CARDIOVASCULAR: peripheral edema, chest pain, thrombophlebitis, hypotension, palpitation, periorbital edema, hypertension, syncope, tachycardia, angina pectoris, bradycardia, peripheral ischemia, edema, dizziness postural
CENTRAL & PERIPHERAL NERVOUS SYSTEM: confusion, paresthesia, vertigo, hypoesthesia, ataxia, convulsions, dysphonia, hypertonia, migraine, involuntary muscle contractions, speech disorder, encephalopathy, abnormal gait, hyperkinesia, hypokinesia, tongue paralysis, abnormal coordination, tremor, dyskinesia
GASTROINTESTINAL: altered bowel habit, constipation, diarrhea-Clostridium difficile, dyspepsia, flatulence, loose stools, gastritis, dysphagia, increased appetite, gastroenteritis, rectal disorder, colitis, pseudomembranous colitis, enteritis, eructation, gastrointestinal disorder, melena, hiccup
ORAL CAVITY: gingivitis, stomatitis, altered saliva, tongue disorder, tongue edema, tooth disorder, cheilitis, halitosis
GENERAL/OTHER: fever, fatigue, pain, asthenia, moniliasis, hot flushes, back pain, chills, infection (bacterial, fungal), malaise, sepsis, alcohol intolerance, allergic reaction, anaphylactoid reaction, drug (other) toxicity/reaction, weight increase, weight decrease
HEMATOPOIETIC: anemia, granulocytopenia, hemorrhage unspecified, leukopenia, prothrombin decreased, thrombocythemia, thrombocytopenia
LIVER/BILIARY: increased hepatic enzymes, hepatic function abnormal, bilirubinemia, discolored feces, jaundice
METABOLIC/NUTRITIONAL: hyperglycemia, thirst
MUSCULOSKELETAL: arthralgia, muscle cramps, myalgia, muscle weakness, skeletal pain, tendinitis, arthropathy
PSYCHIATRIC: anxiety, anorexia, agitation, nervousness, somnolence, insomnia, depression, amnesia, concentration impaired, depersonalization, dreaming abnormal, emotional lability, euphoria, hallucination, impotence, libido decreased-male, paroniria, thinking abnormal
REPRODUCTIVE: Female: leukorrhea, menstrual disorder; Male: balanoposthitis
RESPIRATORY: dyspnea, rhinitis, sinusitis, bronchospasm, coughing, epistaxis, respiratory insufficiency, upper respiratory tract infection, respiratory disorder, asthma, hemoptysis, hypoxia, stridor
SKIN/APPENDAGES: pruritus ani, skin disorder, skin ulceration, angioedema, dermatitis, dermatitis fungal, photosensitivity skin reaction, seborrhea, skin exfoliation, urticaria
SPECIAL SENSES: taste perversion, eye pain, abnormal vision, conjunctivitis, photophobia, conjuctival hemorrhage, hyperacusis, scotoma, tinnitus, visual field defect, diplopia, xerophthalmia
URINARY SYSTEM: dysuria, face edema, micturition frequency, interstitial nephritis, renal failure acute, renal function abnormal, urinary incontinence
LABORATORY CHANGES: Changes in laboratory parameters, without regard to drug relationship, occurring in ≥1% of TROVAN-treated patients were: decreased hemoglobin and hematocrit; increased platelets; decreased and increased WBC; eosinophilia; increased ALT (SGPT), AST (SGOT), and alkaline phosphatase; decreased protein and albumin; increased BUN and creatinine; decreased sodium; and bicarbonate. It is not known whether these abnormalities were caused by the drug or the underlying condition being treated.
The incidence and magnitude of liver function abnormalities with TROVAN were the same as comparator agents except in the only study in which oral TROVAN was administered for 28 days. In this study (chronic bacterial prostatitis) nine percent (13/140) of TROVAN-treated patients experienced elevations of serum transaminases (AST and/or ALT) of ≥3 times the upper limit of normal. These liver function test abnormalities generally developed at the end of, or following completion of, the planned 28-day course of therapy, but were not associated with concurrent elevations of related laboratory measures of hepatic function (such as serum bilirubin, alkaline phosphatase, or lactate dehydrogenase). Patients were asymptomatic with these abnormalities, which generally returned to normal within 1–2 months after discontinuation of therapy. (See ADVERSE REACTIONS: POST-MARKETING EXPERIENCE subsection.)
POST-MARKETING EXPERIENCE
Adverse reactions reported with TROVAN during the post-marketing period include:
GASTROINTESTINAL: symptomatic pancreatitis.
GENERAL/OTHER: anaphylaxis, Stevens-Johnson Syndrome.
HEMATOPOIETIC: agranulocytosis, aplastic anemia, pancytopenia.
LIVER/BILIARY: symptomatic hepatitis (some patients experienced an associated peripheral eosinophilia), liver failure (including acute hepatic necrosis with eosinophilic infiltration). TROVAN-associated liver enzyme abnormalities and/or symptomatic hepatitis have occurred during short-term or long-term therapy. (See WARNINGS.)
-
OVERDOSAGE
Trovafloxacin has a low order of acute toxicity. The minimum lethal oral dose in mice and rats was 2000 mg/kg or greater. The minimum lethal I.V. dose for the prodrug, alatrofloxacin, was 50–125 mg/kg for mice and greater than 75 mg/kg for rats. Clinical signs observed included decreased activity and respiration, ataxia, ptosis, tremors and convulsions.
In the event of acute oral overdosage, the stomach should be emptied by inducing vomiting or by gastric lavage. The patient should be carefully observed and given symptomatic and supportive treatment. Adequate hydration should be maintained. Trovafloxacin is not efficiently removed from the body by hemodialysis.
-
DOSAGE AND ADMINISTRATION
The recommended dosage for TROVAN for the treatment of serious, life- or limb-threatening infections is described in the table below. Doses of TROVAN are administered once every 24 hours. TROVAN should not usually be administered for more than 2 weeks. It should only be administered for longer than 2 weeks if the treating physician believes the benefits to the individual patients clearly outweigh the risks of such longer-term treatment. (See boxed WARNING.)
Oral doses should be administered at least 2 hours before or 2 hours after antacids containing magnesium or aluminum, as well as sucralfate, citric acid buffered with sodium citrate (e.g., Bicitra®), metal cations (e.g., ferrous sulfate) and Videx®, (Didanosine), chewable/buffered tablets or the pediatric powder for oral solution.
Intravenous morphine should be administered at least 2 hours after oral TROVAN dosing in the fasted state and at least 4 hours after oral TROVAN is taken with food.
Patients whose therapy is started with TROVAN I.V. may be switched to TROVAN Tablets to complete the course of therapy, if deemed appropriate by the treating physician. In certain patients with serious and life- or limb-threatening infections as described in the INDICATIONS AND USAGE Section, TROVAN Tablets may be considered appropriate initial therapy, when the treating physician believes that the benefit of the product for the patient outweighs the potential risk.
TROVAN I.V. (alatrofloxacin mesylate injection) should only be administered by INTRAVENOUS infusion. It is not for intramuscular, intrathecal, intraperitoneal, or subcutaneous administration.
Single-use vials require dilution prior to administration. (See PREPARATION OF ALATROFLOXACIN MESYLATE INJECTION FOR ADMINISTRATION.)
DOSAGE GUIDELINES INFECTION*/LOCATION
AND TYPEDAILY UNIT DOSE
AND ROUTE OF
ADMINISTRATIONTOTAL DURATION
(See WARNINGS.)- *
- due to the designated pathogens (See INDICATIONS AND USAGE.)
- †
- Where the 300 mg TROVAN I.V. dose is indicated, therapy should be decreased to the 200 mg dose as soon as clinically indicated.
Nosocomial Pneumonia (See NOTE 1 below.) 300 mg I.V.†followed by 200 mg oral 10–14 days Community Acquired Pneumonia 200 mg oral or 200 mg I.V. followed by 200 mg oral 7–14 days Complicated Intra-Abdominal Infections, including post-surgical infections 300 mg I.V.† followed by 200 mg oral 7–14 days Gynecologic and Pelvic Infections 300 mg I.V.† followed by 200 mg oral 7–14 days Skin and Skin Structure Infections, Complicated, including diabetic foot infections 200 mg oral or 200 mg I.V. followed by 200 mg oral 10–14 days NOTE: As with other antimicrobials, where Pseudomonas aeruginosa is a documented or presumptive pathogen, combination therapy with either an aminoglycoside or aztreonam may be clinically indicated.
IMPAIRED RENAL FUNCTION: No adjustment in the dosage of TROVAN is necessary in patients with impaired renal function. Trovafloxacin is eliminated primarily by biliary excretion. Trovafloxacin is not efficiently removed from the body by hemodialysis.
CHRONIC HEPATIC DISEASE (cirrhosis): The following table provides dosing guidelines for patients with mild or moderate cirrhosis (Child-Pugh Class A and B). There are no data in patients with severe cirrhosis (Child-Pugh Class C).
INDICATED DOSE (Normal hepatic function) CHRONIC HEPATIC DISEASE DOSE 300 mg I.V. 200 mg I.V. 200 mg I.V. or oral 100 mg I.V. or oral INTRAVENOUS ADMINISTRATION
AFTER DILUTION WITH AN APPROPRIATE DILUENT, TROVAN I.V. SHOULD BE ADMINISTERED BY INTRAVENOUS INFUSION OVER A PERIOD OF 60 MINUTES. CAUTION: RAPID OR BOLUS INTRAVENOUS INFUSION SHOULD BE AVOIDED. (See PRECAUTIONS.)
TROVAN I.V. is supplied in single-use vials containing a concentrated solution of alatrofloxacin mesylate in Water for Injection (equivalent of 200 mg or 300 mg as trovafloxacin). Each mL contains alatrofloxacin mesylate equivalent to 5 mg trovafloxacin. (See HOW SUPPLIED for container sizes.) THESE TROVAN I.V. SINGLE-USE VIALS MUST BE FURTHER DILUTED WITH AN APPROPRIATE SOLUTION PRIOR TO INTRAVENOUS ADMINISTRATION. This parenteral drug product should be inspected visually for discoloration and particulate matter prior to dilution and administration. Since no preservative or bacteriostatic agent is present in this product, aseptic technique must be used in preparation of the final parenteral solution.
PREPARATION OF ALATROFLOXACIN MESYLATE INJECTION FOR ADMINISTRATION
The intravenous dose should be prepared by aseptically withdrawing the appropriate volume of concentrate from the vials of TROVAN I.V. This should be diluted with a suitable intravenous solution to a final concentration of 1–2 mg/mL. (SeeCompatible Intravenous Solutions.) The resulting solution should be infused over a period of 60 minutes by direct infusion or through a Y-type intravenous infusion set which may already be in place.
Since the vials are for single-use only, any unused portion should be discarded.
Since only limited data are available on the compatibility of alatrofloxacin intravenous injection with other intravenous substances, additives or other medications should not be added to TROVAN I.V. in single-use vials or infused simultaneously through the same intravenous line.
If the same intravenous line is used for sequential infusion of several different drugs, the line should be flushed before and after infusion of TROVAN I.V. with an infusion solution compatible with TROVAN I.V. and with any other drug(s) administered via this common line.
If TROVAN I.V. is to be given concomitantly with another drug, each drug should be given separately in accordance with the recommended dosage and route of administration for each drug.
The desired dosage of TROVAN I.V. may be prepared according to the following chart:
DOSAGE STRENGTH (mg)
(trovafloxacin equivalent)VOLUME TO WITHDRAW
(mL)DILUENT VOLUME
(mL)TOTAL VOLUME
(mLINFUSION CONC
(mg/mL)100 mg 20 30 50 2 100 mg 20 80 100 1 200 mg 40 60 100 2 200 mg 40 160 200 1 300 mg 60 90 150 2 300 mg 60 240 300 1 For example, to prepare a 200 mg dose at an infusion concentration of 2 mg/mL (as trovafloxacin), 40 mL of TROVAN I.V. is withdrawn from a vial and diluted with 60 mL of a compatible intravenous fluid to produce a total infusion solution volume of 100 mL.
Compatible Intravenous Solutions
5% Dextrose Injection, USP
0.45% Sodium Chloride Injection, USP
5% Dextrose and 0.45% Sodium Chloride Injection, USP
5% Dextrose and 0.2% Sodium Chloride Injection, USP
Lactated Ringer's and 5% Dextrose Injection, USPTROVAN I.V. should not be diluted with 0.9% Sodium Chloride Injection, USP (normal saline), alone or in combination with other diluents. A precipitate may form under these conditions. In addition, TROVAN I.V. should not be diluted with Lactated Ringer's, USP.
Normal saline, 0.9% Sodium Chloride Injection, USP can be used for flushing I.V. lines prior to or after administration of TROVAN I.V.
Stability of TROVAN I.V. as Supplied
When stored under recommended conditions, TROVAN I.V., as supplied in 40 mL or 60 mL vials, is stable through the expiration date printed on the label.
Stability of TROVAN I.V. Following Dilution
TROVAN I.V., when diluted with compatible intravenous solutions to concentrations of 0.5 to 2.0 mg/mL (as trovafloxacin), is physically and chemically stable for up to 7 days when refrigerated or up to 3 days at room temperature stored in glass bottles or plastic (PVC type) intravenous containers.
-
HOW SUPPLIED
Trovan Tablets and Injection are being distributed only to hospitals and long term nursing care facilities for patients initiating therapy in these facilities
Tablets
TROVAN (trovafloxacin mesylate) Tablets are available as blue, film-coated tablets. The 100 mg tablets are round and contain trovafloxacin mesylate equivalent to 100 mg trovafloxacin. The 200 mg tablets are modified oval-shaped and contain trovafloxacin mesylate equivalent to 200 mg trovafloxacin.
TROVAN Tablets are packaged and in unit dose blister strips in the following configurations:
100-mg tablets: color: blue; shape: round; debossing: "PFIZER" on one side and "378" on the other
Bottles of 30 (NDC 0049-3780-30)
Unit Dose/40 tablets (NDC 0049-3780-43)200-mg tablets: color: blue; shape: modified oval; debossing: "PFIZER" on one side and "379" on the other
Bottles of 30 (NDC 0049-3790-30)
Unit Dose/40 tablets (NDC 0049-3790-43)Injection
TROVAN is also available for intravenous administration as the prodrug, TROVAN I.V. (alatrofloxacin mesylate injection), in the following configurations:
Single-use vials containing a clear, colorless to pale-yellow concentrated solution of alatrofloxacin mesylate equivalent to 5 mg trovafloxacin/mL.
5 mg/mL, 40 mL, 200 mg
Unit dose package (NDC 0049-3890-28)5 mg/mL, 60 mL, 300 mg
Unit dose package (NDC 0049-3900-28) -
ANIMAL PHARMACOLOGY
Quinolones have been shown to cause arthropathy in immature animals.
Arthropathy and chondrodysplasia were observed in immature animals given trovafloxacin. (See WARNINGS.)
At doses from 10 to 15 times the human dose based on mg/kg or approximately 3 to 5 times based on mg/m2, trovafloxacin has been shown to cause arthropathy in immature rats and dogs. In addition, these drugs are associated with an increased incidence of chondrodysplasia in rats compared to controls. There is no evidence of arthropathies in fully mature rats and dogs at doses from 40 or 10 times the human dose based on mg/kg or approximately 5 times based on mg/m2 for a 6 month exposure period.
Unlike some other members of the quinolone class, crystalluria and ocular toxicity were not observed in chronic safety studies with rats or dogs with either trovafloxacin or its prodrug, alatrofloxacin.
Quinolones have been reported to have proconvulsant activity that is exacerbated with concomitant use of non-steroidal anti-inflammatory drugs (NSAIDS). Neither trovafloxacin administered orally at 500 mg/kg, nor alatrofloxacin administered intravenously at 75 mg/kg, showed an increase in measures of seizure activity in mice at doses when used in combination with the active metabolite of the NSAID, fenbufen.
As with other members of the quinolone class, trovafloxacin at doses 5 to 10 times the human dose based on mg/kg or 1 to 5 times the human dose based on mg/m2 produces testicular degeneration in rats and dogs dosed for 6 months.
At a dose of trovafloxacin 10 times the highest human dose based on mg/kg or approximately 5 times based on mg/m2, elevated liver enzyme levels which correlated with centrilobular hepatocellular vacuolar degeneration and necrosis were observed in dogs in a 6 month study. A subsequent study demonstrated reversibility of these effects when trovafloxacin was discontinued.
-
CLINICAL STUDIES
Hospitalized Community Acquired Pneumonia
Adult patients with clinically and radiologically documented community acquired pneumonia, requiring hospitalization and initial intravenous therapy, participated in two randomized, multicenter, double-blind, double-dummy trials. The first trial compared intravenous alatrofloxacin (200 mg once daily for 2 to 7 days) followed by oral trovafloxacin (200 mg once daily) for a total of 7 to 14 days of therapy to intravenous ciprofloxacin (400 mg BID) plus ampicillin (500 mg QID) for 2 to 7 days followed by oral ciprofloxacin (500 mg BID) plus amoxicillin (500 mg TID) for a total of 7 to 14 days of therapy. The second study compared intravenous alatrofloxacin (200 mg once daily for 2 to 7 days) followed by oral trovafloxacin (200 mg once daily) for a total of 7 to 14 days of therapy to intravenous ceftriaxone (1000 mg once daily for 2 to 7 days) followed by oral cefpodoxime (400 mg BID) for 7 to 14 days of total therapy with optional blinded erythromycin added to the ceftriaxone/cefpodoxime arm if an atypical pneumonia was suspected.
The clinical success rate (cure + improvement with no need for further antibiotic therapy) at the End of Treatment was 90% (311/346) and 90% (325/363) for TROVAN and the comparator agents, respectively. The clinical success rate at the End of Study (Day 30) was 86% (256/299) and 85% (283/334) for TROVAN and the comparator agents, respectively. All cause mortality (Day 1–35) was 2.45% (10/408) on TROVAN and 5.45% (23/422) on the comparator agents.
The following outcomes are the clinical success rates for the clinically evaluable patient groups by pathogen in these two studies:
End of Treatment End of Study Pathogen TROVAN Comparators TROVAN Comparators S. pneumoniae 89% (63/71) 95% (62/65) 87% (55/63) 91% (50/55) H. influenzae 97% (35/36) 94% (46/49) 90% (28/31) 94% (44/47) M. catarrhalis 100% (8/8) 100% (4/4) 100% (6/6) 100% (4/4) S. aureus 100% (8/8) 93% (13/14) 100% (6/6) 91% (10/11) K. pneumoniae 100% (3/3) 89% (8/9) 100% (3/3) 86% (6/7) L. pneumophila 77% (10/13) 86% (12/14) 75% (9/12) 86% (12/14) M. pneumoniae 100% (20/20) 87% (13/15) 94% (17/18) 79% (11/14) C. pneumoniae 75% (6/8) 100% (18/18) 67% (4/6) 94% (16/17) Of the above patients with clinical failure at end of treatment or study, only one alatrofloxacin patient (H. influenzae + S. pneumoniae) and one ceftriaxone + erythromycin patient (Legionella) had a microbiologically confirmed persistent pathogen at the time of failure with no emergence of resistance in either study.
Nosocomial Pneumonia
Adult patients with clinically and radiologically documented nosocomial pneumonia participated in a randomized, multicenter, double-blind, double-dummy trial comparing intravenous alatrofloxacin (300 mg once daily for 2 to 7 days) followed by oral trovafloxacin (200 mg once daily) for a total of 7 to 14 days of therapy to intravenous ciprofloxacin (400 mg BID) for 2 to 7 days followed by oral ciprofloxacin (750 mg BID) for a total of 7 to 14 days of therapy with optional blinded clindamycin or metronidazole added to the ciprofloxacin arm if an anaerobic pneumonia was suspected. In subjects with documented Pseudomonas infection or methicillin-resistant S. aureus, aztreonam or vancomycin, respectively, could have been added to either treatment regimen.
The clinical success rate (cure + improvement with no need for further antibiotic therapy) at the End of Treatment was 77% (68/88) and 78% (79/101) for TROVAN and ciprofloxacin, respectively. The clinical success rate at the End of Study (Day 30) was 69% (50/72) and 68% (54/79) for TROVAN and ciprofloxacin, respectively.
The following outcomes are the clinical success rates for the clinically evaluable patient groups by pathogen:
End of Treatment End of Study Pathogen TROVAN Ciprofloxacin TROVAN Ciprofloxacin P. aeruginosa 67% (10/15) 55% (6/11) 62% (8/13) 25% (2/8) H. influenzae 88% (7/8) 89% (8/9) 83% (5/6) 86% (6/7) E. coli 71% (5/7) 80% (4/5)) 50% (3/6) 80% (4/5) S. aureus 64% (7/11) 80% (8/10) 50% (4/8) 67% (4/6) Of the above patients with clinical failure at end of treatment or study, 2 alatrofloxacin patients (S. aureus, P. aeruginosa) and 4 ciprofloxacin patients (all P. aeruginosa) had a microbiologically confirmed persistent pathogen at the time of failure. Three of the 4 ciprofloxacin patients with clinical failure and persistence had emergence of resistance with none on alatrofloxacin.
Complicated Intra-Abdominal Infections
Patients hospitalized with clinically documented, complicated intra-abdominal infections, including post-surgical infections, participated in a randomized, double-blind, multicenter trial comparing intravenous alatrofloxacin (300 mg once daily) followed by oral trovafloxacin (200 mg once daily) to intravenous imipenem/cilastatin (1g q8h) followed by oral amoxicillin/clavulanic acid (500 mg TID) for a maximum of 14 days of therapy. The clinical success rate (cure + improvement) at the End of Treatment was 88% (136/155) and 86% (122/142) for alatrofloxacin→trovafloxacin and imipenem/cilastatin→amoxicillin/clavulanic acid, respectively. The clinical success rate at the End of Study (Day 30) was 83% (129/156) and 84% (127/152) for alatrofloxacin→trovafloxacin and imipenem/cilastatin→amoxicillin/clavulanic acid, respectively.
The following are the clinical success rates for the clinically evaluable patient groups by pathogen:
End of Treatment End of Study Pathogen TROVAN Imipenem/Cila Amox/Clav TROVAN Imipenem/Cila Amox/Clav E. coli 94% (72/77) 90% (52/58) 86% (66/77) 86% (51/59) Bacteroides fragilis 97% (30/31) 82% (28/34) 84% (26/31) 75% (27/36) viridans group streptococci 90% (18/20) 83% (19/23) 90% (18/20) 78% (18/23) Pseudomonas aeruginosa 94% (15/16) 82% (14/17) 88% (14/16) 83% (15/18) Klebsiella pneumoniae 80% (12/15) 71% (10/14) 67% (10/15) 71% (10/14) Peptostreptococcus spp. 86% (12/14) 88% (7/8) 79% (11/14) 75% (6/8) Prevotella spp. 77% (10/13) 50% (2/4) 77% (10/13) 60% (3/5) Of patients with a baseline pathogen and a clinical response of failure at the End of Study, 9 of 26 on TROVAN and 10 of 21 on imipenem/cilastatin had microbiologically-confirmed persistence of the baseline pathogen with no emergence of resistance in either group.
-
REFERENCES
- National Committee for Clinical Laboratory Standards, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically – Fourth Edition; Approved Standard, NCCLS Document M7-A4, Vol. 17, No. 2, NCCLS, Wayne, PA, January, 1997.
- National Committee for Clinical Laboratory Standards. Performance Standards for Antimicrobial Disk Susceptibility Tests – Sixth Edition; Approved Standard, NCCLS Document M2-A6, Vol. 17, No. 1, NCCLS, Wayne, PA, January, 1997.
- National Committee for Clinical Laboratory Standards. Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria – Fourth Edition; Approved Standard, NCCLS Document M11-A4, Vol. 17, No. 22, NCCLS, Wayne, PA, December, 1997.
- SPL UNCLASSIFIED SECTION
-
INGREDIENTS AND APPEARANCE
TROVAN
trovafloxacin mesylate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0049-3780 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength trovafloxacin mesylate (UNII: 0P1LKO80WN) (trovafloxacin - UNII:9F388J00UK) 100 mg Inactive Ingredients Ingredient Name Strength microcrystalline cellulose () cross linked sodium carboxymethylcellulose () magnesium stearate (UNII: 70097M6I30) hydroxypropylcellulose () hydroxypropylmethylcellulose () titanium dioxide (UNII: 15FIX9V2JP) polyethylene glycol () FD&C blue #2 aluminum lake () Product Characteristics Color BLUE (BLUE) Score no score Shape ROUND (ROUND) Size 8mm Flavor Imprint Code Pfizer;378 Contains Coating true Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0049-3780-30 30 in 01 BOTTLE 2 NDC:0049-3780-43 40 in 01 BLISTER PACK TROVAN
trovafloxacin mesylate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0049-3790 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength trovafloxacin mesylate (UNII: 0P1LKO80WN) (trovafloxacin - UNII:9F388J00UK) 200 mg Inactive Ingredients Ingredient Name Strength microcrystalline cellulose () cross linked sodium carboxymethylcellulose () magnesium stearate (UNII: 70097M6I30) hydroxypropylcellulose () hydroxypropylmethylcellulose () titanium dioxide (UNII: 15FIX9V2JP) polyethylene glycol () FD&C blue #2 aluminum lake () Product Characteristics Color BLUE (BLUE) Score no score Shape OVAL (OVAL) Size 14mm Flavor Imprint Code Pfizer;379 Contains Coating true Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0049-3790-30 30 in 01 BOTTLE 2 NDC:0049-3790-43 40 in 01 BLISTER PACK TROVAN
trovafloxacin mesylate injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0049-3890 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength trovafloxacin mesylate (UNII: 0P1LKO80WN) (trovafloxacin - UNII:9F388J00UK) 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength Water () sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0049-3890-28 40 mL in 1 VIAL, SINGLE-USE TROVAN
trovafloxacin mesylate injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0049-3900 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength trovafloxacin mesylate (UNII: 0P1LKO80WN) (trovafloxacin - UNII:9F388J00UK) 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength Water () sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0049-3900-28 60 mL in 1 VIAL, SINGLE-USE Labeler - Roerig
