Label: TELMISARTAN- telmisartan tablet



-
NDC Code(s):
42571-226-05,
42571-226-10,
42571-226-11,
42571-226-30, view more42571-226-32, 42571-226-41, 42571-226-90, 42571-227-05, 42571-227-10, 42571-227-11, 42571-227-30, 42571-227-32, 42571-227-41, 42571-227-90, 42571-228-05, 42571-228-10, 42571-228-11, 42571-228-30, 42571-228-32, 42571-228-41, 42571-228-90
- Packager: Micro Labs Limited
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated January 19, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TELMISARTAN TABLETS safely and effectively. See full prescribing information for TELMISARTAN TABLETS.
TELMISARTAN tablets, for oral use
Initial U.S. Approval: 1998
INDICATIONS AND USAGE
Telmisartan is an angiotensin II receptor blocker (ARB) indicated for:
- Treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. ( 1.1)
- Cardiovascular (CV) risk reduction in patients unable to take ACE inhibitors ( 1.2)
DOSAGE AND ADMINISTRATION
- May be administered with or without food ( 2.1)
- When used for cardiovascular risk reduction, monitoring of blood pressure is recommended, and if appropriate, adjustment of medications that lower blood pressure may be necessary ( 2.2)
Indication
Starting Dose
Dose Range
Hypertension ( 2.1)
40 mg once daily
40 to 80 mg once daily
Cardiovascular Risk Reduction ( 2.2)
80 mg once daily
80 mg once daily
DOSAGE FORMS AND STRENGTHS
- Tablets: 20 mg, 40 mg, 80 mg ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Avoid fetal or neonatal exposure ( 5.1)
- Hypotension: Correct any volume or salt depletion before initiating therapy. Observe for signs and symptoms of hypotension ( 5.2)
- Monitor carefully in patients with impaired hepatic (5.4) or renal function ( 5.5)
- Avoid concomitant use of an ACE inhibitor and angiotensin receptor blocker ( 5.6)
ADVERSE REACTIONS
- Hypertension: The most common adverse events (≥1%) reported in hypertension trials are back pain, sinusitis, and diarrhea ( 6.1)
- Cardiovascular risk reduction: The serious adverse events (≥1%) reported in cardiovascular risk reduction trials were intermittent claudication and skin ulcer ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Micro Labs USA Inc. at 1-855-839-8195, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: FETAL TOXICITY
1 INDICATIONS AND USAGE
1.1 Hypertension
1.2 Cardiovascular Risk Reduction
2 DOSAGE AND ADMINISTRATION
2.1 Hypertension
2.2 Cardiovascular Risk Reduction
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
5.2 Hypotension
5.3 Hyperkalemia
5.4 Impaired Hepatic Function
5.5 Impaired Renal Function
5.6 Dual Blockade of the Renin-Angiotensin-Aldosterone System (RAS)
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Insufficiency
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Hypertension
14.2 Cardiovascular Risk Reduction
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- BOXED WARNING (What is this?)
-
1 INDICATIONS AND USAGE
1.1 Hypertension
Telmisartan tablets are indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including the class to which this drug principally belongs.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
Telmisartan may be used alone or in combination with other antihypertensive agents [see Clinical Studies ( 14.1)] .
1.2 Cardiovascular Risk Reduction
Telmisartan is indicated for reduction of the risk of myocardial infarction, stroke, or death from cardiovascular causes in patients 55 years of age or older at high risk of developing major cardiovascular events who are unable to take ACE inhibitors.
High risk for cardiovascular events can be evidenced by a history of coronary artery disease, peripheral arterial disease, stroke, transient ischemic attack, or high-risk diabetes (insulin-dependent or non-insulin dependent) with evidence of end-organ damage [see Clinical Studies ( 14.2)] . Telmisartan can be used in addition to other needed treatment (such as antihypertensive, antiplatelet or lipid-lowering therapy) [see Clinical Studies ( 14.2)] .
Studies of telmisartan in this setting do not exclude the possibility that telmisartan may not preserve a meaningful fraction of the effect of the ACE inhibitor to which it was compared. Consider using the ACE inhibitor first, and, if it is stopped for cough only, consider re-trying the ACE inhibitor after the cough resolves.
Use of telmisartan with an ACE inhibitor is not recommended [see Warnings and Precautions ( 5.6)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Hypertension
Dosage must be individualized. The usual starting dose of telmisartan tablets is 40 mg orally once a day. Blood pressure response is dose-related over the range of 20 to 80 mg [see Clinical Studies ( 14.1)] .
Most of the antihypertensive effect is apparent within 2 weeks and maximal reduction is generally attained after 4 weeks.
No initial dosage adjustment is necessary for elderly patients or patients with renal impairment, including those on hemodialysis. Patients on dialysis may develop orthostatic hypotension; their blood pressure should be closely monitored.
Telmisartan tablets may be administered with other antihypertensive agents.
Telmisartan tablets may be administered with or without food.
2.2 Cardiovascular Risk Reduction
The recommended dose of telmisartan tablets is 80 mg once a day and can be administered with or without food. It is not known whether doses lower than 80 mg of telmisartan are effective in reducing the risk of cardiovascular morbidity and mortality.
When initiating telmisartan therapy for cardiovascular risk reduction, monitoring of blood pressure is recommended, and if appropriate, adjustment of medications that lower blood pressure may be necessary.
-
3 DOSAGE FORMS AND STRENGTHS
- 20 mg, white to off-white, circular, flat face bevel edged, uncoated tablets engraved with ‘T’ on one face and ‘20’ on other face.
- 40 mg, white to off-white, oval shaped, biconvex, uncoated tablets engraved with ‘T40’ on one face and plain on other face.
- 80 mg, white to off-white, capsule shaped, biconvex, uncoated tablets engraved with ‘T80’ on one face and plain on other face.
-
4 CONTRAINDICATIONS
Telmisartan tablets are contraindicated in patients with known hypersensitivity (e.g., anaphylaxis or angioedema) to telmisartan or any other component of this product [see Adverse Reactions ( 6.2)].
Do not co-administer aliskiren with telmisartan tablets in patients with diabetes [see Drug Interactions ( 7)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue telmisartan as soon as possible [see Use in Specific Populations ( 8.1)].
5.2 Hypotension
In patients with an activated renin-angiotensin system, such as volume- or salt-depleted patients (e.g., those being treated with high doses of diuretics), symptomatic hypotension may occur after initiation of therapy with telmisartan. Either correct this condition prior to administration of telmisartan, or start treatment under close medical supervision with a reduced dose.
If hypotension does occur, the patient should be placed in the supine position and, if necessary, given an intravenous infusion of normal saline. A transient hypotensive response is not a contraindication to further treatment, which usually can be continued without difficulty once the blood pressure has stabilized.
5.3 Hyperkalemia
Hyperkalemia may occur in patients on ARBs, particularly in patients with advanced renal impairment, heart failure, on renal replacement therapy, or on potassium supplements, potassium-sparing diuretics, potassium-containing salt substitutes or other drugs that increase potassium levels. Consider periodic determinations of serum electrolytes to detect possible electrolyte imbalances, particularly in patients at risk.
5.4 Impaired Hepatic Function
As the majority of telmisartan is eliminated by biliary excretion, patients with biliary obstructive disorders or hepatic insufficiency can be expected to have reduced clearance. Initiate telmisartan at low doses and titrate slowly in these patients [see Use in Specific Populations ( 8.6) and Clinical Pharmacology ( 12.3)] .
5.5 Impaired Renal Function
As a consequence of inhibiting the renin-angiotensin-aldosterone system, anticipate changes in renal function in susceptible individuals. In patients whose renal function may depend on the activity of the renin-angiotensin-aldosterone system (e.g., patients with severe congestive heart failure or renal dysfunction), treatment with angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor antagonists has been associated with oliguria and/or progressive azotemia and (rarely) with acute renal failure and/or death. Similar results have been reported with telmisartan [see Clinical Pharmacology ( 12.3)] .
In studies of ACE inhibitors in patients with unilateral or bilateral renal artery stenosis, increases in serum creatinine or blood urea nitrogen were observed. There has been no long-term use of telmisartan in patients with unilateral or bilateral renal artery stenosis, but anticipate an effect similar to that seen with ACE inhibitors.
5.6 Dual Blockade of the Renin-Angiotensin-Aldosterone System (RAS)
Dual blockade of the RAS with angiotensin-receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy.
The ONTARGET trial enrolled 25,620 patients ≥55 years old with atherosclerotic disease or diabetes with end-organ damage, randomizing them to telmisartan only, ramipril only, or the combination, and followed them for a median of 56 months. Patients receiving the combination of telmisartan and ramipril did not obtain any additional benefit compared to monotherapy, but experienced an increased incidence of renal dysfunction (e.g., acute renal failure) compared with groups receiving telmisartan alone or ramipril alone.
In most patients no benefit has been associated with using two RAS inhibitors concomitantly. In general, avoid combined use of RAS inhibitors. Closely monitor blood pressure, renal function, and electrolytes in patients on telmisartan and other agents that affect the RAS.
Do not co-administer aliskiren with telmisartan in patients with diabetes. Avoid concomitant use of aliskiren with telmisartan in patients with renal impairment (GFR <60 mL/min/1.73 m 2).
-
6 ADVERSE REACTIONS
The following adverse reaction is described elsewhere in labeling:
- Renal dysfunction upon use with ramipril [see Warnings and Precautions ( 5.6)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reactions rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
Hypertension
Telmisartan has been evaluated for safety in more than 3700 patients, including 1900 treated for over 6 months and more than 1300 for over one year. Adverse experiences have generally been mild and transient in nature and have infrequently required discontinuation of therapy.
In placebo-controlled trials involving 1041 patients treated with various doses of telmisartan (20 to 160 mg) monotherapy for up to 12 weeks, the overall incidence of adverse events was similar to that in patients treated with placebo.
Adverse events occurring at an incidence of ≥1% in patients treated with telmisartan and at a greater rate than in patients treated with placebo, irrespective of their causal association, are presented in Table 1.
Table 1 Adverse Events Occurring at an Incidence of ≥1% in Patients Treated with Telmisartan and at a Greater Rate Than Patients Treated with Placebo
Telmisartan
n=1455
%
Placebo
n=380
%
Upper respiratory tract infection
7
6
Back pain
3
1
Sinusitis
3
2
Diarrhea
3
2
Pharyngitis
1
0
In addition to the adverse events in the table, the following events occurred at a rate of ≥1% but were at least as frequent in the placebo group: influenza-like symptoms, dyspepsia, myalgia, urinary tract infection, abdominal pain, headache, dizziness, pain, fatigue, coughing, hypertension, chest pain, nausea, and peripheral edema. Discontinuation of therapy because of adverse events was required in 2.8% of 1455 patients treated with telmisartan tablets and 6.1% of 380 placebo patients in placebo-controlled clinical trials.
The incidence of adverse events was not dose-related and did not correlate with gender, age, or race of patients.
The incidence of cough occurring with telmisartan in 6 placebo-controlled trials was identical to that noted for placebo-treated patients (1.6%).
In addition to those listed above, adverse events that occurred in more than 0.3% of 3500 patients treated with telmisartan monotherapy in controlled or open trials are listed below. It cannot be determined whether these events were causally related to telmisartan tablets:
Autonomic Nervous System: impotence, increased sweating, flushing; Body as a Whole: allergy, fever, leg pain, malaise; Cardiovascular: palpitation, dependent edema, angina pectoris, tachycardia, leg edema, abnormal ECG; CNS: insomnia, somnolence, migraine, vertigo, paresthesia, involuntary muscle contractions, hypoesthesia; Gastrointestinal: flatulence, constipation, gastritis, vomiting, dry mouth, hemorrhoids, gastroenteritis, enteritis, gastroesophageal reflux, toothache, non-specific gastrointestinal disorders; Metabolic: gout, hypercholesterolemia, diabetes mellitus; Musculoskeletal: arthritis, arthralgia, leg cramps; Psychiatric: anxiety, depression, nervousness; Resistance Mechanism: infection, fungal infection, abscess, otitis media; Respiratory: asthma, bronchitis, rhinitis, dyspnea, epistaxis; Skin: dermatitis, rash, eczema, pruritus; Urinary: micturition frequency, cystitis; Vascular: cerebrovascular disorder; and Special Senses: abnormal vision, conjunctivitis, tinnitus, earache.
During initial clinical studies, a single case of angioedema was reported (among a total of 3781 patients treated).
Clinical Laboratory Findings
In placebo-controlled clinical trials, clinically relevant changes in standard laboratory test parameters were rarely associated with administration of telmisartan tablets.
Hemoglobin: A greater than 2 g/dL decrease in hemoglobin was observed in 0.8% telmisartan patients compared with 0.3% placebo patients. No patients discontinued therapy because of anemia.
Creatinine: A 0.5 mg/dL rise or greater in creatinine was observed in 0.4% telmisartan patients compared with 0.3% placebo patients. One telmisartan-treated patient discontinued therapy because of increases in creatinine and blood urea nitrogen.
Liver Enzymes: Occasional elevations of liver chemistries occurred in patients treated with telmisartan; all marked elevations occurred at a higher frequency with placebo. No telmisartan-treated patients discontinued therapy because of abnormal hepatic function.
Cardiovascular Risk Reduction
Because common adverse reactions were well characterized in studies of telmisartan in hypertension, only adverse events leading to discontinuation and serious adverse events were recorded in subsequent studies of telmisartan for cardiovascular risk reduction. In TRANSCEND (N=5926, 4 years and 8 months of follow-up), discontinuations for adverse events were 8.4% on telmisartan and 7.6% on placebo. The only serious adverse events at least 1% more common on telmisartan than placebo were intermittent claudication (7% vs 6%) and skin ulcer (3% vs 2%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of telmisartan. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate reliably their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: Anemia , eosinophilia, thrombocytopenia Gastrointestinal Disorders: Abdominal pain, diarrhea, dyspepsia, nausea
General Disorders and Administration Site Conditions: Asthenia, chest pain, edema, face edema, fatigue, lower limb edema, pain, weakness
Hepato-biliary: Abnormal hepatic function/liver disorder
Immune System Disorders: Anaphylactic reaction, hypersensitivity
Investigations: Increased CPK, uric acid increased
Metabolism and Nutrition Disorders: Hyperkalemia, hypoglycemia (in diabetic patients), hyponatremia
Musculoskeletal and Connective Tissue Disorders: Myalgia
Nervous System Disorders: Dizziness, headache, syncope
Renal and Urinary Disorders: Renal impairment including acute renal failure
Reproductive System and Breast Disorders: Erectile dysfunction Respiratory,
Thoracic and Mediastinal Disorders: Coughing
Skin and Subcutaneous Tissue Disorders: Angioedema (with fatal outcome), angioneurotic edema, drug eruption (toxic skin eruption mostly reported as toxicoderma, rash, and urticaria), erythema, sweating increased, urticaria
Vascular Disorder: Hypotension (including postural hypotension)
Rare cases of rhabdomyolysis have been reported in patients receiving angiotensin II receptor blockers, including telmisartan.
-
7 DRUG INTERACTIONS
Aliskiren: Do not co-administer aliskiren with telmisartan in patients with diabetes. Avoid use of aliskiren with telmisartan in patients with renal impairment (GFR <60 mL/min).
Digoxin: When telmisartan was co-administered with digoxin, median increases in digoxin peak plasma concentration (49%) and in trough concentration (20%) were observed. Therefore, monitor digoxin levels when initiating, adjusting, and discontinuing telmisartan for the purpose of keeping the digoxin level within the therapeutic range.
Lithium: Reversible increases in serum lithium concentrations and toxicity have been reported during concomitant administration of lithium with angiotensin II receptor antagonists including telmisartan. Therefore, monitor serum lithium levels during concomitant use.
Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors): In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, co-administration of NSAIDs, including selective COX-2 inhibitors, with angiotensin II receptor antagonists, including telmisartan, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving telmisartan and NSAID therapy.
The antihypertensive effect of angiotensin II receptor antagonists, including telmisartan may be attenuated by NSAIDs including selective COX-2 inhibitors.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Telmisartan can cause fetal harm when administered to a pregnant woman. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death (see Clinical Considerations). Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Studies in rats and rabbits with telmisartan showed fetotoxicity only at maternally toxic doses (see Data). When pregnancy is detected, discontinue telmisartan as soon as possible.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Hypertension in pregnancy increases the maternal risk for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage). Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death. Pregnant women with hypertension should be carefully monitored and managed accordingly.
Fetal/Neonatal adverse reactions
Use of drugs that act on the RAS in the second and third trimesters of pregnancy can result in the following: oligohydramnios, reduced fetal renal function leading to anuria and renal failure, fetal lung hypoplasia, skeletal deformations, including skull hypoplasia, hypotension, and death. In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus.
In patients taking telmisartan during pregnancy, perform serial ultrasound examinations to assess the intra-amniotic environment. Fetal testing may be appropriate, based on the week of gestation. If oligohydramnios is observed, discontinue telmisartan, unless it is considered lifesaving for the mother. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury.
Closely observe infants with histories of in utero exposure to telmisartan for hypotension, oliguria, and hyperkalemia. If oliguria or hypotension occurs, support blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function [see Use in Specific Populations ( 8.4)] .
Data
Animal Data
No teratogenic effects were observed when telmisartan was administered to pregnant rats at oral doses of up to 50 mg/kg/day and to pregnant rabbits at oral doses up to 45 mg/kg/day. In rabbits, embryolethality associated with maternal toxicity (reduced body weight gain and food consumption) was observed at 45 mg/kg/day [about 12 times the maximum recommended human dose (MRHD) of 80 mg on a mg/m 2 basis]. In rats, maternally toxic (reduction in body weight gain and food consumption) telmisartan doses of 15 mg/kg/day (about 1.9 times the MRHD on a mg/m 2 basis), administered during late gestation and lactation, were observed to produce adverse effects in neonates, including reduced viability, low birth weight, delayed maturation, and decreased weight gain. The no-observed-effect doses for developmental toxicity in rats and rabbits, 5 and 15 mg/kg/day, respectively, are about 0.64 and 3.7 times, on a mg/m 2 basis, the maximum recommended human dose of telmisartan (80 mg/day).
8.2 Lactation
Risk Summary
There is no information regarding the presence of telmisartan in human milk, the effects on the breastfed infant, or the effects on milk production. Telmisartan is present in the milk of lactating rats (see Data). Because of the potential for serious adverse reactions in the breastfed infant including hypotension, hyperkalemia and renal impairment, advise a nursing woman not to breastfeed during treatment with telmisartan.
Data
Telmisartan was present in the milk of lactating rats at concentrations 1.5 to 2 times those found in plasma from 4 to 8 hours after administration.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established [see Clinical Pharmacology ( 12.3)].
Neonates with a history of in utero exposure to telmisartan:
If oliguria or hypotension occurs, support blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function.
8.5 Geriatric Use
Of the total number of patients receiving telmisartan in hypertension clinical studies, 551 (19%) were 65 to 74 years of age and 130 (4%) were 75 years or older. No overall differences in effectiveness and safety were observed in these patients compared to younger patients and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Of the total number of patients receiving telmisartan in the cardiovascular risk reduction study (ONTARGET), the percentage of patients ≥65 to <75 years of age was 42%; 15% of patients were ≥75 years old. No overall differences in effectiveness and safety were observed in these patients compared to younger patients and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Insufficiency
Monitor carefully and uptitrate slowly in patients with biliary obstructive disorders or hepatic insufficiency [see Warnings and Precautions ( 5.4)] .
-
10 OVERDOSAGE
Limited data are available with regard to overdosage in humans. The most likely manifestation of overdosage with telmisartan tablets would be hypotension, dizziness and tachycardia; bradycardia could occur from parasympathetic (vagal) stimulation. If symptomatic hypotension should occur, supportive treatment should be instituted. Telmisartan is not removed by hemofiltration and is not dialyzable.
-
11 DESCRIPTION
Telmisartan tablets, USP is a non-peptide angiotensin II receptor (type AT 1) antagonist.
Telmisartan is chemically described as 4'-[(1,4’-dimethyl-2'-propyl [2,6'-bi-1 H-benzimidazol]-1'-yl)methyl]-[1,1'-biphenyl]-2-carboxylic acid. Its molecular formula is C 33H 30N 4O 2, its molecular weight is 514.62, and its structural formula is:
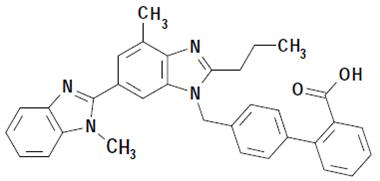
Telmisartan USP is a white to slightly yellowish crystalline powder. It is practically insoluble in water and in the pH range of 3 to 9, sparingly soluble in strong acid (except insoluble in hydrochloric acid), and soluble in strong base.
Telmisartan USP is available as tablets for oral administration, containing 20 mg, 40 mg or 80 mg of telmisartan USP. The tablets contain the following inactive ingredients: crospovidone Type A/kollidon CL, mannitol, magnesium stearate, meglumine, povidone K-30, and sodium hydroxide. Telmisartan tablets USP are hygroscopic and require protection from moisture.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Angiotensin II is formed from angiotensin I in a reaction catalyzed by angiotensin-converting enzyme (ACE, kininase II). Angiotensin II is the principal pressor agent of the renin-angiotensin system, with effects that include vasoconstriction, stimulation of synthesis and release of aldosterone, cardiac stimulation, and renal reabsorption of sodium. Telmisartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT 1 receptor in many tissues, such as vascular smooth muscle and the adrenal gland. Its action is therefore independent of the pathways for angiotensin II synthesis.
There is also an AT 2 receptor found in many tissues, but AT 2 is not known to be associated with cardiovascular homeostasis. Telmisartan has much greater affinity (>3,000 fold) for the AT1 receptor than for the AT 2 receptor.
Blockade of the renin-angiotensin system with ACE inhibitors, which inhibit the biosynthesis of angiotensin II from angiotensin I, is widely used in the treatment of hypertension. ACE inhibitors also inhibit the degradation of bradykinin, a reaction also catalyzed by ACE. Because telmisartan does not inhibit ACE (kininase II), it does not affect the response to bradykinin. Whether this difference has clinical relevance is not yet known. Telmisartan does not bind to or block other hormone receptors or ion channels known to be important in cardiovascular regulation.
Blockade of the angiotensin II receptor inhibits the negative regulatory feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and angiotensin II circulating levels do not overcome the effect of telmisartan on blood pressure.
12.2 Pharmacodynamics
In normal volunteers, a dose of telmisartan 80 mg inhibited the pressor response to an intravenous infusion of angiotensin II by about 90% at peak plasma concentrations with approximately 40% inhibition persisting for 24 hours.
Plasma concentration of angiotensin II and plasma renin activity (PRA) increased in a dose-dependent manner after single administration of telmisartan to healthy subjects and repeated administration to hypertensive patients. The once-daily administration of up to 80 mg telmisartan to healthy subjects did not influence plasma aldosterone concentrations. In multiple dose studies with hypertensive patients, there were no clinically significant changes in electrolytes (serum potassium or sodium), or in metabolic function (including serum levels of cholesterol, triglycerides, HDL, LDL, glucose, or uric acid).
In 30 hypertensive patients with normal renal function treated for 8 weeks with telmisartan 80 mg or telmisartan 80 mg in combination with hydrochlorothiazide 12.5 mg, there were no clinically significant changes from baseline in renal blood flow, glomerular filtration rate, filtration fraction, renovascular resistance, or creatinine clearance.
12.3 Pharmacokinetics
Absorption:
Following oral administration, peak concentrations (C max) of telmisartan are reached in 0.5 to 1 hour after dosing. Food slightly reduces the bioavailability of telmisartan, with a reduction in the area under the plasma concentration-time curve (AUC) of about 6% with the 40 mg tablet and about 20% after a 160 mg dose. The absolute bioavailability of telmisartan is dose dependent. At 40 mg and 160 mg, the bioavailability was 42% and 58%, respectively. The pharmacokinetics of orally administered telmisartan are nonlinear over the dose range 20 mg to 160 mg, with greater than proportional increases of plasma concentrations (C max and AUC) with increasing doses. Telmisartan shows bi-exponential decay kinetics with a terminal elimination half-life of approximately 24 hours. Trough plasma concentrations of telmisartan with once daily dosing are about 10% to 25% of peak plasma concentrations. Telmisartan has an accumulation index in plasma of 1.5 to 2 upon repeated once daily dosing.
Distribution
Telmisartan is highly bound to plasma proteins (>99.5%), mainly albumin and α 1 - acid glycoprotein. Plasma protein binding is constant over the concentration range achieved with recommended doses. The volume of distribution for telmisartan is approximately 500 liters indicating additional tissue binding.
Metabolism and Elimination
Following either intravenous or oral administration of 14C-labeled telmisartan, most of the administered dose (>97%) was eliminated unchanged in feces via biliary excretion; only minute amounts were found in the urine (0.91% and 0.49% of total radioactivity, respectively).
Telmisartan is metabolized by conjugation to form a pharmacologically inactive acyl glucuronide; the glucuronide of the parent compound is the only metabolite that has been identified in human plasma and urine. After a single dose, the glucuronide represents approximately 11% of the measured radioactivity in plasma. The cytochrome P450 isoenzymes are not involved in the metabolism of telmisartan.
Total plasma clearance of telmisartan is >800 mL/min. Terminal half-life and total clearance appear to be independent of dose.
Specific Populations
Renal Insufficiency
No dosage adjustment is necessary in patients with decreased renal function. Telmisartan is not removed from blood by hemofiltration and is not dialyzable [see Warnings and Precautions ( 5.5) and Dosage and Administration ( 2.1)].
Hepatic Insufficiency
In patients with hepatic insufficiency, plasma concentrations of telmisartan are increased, and absolute bioavailability approaches 100% [see Warnings and Precautions ( 5.4) and Use in Specific Populations ( 8.6)].
Gender
Plasma concentrations of telmisartan are generally 2 to 3 times higher in females than in males. In clinical trials, however, no significant increases in blood pressure response or in the incidence of orthostatic hypotension were found in women. No dosage adjustment is necessary.
Geriatric Patients
The pharmacokinetics of telmisartan do not differ between the elderly and those younger than 65 years [see Dosage and Administration ( 2.1)].
Pediatric Patients
Telmisartan pharmacokinetics have not been investigated in patients <18 years of age.
Drug Interaction Studies
Telmisartan
Ramipril and Ramiprilat: Co-administration of telmisartan 80 mg once daily and ramipril 10 mg once daily to healthy subjects increases steady-state C max and AUC of ramipril 2.3-and 2.1-fold, respectively, and C max and AUC of ramiprilat 2.4-and 1.5-fold, respectively. In contrast, C max and AUC of telmisartan decrease by 31% and 16%, respectively. When co-administering telmisartan and ramipril, the response may be greater because of the possibly additive pharmacodynamic effects of the combined drugs, and also because of the increased exposure to ramipril and ramiprilat in the presence of telmisartan.
Other Drugs: Co-administration of telmisartan did not result in a clinically significant interaction with acetaminophen, amlodipine, glyburide, simvastatin, hydrochlorothiazide, warfarin, or ibuprofen. Telmisartan is not metabolized by the cytochrome P450 system and had no effects in vitro on cytochrome P450 enzymes, except for some inhibition of CYP2C19. Telmisartan is not expected to interact with drugs that inhibit cytochrome P450 enzymes; it is also not expected to interact with drugs metabolized by cytochrome P450 enzymes, except for possible inhibition of the metabolism of drugs metabolized by CYP2C19.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
There was no evidence of carcinogenicity when telmisartan was administered in the diet to mice and rats for up to 2 years. The highest doses administered to mice (1000 mg/kg/day) and rats (100 mg/kg/day) are, on a mg/m 2 basis, about 59 and 13 times, respectively, the maximum recommended human dose (MRHD) of telmisartan. These same doses have been shown to provide average systemic exposures to telmisartan >100 times and >25 times, respectively, the systemic exposure in humans receiving the MRHD (80 mg/day).
Genotoxicity assays did not reveal any telmisartan-related effects at either the gene or chromosome level. These assays included bacterial mutagenicity tests with Salmonella and E. coli (Ames), a gene mutation test with Chinese hamster V79 cells, a cytogenetic test with human lymphocytes, and a mouse micronucleus test.
No drug-related effects on the reproductive performance of male and female rats were noted at 100 mg/kg/day (the highest dose administered), about 13 times, on a mg/m 2 basis, the MRHD of telmisartan. This dose in the rat resulted in an average systemic exposure (telmisartan AUC as determined on day 6 of pregnancy) at least 50 times the average systemic exposure in humans at the MRHD (80 mg/day).
-
14 CLINICAL STUDIES
14.1 Hypertension
The antihypertensive effects of telmisartan have been demonstrated in six principal placebo-controlled clinical trials, studying a range of 20 to 160 mg; one of these examined the antihypertensive effects of telmisartan and hydrochlorothiazide in combination. The studies involved a total of 1773 patients with mild to moderate hypertension (diastolic blood pressure of 95 to 114 mmHg), 1031 of whom were treated with telmisartan. Following once daily administration of telmisartan, the magnitude of blood pressure reduction from baseline after placebo subtraction was approximately (SBP/DBP) 6 to 8/6 mmHg for 20 mg, 9 to 13/6 to 8 mmHg for 40 mg, and 12 to 13/7 to 8 mmHg for 80 mg. Larger doses (up to 160 mg) did not appear to cause a further decrease in blood pressure.
Upon initiation of antihypertensive treatment with telmisartan, blood pressure was reduced after the first dose, with a maximal reduction by about 4 weeks. With cessation of treatment with telmisartan tablets, blood pressure gradually returned to baseline values over a period of several days to one week. During long-term studies (without placebo control) the effect of telmisartan appeared to be maintained for up to at least one year. The antihypertensive effect of telmisartan is not influenced by patient age, gender, weight, or body mass index. Blood pressure response in black patients (usually a low-renin population) is noticeably less than that in Caucasian patients. This has been true for most, but not all, angiotensin II antagonists and ACE inhibitors.
In a controlled study, the addition of telmisartan to hydrochlorothiazide produced an additional dose-related reduction in blood pressure that was similar in magnitude to the reduction achieved with telmisartan monotherapy. Hydrochlorothiazide also had an added blood pressure effect when added to telmisartan.
The onset of antihypertensive activity occurs within 3 hours after administration of a single oral dose. At doses of 20, 40, and 80 mg, the antihypertensive effect of once daily administration of telmisartan is maintained for the full 24-hour dose interval. With automated ambulatory blood pressure monitoring and conventional blood pressure measurements, the 24-hour trough-to-peak ratio for 40 to 80 mg doses of telmisartan was 70% to 100% for both systolic and diastolic blood pressure. The incidence of symptomatic orthostasis after the first dose in all controlled trials was low (0.04%).
There were no changes in the heart rate of patients treated with telmisartan in controlled trials.
There are no trials of telmisartan tablets demonstrating reductions in cardiovascular risk in patients with hypertension, but at least one pharmacologically similar drug has demonstrated such benefits.
14.2 Cardiovascular Risk Reduction
Support for use to reduce the risk of cardiovascular events was obtained in a pair of studies. Both enrolled subjects age ≥55 years, at high cardiovascular risk as evidenced by coronary artery disease (75%), diabetes mellitus (27%) accompanied with end-organ damage (e.g., retinopathy, left ventricular hypertrophy, and, in ONTARGET only, macro-or microalbuminuria), stroke (16%), peripheral vascular disease (13%), or transient ischemic attack (4%). Patients without a history of intolerance to ACE inhibitors entered ONTARGET, and those with such a history, usually cough (90%), entered TRANSCEND, but patients with >1+ proteinuria on dipstick were excluded from TRANSCEND. For both ONTARGET and TRANSCEND trials, the primary 4-component composite endpoint was death from cardiovascular causes, myocardial infarction, stroke, and hospitalization for heart failure. The secondary 3-component composite endpoint was death from cardiovascular causes, myocardial infarction, and stroke.
ONTARGET was a randomized, active-controlled, multinational, double-blind study in 25,620 patients who were randomized to telmisartan 80 mg, ramipril 10 mg, or their combination. The population studied was 73% male, 74% Caucasian, 14% Asian, and 57% were 65 years of age or older. Baseline therapy included acetylsalicylic acid (76%), lipid lowering agents (64%), beta-blockers (57%), calcium channel blockers (34%), nitrates (29%), and diuretics (28%). Mean blood pressure at randomization was 134/77 mmHg. The mean duration of follow up was about 4 years and 6 months. During the study, 22% (n=1878) of telmisartan patients discontinued the active treatment, compared to 24.4% (n=2095) of ramipril patients and 25.3% (n=2152) of telmisartan/ramipril patients.
TRANSCEND randomized patients to telmisartan 80 mg (n=2954) or placebo (n=2972). The mean duration of follow up was 4 years and 8 months. The population studied was 57% male, 62% Caucasian, 21% Asian, and 60% were 65 years of age or older. Baseline therapy included acetylsalicylic acid (75%), lipid lowering agents (58%), beta-blockers (58%), calcium channel blockers (41%), nitrates (34%) and diuretics (33%). Mean blood pressure at randomization was 135/78 mmHg. During the study, 17.7% (n=523) of telmisartan patients discontinued the active treatment, compared to 19.4% (n=576) of placebo patients.
The results for the TRANSCEND trial are summarized in Table 2, and the results for ONTARGET are summarized in Table 3, below:
Table 2 Incidence of the Primary and Secondary Outcomes from TRANSCEND
Telmisartan vs. Placebo
(n=2954) (n=2972)
No. of Events Telmisartan / Placebo
Hazard Ratio
95% CI
p-value
*Composite of CV death, myocardial infarction, stroke, or hospitalization for heart failure
465 (15.7%) / 504 (17%)
0.92 (0.81 – 1.05)
0.2129
*Composite of CV death, myocardial infarction, or stroke
384 (13%) / 440 (14.8%)
0.87 (0.76 – 1)
0.0483
Individual components of the primary composite endpoint
No. of Events Telmisartan / Placebo
Hazard Ratio 95% CI
p-value
**All non-fatal MI
114 (3.9%) / 145 (4.9%)
0.79 (0.62 – 1.01)
0.0574
**All non-fatal strokes
112 (3.8%) / 136 (4.6%)
0.83 (0.64 – 1.06)
0.1365
*The primary endpoint was defined as the time to first event. In case of multiple simultaneous events, all individual events were considered; the sum of patients with individual outcomes may exceed the number of patients with composite (primary or secondary) outcomes.
**For individual components of the primary composite endpoints, all events, regardless whether or not they were the first event, were considered. Therefore, they are more than the first events considered for the primary or secondary composite endpoint.
Table 3 Incidence of the Primary and Secondary Outcomes from ONTARGET
Telmisartan vs. Ramipril
(n=8542) (n=8576)
No. of Events Telmisartan / Ramipril
Hazard Ratio
97.5% CI
Composite of CV death, myocardial infarction, stroke, or hospitalization for heart failure
1423 (16.7%) / 1412 (16.5%)
1.01 (0.93 – 1.10)
Composite of CV death, myocardial infarction, or stroke
1190 (13.9%) / 1210 (14.1%)
0.99 (0.90 – 1.08)
Although the event rates in ONTARGET were similar on telmisartan and ramipril, the results did not unequivocally rule out that telmisartan may not preserve a meaningful fraction of the effect of ramipril in reducing cardiovascular events. However, the results of both ONTARGET and TRANSCEND do adequately support telmisartan being more effective than placebo would be in this setting, particularly for the endpoint of time to cardiovascular death, myocardial infarction, or stroke.
In ONTARGET, there was no evidence that combining ramipril and telmisartan reduced the risk of death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for heart failure greater than ramipril alone; instead, patients who received the combination of ramipril and telmisartan in ONTARGET experienced an increased incidence of clinically important renal dysfunction (e.g., acute renal failure) compared to patients receiving telmisartan or ramipril alone.
Multiple sub-group analyses did not demonstrate any differences in the 4-component composite primary endpoint based on age, gender, or ethnicity for either ONTARGET or TRANSCEND trial.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Telmisartan tablets USP, 20 mg are white to off-white, circular, flat face bevel edged, uncoated tablets, engraved with ‘T’ on one face and ‘20’ on other face. They are available as follows.
Bottles of 30 NDC 42571-226-30
Bottles of 90 NDC 42571-226-90
Bottles of 500 NDC 42571-226-05
Bottles of 1000 NDC 42571-226-10
Carton of 30 (3x10) Unit-dose Tablets NDC 42571-226-41
Carton of 100(10x10) Unit-dose Tablets NDC 42571-226-11
Telmisartan tablets USP, 40 mg are white to off-white, oval shaped, biconvex, uncoated tablets, engraved with ‘T40’ on one face and plain on other face. They are available as follows.
Bottles of 30 NDC 42571-227-30
Bottles of 90 NDC 42571-227-90
Bottles of 500 NDC 42571-227-05
Bottles of 1000 NDC 42571-227-10
Carton of 30 (3x10) Unit-dose Tablets NDC 42571-227-41
Carton of 100(10x10) Unit-dose Tablets NDC 42571-227-11
Telmisartan tablets USP, 80 mg are white to off-white, capsule shaped, biconvex, uncoated tablets, engraved with ‘T80’ on one face and plain on other face. They are available as follows.
Bottles of 30 NDC 42571-228-30
Bottles of 90 NDC 42571-228-90
Bottles of 500 NDC 42571-228-05
Bottles of 1000 NDC 42571-228-10
Carton of 30 (3x10) Unit-dose Tablets NDC 42571-228-41
Carton of 100(10x10) Unit-dose Tablets NDC 42571-228-11
Storage
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Tablets should not be removed from blisters or bottles until immediately before administration.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information)
Pregnancy
Advise female patients of childbearing age about the consequences of exposure to telmisartan during pregnancy. Discuss treatment options with women planning to become pregnant. Tell patients to report pregnancies to their physicians as soon as possible [see Warnings and Precautions ( 5.1) and Use in Specific Populations ( 8.1)].
Lactation
Advise nursing women not to breastfeed during treatment with telmisartan [see Use in Specific Populations ( 8.2)].
Symptomatic Hypotension and Syncope
Advise patients that lightheadedness can occur, especially during the first days of therapy, and to report it to their healthcare provider. Inform patients that inadequate fluid intake, excessive perspiration, diarrhea, or vomiting can lead to an excessive fall in blood pressure, with the same consequences of lightheadedness and possible syncope. Advise patients to contact their healthcare provider if syncope occurs [see Warnings and Precautions ( 5.2)].
Potassium Supplements
Advise patients not to use potassium supplements or salt substitutes that contain potassium without consulting the prescribing healthcare provider [see Warnings and Precautions ( 5.3)].
Manufactured by:
Micro Labs Limited
Goa-403 722, INDIA.
Manufactured for:
Micro Labs USA, Inc.
Somerset, NJ 08873
Rev.01/2023
-
Patient Information
Telmisartan (tel" mi sar' tan) Tablets, USP
Read this Patient Information before you start taking telmisartan tablets and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or your treatment.
What is the most important information I should know about telmisartan tablets?
Telmisartan tablets can cause harm or death to an unborn baby. Talk to your doctor about other ways to lower your blood pressure if you plan to become pregnant. If you get pregnant while taking telmisartan tablets, tell your doctor right away.
What are telmisartan tablets?
Telmisartan tablets are a prescription medicine used:
- to treat high blood pressure (hypertension)
- in certain high risk people aged 55 years and older to help lower their risk of having certain cardiovascular problems such as stroke, heart attack, or death.
It is not known if telmisartan tablets are safe and effective in children.
Who should not take telmisartan tablets?
You should not take telmisartan tablets if you are allergic (hypersensitive) to the active ingredient (telmisartan) or any of the other ingredients listed at the end of this leaflet.
For patients with diabetes, if you are taking telmisartan tablets you should not take aliskiren.
What should I tell my doctor before taking telmisartan tablets?
Before you take telmisartan tablets, tell your doctor if you:
- are pregnant or are planning to become pregnant. See “What is the most important information I should know about telmisartan tablets?”
- are breast-feeding or plan to breast-feed. It is not known if telmisartan passes into your breast milk. You and your doctor should decide if you will take telmisartan tablets or breast-feed. You should not do both. Talk with your doctor about the best way to feed your baby if you take telmisartan tablets.
- have liver problems
- have kidney problems
- have heart problems
- have any other medical conditions
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
For patients with diabetes, if you are taking telmisartan tablets you should not take aliskiren.
Telmisartan tablets may affect the way other medicines work, and other medicines may affect how telmisartan tablets works. Especially tell your doctor if you take:
- aliskiren
- digoxin (Lanoxin ®)
- lithium (Lithobid ®, lithium carbonate, lithium citrate)
- aspirin or other non-steroidal anti-inflammatory drugs (NSAIDs)
- other medicines used to treat your high blood pressure or heart problem
- water pills (diuretic)
Know the medicines you take. Keep a list of them and show it to your doctor or pharmacist when you get a new medicine.
How should I take telmisartan tablets?
- Take telmisartan tablets exactly as your doctor tells you to take it.
- Your doctor will tell you how many telmisartan tablets to take and when to take it.
- Do not change your dose unless your doctor tells you to.
- Take telmisartan tablets one time each day at the same time.
- Take telmisartan tablets with or without food.
- If you miss a dose, take it as soon as you remember. If it is close to your next dose, do not take the missed dose. Take the next dose at your regular time.
- If you take too many telmisartan tablets, call your doctor, or go to the nearest hospital emergency room right away.
- Read the “How to Open the Blister” at the end of this leaflet before you use telmisartan tablets. Talk with your doctor if you do not understand the instructions.
What are the possible side effects of telmisartan tablets?
Telmisartan tablets may cause serious side effects, including:
- Injury or death to your unborn baby. See “What is the most important information I should know about telmisartan tablets?”
- Low blood pressure (hypotension) is most likely to happen if you also:
- take water pills (diuretics)
- are on a low-salt diet
- get dialysis treatments
- have heart problems
- get sick with vomiting or diarrhea
If you feel faint or dizzy, lie down and call your doctor right away.
- Kidney problems, which may get worse if you already have kidney disease. You may have changes in your kidney test results, and you may need a lower dose of telmisartan tablets. Call your doctor if you get:
• swelling in your feet, ankles, or hands
• unexplained weight gainCall your doctor right away if you get any of the symptoms listed above.
- High potassium in the blood (hyperkalemia). Your doctor may check your potassium levels as needed.
Rare, serious allergic reactions may happen. Tell your doctor right away if you get any of these symptoms:
- swelling of the face, tongue, throat
- difficulty breathing
- skin rash
The most common side effects of telmisartan tablets include:
- sinus pain and congestion (sinusitis)
- back pain
- diarrhea
These are not all the possible side effects with telmisartan tablets. Tell your doctor if you have any side effect that bothers you or that does not go away. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store telmisartan tablets?
- Store telmisartan tablets at room temperature between 68°F to 77°F (20°C to 25°C).
- Do not remove telmisartan tablets from blisters or bottles until right before you take them.
Keep telmisartan tablets and all medicines out of the reach of children.
General information about telmisartan tablets
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use telmisartan tablets for a condition for which it was not prescribed. Do not give telmisartan tablets to other people, even if they have the same condition you have. It may harm them.
This Patient Information leaflet summarizes the most important information about telmisartan tablets. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about telmisartan tablets that is written for health professionals.
For more information, call Micro Labs USA Inc. at 1-855-839-8195.
What are the ingredients in telmisartan tablets?
Active Ingredient: telmisartan
Inactive Ingredients: crospovidone Type A/kollidon CL, mannitol, magnesium stearate, meglumine, povidone K-30, and sodium hydroxide.
What is High Blood Pressure (Hypertension)?
Blood pressure is the force in your blood vessels when your heart beats and when your heart rests. You have high blood pressure when the force is too much. Telmisartan tablets can help your blood vessels relax so your blood pressure is lower. Medicines that lower your blood pressure lower your chance of having a stroke or heart attack.
High blood pressure makes the heart work harder to pump blood throughout the body and causes damage to the blood vessels. If high blood pressure is not treated, it can lead to stroke, heart attack, heart failure, kidney failure, and vision problems.
What is Cardiovascular Risk?
Patients older than 55 years of age who have been diagnosed with blood vessel disease in the heart, legs, or brain (coronary, peripheral, or cerebral vascular disease) or diabetes with end organ damage (for example: kidney, heart, and brain) are at higher risk of cardiovascular events (for example: death from cardiovascular causes, stroke, and/or heart attack).
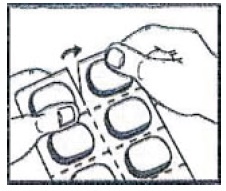
How to open the blister:
1. Tear (You may also use scissors to tear the blister apart)
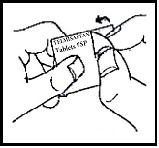
2. Peel (Peel off the paper layer from the aluminum foil)
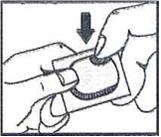
3. Push (Push the tablet through the aluminum foil)
This Patient Information has been approved by the U.S. Food and Drug Administration
The brands listed are trademarks of their respective owners and are not trademarks of Micro Labs. The makers of these brands are not affiliated with and do not endorse Micro Labs or its products.
Manufactured by:
Micro Labs Limited
Goa-403 722, INDIA.
Manufactured for:
Micro Labs USA, Inc.
Somerset, NJ 08873
Rev.01/2023
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TELMISARTAN
telmisartan tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:42571-226 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TELMISARTAN (UNII: U5SYW473RQ) (TELMISARTAN - UNII:U5SYW473RQ) TELMISARTAN 20 mg Inactive Ingredients Ingredient Name Strength CROSPOVIDONE (UNII: 68401960MK) MANNITOL (UNII: 3OWL53L36A) MAGNESIUM STEARATE (UNII: 70097M6I30) MEGLUMINE (UNII: 6HG8UB2MUY) POVIDONE K30 (UNII: U725QWY32X) SODIUM HYDROXIDE (UNII: 55X04QC32I) Product Characteristics Color white (white to off white) Score no score Shape ROUND (flat face bevel edged) Size 7mm Flavor Imprint Code T;20 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:42571-226-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 2 NDC:42571-226-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 3 NDC:42571-226-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 4 NDC:42571-226-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 5 NDC:42571-226-11 10 in 1 CARTON 01/01/2018 5 NDC:42571-226-32 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 6 NDC:42571-226-41 3 in 1 CARTON; Type 0: Not a Combination Product 01/01/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA207016 01/01/2018 TELMISARTAN
telmisartan tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:42571-227 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TELMISARTAN (UNII: U5SYW473RQ) (TELMISARTAN - UNII:U5SYW473RQ) TELMISARTAN 40 mg Inactive Ingredients Ingredient Name Strength CROSPOVIDONE (UNII: 68401960MK) MANNITOL (UNII: 3OWL53L36A) MAGNESIUM STEARATE (UNII: 70097M6I30) MEGLUMINE (UNII: 6HG8UB2MUY) POVIDONE K30 (UNII: U725QWY32X) SODIUM HYDROXIDE (UNII: 55X04QC32I) Product Characteristics Color white (white to off white) Score no score Shape OVAL (biconvex) Size 12mm Flavor Imprint Code T40 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:42571-227-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 2 NDC:42571-227-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 3 NDC:42571-227-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 4 NDC:42571-227-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 5 NDC:42571-227-11 10 in 1 CARTON 01/01/2018 5 NDC:42571-227-32 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 6 NDC:42571-227-41 3 in 1 CARTON; Type 0: Not a Combination Product 01/01/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA207016 01/01/2018 TELMISARTAN
telmisartan tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:42571-228 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TELMISARTAN (UNII: U5SYW473RQ) (TELMISARTAN - UNII:U5SYW473RQ) TELMISARTAN 80 mg Inactive Ingredients Ingredient Name Strength POVIDONE K30 (UNII: U725QWY32X) SODIUM HYDROXIDE (UNII: 55X04QC32I) CROSPOVIDONE (UNII: 68401960MK) MANNITOL (UNII: 3OWL53L36A) MAGNESIUM STEARATE (UNII: 70097M6I30) MEGLUMINE (UNII: 6HG8UB2MUY) Product Characteristics Color white (white to off white) Score no score Shape CAPSULE (biconvex) Size 16mm Flavor Imprint Code T80 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:42571-228-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 2 NDC:42571-228-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 3 NDC:42571-228-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 4 NDC:42571-228-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 01/01/2018 5 NDC:42571-228-11 10 in 1 CARTON 01/01/2018 5 NDC:42571-228-32 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 6 NDC:42571-228-41 3 in 1 CARTON; Type 0: Not a Combination Product 01/01/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA207016 01/01/2018 Labeler - Micro Labs Limited (862174955) Establishment Name Address ID/FEI Business Operations Micro Labs Limited 915793658 analysis(42571-226, 42571-227, 42571-228) , label(42571-226, 42571-227, 42571-228) , manufacture(42571-226, 42571-227, 42571-228) , pack(42571-226, 42571-227, 42571-228)


