Label: BROMOCRIPTINE MESYLATE capsule

- NDC Code(s): 68382-110-01, 68382-110-06
- Packager: Zydus Pharmaceuticals USA Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated September 11, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
Bromocriptine mesylate is an ergot derivative with potent dopamine receptor agonist activity. Bromocriptine mesylate is chemically designated as Ergotaman-3΄, 6΄, 18-trione, 2-bromo-12΄-hydroxy-2΄-(1-methylethyl)-5΄-(2-methylpropyl)-, (5΄α)-mono-methanesulfonate (salt).
The structural formula is:
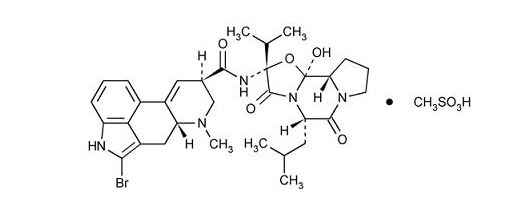
C32H40BrN5O5.CH4SO3
Mol. wt. 750.70
Bromocriptine mesylate, USP is white or slightly colored, fine crystalline powder and odorless or having a weak, characteristic odor.
Each bromocriptine mesylate capsule USP, 5 mg intended for oral administration contains bromocriptine mesylate equivalent to 5 mg of bromocriptine. In addition, each capsule contains the following inactive ingredients: carrageenan, colloidal silicon dioxide, hypromellose, iron oxide red, lactose monohydrate, magnesium stearate, maleic acid, potassium hydroxide and titanium dioxide. Each capsule is printed with black pharmaceutical ink and has following inactive ingredients: black iron oxide, potassium hydroxide, propylene glycol, purified water, shellac and strong ammonia solution.
-
CLINICAL PHARMACOLOGY
Bromocriptine mesylate is a dopamine receptor agonist, which activates post-synaptic dopamine receptors. The dopaminergic neurons in the tuberoinfundibular process modulate the secretion of prolactin from the anterior pituitary by secreting a prolactin inhibitory factor (thought to be dopamine); in the corpus striatum the dopaminergic neurons are involved in the control of motor function. Clinically, bromocriptine significantly reduces plasma levels of prolactin in patients with physiologically elevated prolactin as well as in patients with hyperprolactinemia. The inhibition of physiological lactation as well as galactorrhea in pathological hyperprolactinemic states is obtained at dose levels that do not affect secretion of other tropic hormones from the anterior pituitary. Experiments have demonstrated that bromocriptine induces long-lasting stereotyped behavior in rodents and turning behavior in rats having unilateral lesions in the substantia nigra. These actions, characteristic of those produced by dopamine, are inhibited by dopamine antagonists and suggest a direct action of bromocriptine on striatal dopamine receptors.
Bromocriptine mesylate is a nonhormonal, nonestrogenic agent that inhibits the secretion of prolactin in humans, with little or no effect on other pituitary hormones, except in patients with acromegaly, where it lowers elevated blood levels of growth hormone in the majority of patients.
Bromocriptine mesylate produces its therapeutic effect in the treatment of Parkinson's disease, a clinical condition characterized by a progressive deficiency in dopamine synthesis in the substantia nigra, by directly stimulating the dopamine receptors in the corpus striatum. In contrast, levodopa exerts its therapeutic effect only after conversion to dopamine by the neurons of the substantia nigra, which are known to be numerically diminished in this patient population.
-
PHARMACOKINETICS
Following single dose administration of bromocriptine mesylate tablets, 2 x 2.5 mg to 5 healthy volunteers under fasted conditions, the mean peak plasma levels of bromocriptine, time to reach peak plasma concentrations and elimination half-life were 465 pg/mL ± 226, 2.5 hrs ± 2 and 4.85 hr, respectively.1 Linear relationship was found between single doses of bromocriptine and Cmax and AUC in the dose range of 1 to 7.5 mg.2 The pharmacokinetics of bromocriptine metabolites have not been reported.
Food did not significantly affect the systemic exposure of bromocriptine following administration of bromocriptine mesylate tablets, 2.5 mg.3 It is recommended that bromocriptine be taken with food because of the high percentage of subjects who vomit upon receiving bromocriptine under fasting conditions.
Following bromocriptine mesylate 5 mg administered twice daily for 14 days, the bromocriptine Cmax and AUC at steady-state were 628 ± 375 pg/mL and 2377 ± 1186 pg*hr/mL, respectively.4
Distribution
In vitro experiments showed that bromocriptine was 90% to 96% bound to serum albumin.
Metabolism
Bromocriptine undergoes extensive first-pass biotransformation, reflected by complex metabolite profiles and by almost complete absence of parent drug in urine and feces.
In vitro studies using human liver microsomes showed that bromocriptine has a high affinity for CYP3A and hydroxylations at the proline ring of the cyclopeptide moiety constituted a main metabolic pathway.5 Inhibitors and/or potent substrates for CYP3A4 might therefore inhibit the clearance of bromocriptine and lead to increased levels. (see PRECAUTIONS, drug interactions section). The participation of other major CYP enzymes such as 2D6, 2C8, and 2C19 on the metabolism of bromocriptine has not been evaluated. Bromocriptine is also an inhibitor of CYP3A4 with a calculated IC50 value of 1.69 μM.6 Given the low therapeutic concentrations of bromocriptine in patients (Cmax=0.82 nM), a significant alteration of the metabolism of a second drug whose clearance is mediated by CYP3A4 should not be expected. The potential effect of bromocriptine and its metabolites to act as inducers of CYP enzymes has not been reported.
Excretion
About 82% and 5.6% of the radioactive dose orally administered was recovered in feces and urine, respectively. Bromolysergic acid and bromoisolysergic acid accounted for half of the radioactivity in urine.5
1 Nelson, M. et. al. (1990). Pharmacokinetic evaluation of erythromycin and caffeine administered with bromocriptine. Clin Pharmacol Ther; 47(6):694-7.
2 Schran, H.F., Bhuta, S.I., Schwartz, et al. (1980). The pharmacokinetics of bromocriptine in man. In: Golstein, M. Calne, D.B., et. Al (eds). Ergot compound and brain function: Neuroendocrine and neuropsychiatric aspects, pp. 125-139, New York, Rave Press.
3 Kopitar, Z., Vrhovac, B., Povsic, L., Plavsic, F., Francetic, I., Urbancic, J. (1991). The effect of food and metoclopramide on the pharmacokinetics and side effects of bromocriptine. Eur J Drug Metab Pharmacokinet; 16(3):177-81
4 Flogstad, A.K., Halse, J., Grass, P., Abisch, E., Djoseland, O., Kutz, K., Bodd, E., and Jervell, J., (1994). A comparison of octreotide, bromocriptine, or a combination of both drugs in acromegaly. Journal of Clinical Endocrinology & Metabolism; Vol 79, 461-465
5 Peyronneau MA, Delaforge M, Riviere R et al. 1994. High affinity of ergopeptides for CYP P450 3A. Importance of their peptide moiety for P450 recognition and hydroxylation of bromocriptine. Eur J Biochem 223:947-56.
6 Wynalda, M.A., Wienkers, L.C. (1997). Assessment of potential interactions between dopamine receptor agonists and various human cytochrome P450 enzymes using a simple in vitro inhibition screen. Drug Metab Dispos; 25:1211-14.
Specific Populations
Effect of Renal Impairment
The effect of renal function on the pharmacokinetics of bromocriptine has not been evaluated.
Since parent drug and metabolites are almost completely excreted via metabolism, and only 6% eliminated via the kidney, renal impairment may not have a significant impact on the PK of bromocriptine and its metabolites (see PRECAUTIONS, general).
Effect of Hepatic Impairment
The effect of liver impairment on the PK of bromocriptine mesylate and its metabolites has not been evaluated. Since bromocriptine mesylate is mainly eliminated by metabolism, liver impairment may increase the plasma levels of bromocriptine, therefore, caution may be necessary (see PRECAUTIONS, general).
The effect of age, race, and gender on the pharmacokinetics of bromocriptine and its metabolites has not been evaluated.
Clinical Studies
In about 75% of cases of amenorrhea and galactorrhea, bromocriptine mesylate capsule therapy suppresses the galactorrhea completely, or almost completely, and reinitiates normal ovulatory menstrual cycles.
Menses are usually reinitiated prior to complete suppression of galactorrhea; the time for this on average is 6 weeks to 8 weeks. However, some patients respond within a few days, and others may take up to 8 months.
Galactorrhea may take longer to control depending on the degree of stimulation of the mammary tissue prior to therapy. At least a 75% reduction in secretion is usually observed after 8 weeks to 12 weeks. Some patients may fail to respond even after 12 months of therapy.
In many acromegalic patients, bromocriptine produces a prompt and sustained reduction in circulating levels of serum growth hormone.
-
INDICATIONS AND USAGE
Hyperprolactinemia-Associated Dysfunctions
Bromocriptine mesylate capsules are indicated for the treatment of dysfunctions associated with hyperprolactinemia including amenorrhea with or without galactorrhea, infertility or hypogonadism. Bromocriptine treatment is indicated in patients with prolactin-secreting adenomas, which may be the basic underlying endocrinopathy contributing to the above clinical presentations. Reduction in tumor size has been demonstrated in both male and female patients with macroadenomas. In cases where adenectomy is elected, a course of bromocriptine mesylate capsule therapy may be used to reduce the tumor mass prior to surgery.
Acromegaly
Bromocriptine mesylate capsule therapy is indicated in the treatment of acromegaly. Bromocriptine mesylate capsule therapy, alone or as adjunctive therapy with pituitary irradiation or surgery, reduces serum growth hormone by 50% or more in approximately ½ of patients treated, although not usually to normal levels.
Since the effects of external pituitary radiation may not become maximal for several years, adjunctive therapy with bromocriptine mesylate capsule offers potential benefit before the effects of irradiation are manifested.
Parkinson's Disease
Bromocriptine mesylate capsules are indicated in the treatment of the signs and symptoms of idiopathic or postencephalitic Parkinson's disease. As adjunctive treatment to levodopa (alone or with a peripheral decarboxylase inhibitor), bromocriptine mesylate capsule therapy may provide additional therapeutic benefits in those patients who are currently maintained on optimal dosages of levodopa, those who are beginning to deteriorate (develop tolerance) to levodopa therapy, and those who are experiencing "end of dose failure" on levodopa therapy. Bromocriptine mesylate capsule therapy may permit a reduction of the maintenance dose of levodopa and, thus may ameliorate the occurrence and/or severity of adverse reactions associated with long-term levodopa therapy such as abnormal involuntary movements (e.g., dyskinesias) and the marked swings in motor function ("on-off" phenomenon). Continued efficacy of bromocriptine mesylate capsule therapy during treatment of more than 2 years has not been established.
Data are insufficient to evaluate potential benefit from treating newly diagnosed Parkinson's disease with bromocriptine mesylate capsules. Studies have shown, however, significantly more adverse reactions (notably nausea, hallucinations, confusion and hypotension) in bromocriptine-treated patients than in levodopa/carbidopa-treated patients. Patients unresponsive to levodopa are poor candidates for bromocriptine mesylate capsule therapy.
-
CONTRAINDICATIONS
Hypersensitivity to bromocriptine or to any of the excipients of bromocriptine mesylate capsules, uncontrolled hypertension and sensitivity to any ergot alkaloids. In patients being treated for hyperprolactinemia, bromocriptine mesylate capsules should be withdrawn when pregnancy is diagnosed (see PRECAUTIONS, Hyperprolactinemic States). In the event that bromocriptine is reinstituted to control a rapidly expanding macroadenoma (see PRECAUTIONS, Hyperprolactinemic States) and a patient experiences a hypertensive disorder of pregnancy, the benefit of continuing bromocriptine mesylate capsules must be weighed against the possible risk of its use during a hypertensive disorder of pregnancy. When bromocriptine mesylate capsules are being used to treat acromegaly, prolactinoma, or Parkinson's disease in patients who subsequently become pregnant, a decision should be made as to whether the therapy continues to be medically necessary or can be withdrawn. If it is continued, the drug should be withdrawn in those who may experience hypertensive disorders of pregnancy (including eclampsia, preeclampsia, or pregnancy-induced hypertension) unless withdrawal of bromocriptine mesylate capsules are considered to be medically contraindicated.
The drug should not be used during the postpartum period in women with a history of coronary artery disease and other severe cardiovascular conditions unless withdrawal is considered medically contraindicated. If the drug is used in the postpartum period, the patient should be observed with caution.
-
WARNINGS
Since hyperprolactinemia with amenorrhea/galactorrhea and infertility has been found in patients with pituitary tumors, a complete evaluation of the pituitary is indicated before treatment with bromocriptine mesylate capsules.
If pregnancy occurs during bromocriptine administration, careful observation of these patients is mandatory. Prolactin-secreting adenomas may expand and compression of the optic or other cranial nerves may occur, emergency pituitary surgery becoming necessary. In most cases, the compression resolves following delivery. Reinitiation of bromocriptine treatment has been reported to produce improvement in the visual fields of patients in whom nerve compression has occurred during pregnancy. The safety of bromocriptine treatment during pregnancy to the mother and fetus has not been established.
Bromocriptine mesylate has been associated with somnolence, and episodes of sudden sleep onset, particularly in patients with Parkinson's disease. Sudden onset of sleep during daily activities, in some cases without awareness or warning signs, has been reported. Patients must be informed of this and advised not to drive or operate machines during treatment with bromocriptine. Patients who have experienced somnolence and/or an episode of sudden sleep onset must not drive or operate machines. Furthermore, a reduction of dosage or termination of therapy may be considered.
Symptomatic hypotension can occur in patients treated with bromocriptine for any indication. In postpartum studies with bromocriptine, decreases in supine systolic and diastolic pressures of greater than 20 mm and 10 mm Hg, respectively, have been observed in almost 30% of patients receiving bromocriptine. On occasion, the drop in supine systolic pressure was as much as 50-59 mm of Hg.
Since, especially during the first days of treatment, hypotensive reactions may occasionally occur and result in reduced alertness, particular care should be exercised when driving a vehicle or operating machinery.
While hypotension during the start of therapy with bromocriptine occurs in some patients, in rare cases serious adverse events, including hypertension, myocardial infarction, seizures, stroke, have been reported in postpartum women treated with bromocriptine for the inhibition of lactation. Hypertension have been reported, sometimes at the initiation of therapy, but often developing in the second week of therapy; seizures have also been reported both with and without the prior development of hypertension; stroke have been reported mostly in postpartum patients whose prenatal and obstetric courses had been uncomplicated. Many of these patients experiencing seizures (including cases of status epilepticus) and/or strokes reported developing a constant and often progressively severe headache hours to days prior to the acute event. Some cases of strokes and seizures were also preceded by visual disturbances (blurred vision, and transient cortical blindness). Cases of acute myocardial infarction have also been reported.
Although a causal relationship between bromocriptine administration and hypertension, seizures, strokes, and myocardial infarction in postpartum women has not been established, use of the drug for prevention of physiological lactation, or in patients with uncontrolled hypertension is not recommended. In patients being treated for hyperprolactinemia bromocriptine should be withdrawn when pregnancy is diagnosed (see PRECAUTIONS, Hyperprolactinemic States). In the event that bromocriptine is reinstituted to control a rapidly expanding macroadenoma (see PRECAUTIONS, Hyperprolactinemic States) and a patient experiences a hypertensive disorder of pregnancy, the benefit of continuing bromocriptine mesylate capsules must be weighed against the possible risk of its use during a hypertensive disorder of pregnancy. When bromocriptine is being used to treat acromegaly or Parkinson's disease in patients who subsequently become pregnant, a decision should be made as to whether the therapy continues to be medically necessary or can be withdrawn. If it is continued, the drug should be withdrawn in those who may experience hypertensive disorders of pregnancy (including eclampsia, preeclampsia, or pregnancy-induced hypertension) unless withdrawal of bromocriptine is considered to be medically contraindicated. Because of the possibility of an interaction between bromocriptine and other ergot alkaloids, the concomitant use of these medications is not recommended. Periodic monitoring of the blood pressure, particularly during the first weeks of therapy is prudent. If hypertension, severe, progressive, or unremitting headache (with or without visual disturbance), or evidence of CNS toxicity develops, drug therapy should be discontinued and the patient should be evaluated promptly. Particular attention should be paid to patients who have recently been treated or are on concomitant therapy with drugs that can alter blood pressure. Their concomitant use in the puerperium is not recommended.
Among patients on bromocriptine, particularly on long-term and high-dose treatment, pleural and pericardial effusions, as well as pleural and pulmonary fibrosis and constrictive pericarditis, have been reported. Patients with unexplained pleuropulmonary disorders should be examined thoroughly and discontinuation of bromocriptine mesylate capsules therapy should be considered. In those instances in which bromocriptine treatment was terminated, the changes slowly reverted towards normal.
In a few patients on bromocriptine, particularly on long-term and high-dose treatment, retroperitoneal fibrosis has been reported. To ensure recognition of retroperitoneal fibrosis at an early reversible stage it is recommended that its manifestations (e.g., back pain, edema of the lower limbs, impaired kidney function) should be watched in this category of patients. Bromocriptine medication should be withdrawn if fibrotic changes in the retroperitoneum are diagnosed or suspected.
-
PRECAUTIONS
There have been reports of patients experiencing intense urges to gamble, increased sexual urges, intense urges to spend money uncontrollably, and/or other intense urges, and the inability to control these urges while taking one or more of the medications, including bromocriptine, that increase central dopaminergic tone. In some cases, although not all, these urges were reported to have stopped when the dose was reduced, or the medication was discontinued. Because patients may not recognize these behaviors as abnormal, it is important for prescribers to specifically ask patients or their caregivers about the development of new or increased gambling urges, sexual urges, uncontrolled spending, or other urges while being treated with bromocriptine for Parkinson's disease or hyperprolactinemia-associated dysfunctions. Physicians should consider dose reduction or stopping the medication if a patient develops such urges while taking bromocriptine.
Safety and efficacy of bromocriptine have not been established in patients with renal or hepatic disease. Care should be exercised when administering bromocriptine mesylate capsule therapy concomitantly with other medications known to lower blood pressure.
The drug should be used with caution in patients with a history of psychosis or cardiovascular disease. If acromegalic patients or patients with prolactinoma or Parkinson's disease are being treated with bromocriptine during pregnancy, they should be cautiously observed, particularly during the postpartum period if they have a history of cardiovascular disease.
Patients with rare hereditary problems of galactose intolerance, severe lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Hyperprolactinemic States
Visual field impairment is a known complication of macroprolactinoma. Effective treatment with bromocriptine leads to a reduction in hyperprolactinemia and often to a resolution of the visual impairment. In some patients, however, a secondary deterioration of visual fields may subsequently develop despite normalized prolactin levels and tumor shrinkage, which may result from traction on the optic chiasm which is pulled down into the now partially empty sella. In these cases, the visual field defect may improve on reduction of bromocriptine dosage while there is some elevation of prolactin and some tumor re-expansion. Monitoring of visual fields in patients with macroprolactinoma is therefore recommended for an early recognition of secondary field loss due to chiasmal herniation and adaptation of drug dosage.
The relative efficacy of bromocriptine versus surgery in preserving visual fields is not known. Patients with rapidly progressive visual field loss should be evaluated by a neurosurgeon to help decide on the most appropriate therapy.
Since pregnancy is often the therapeutic objective in many hyperprolactinemic patients presenting with amenorrhea/galactorrhea and hypogonadism (infertility), a careful assessment of the pituitary is essential to detect the presence of a prolactin-secreting adenoma. Patients not seeking pregnancy, or those harboring large adenomas, should be advised to use contraceptive measures, other than oral contraceptives, during treatment with bromocriptine. Since pregnancy may occur prior to reinitiation of menses, a pregnancy test is recommended at least every 4 weeks during the amenorrheic period, and, once menses are reinitiated, every time a patient misses a menstrual period. Treatment with bromocriptine mesylate capsules should be discontinued as soon as pregnancy has been established. Patients must be monitored closely throughout pregnancy for signs and symptoms that may signal the enlargement of a previously undetected or existing prolactin-secreting tumor. Discontinuation of bromocriptine treatment in patients with known macroadenomas has been associated with rapid regrowth of tumor and increase in serum prolactin in most cases.
Cerebrospinal fluid rhinorrhea has been observed in some patients with prolactin-secreting adenomas treated with bromocriptine.
Acromegaly
Cold-sensitive digital vasospasm has been observed in some acromegalic patients treated with bromocriptine. The response, should it occur, can be reversed by reducing the dose of bromocriptine and may be prevented by keeping the fingers warm. Cases of severe gastrointestinal bleeding from peptic ulcers have been reported, some fatal. Although there is no evidence that bromocriptine increases the incidence of peptic ulcers in acromegalic patients, symptoms suggestive of peptic ulcer should be investigated thoroughly and treated appropriately. Patients with a history of peptic ulcer or gastrointestinal bleeding should be observed carefully during treatment with bromocriptine.
Possible tumor expansion while receiving bromocriptine mesylate capsule therapy has been reported in a few patients. Since the natural history of growth hormone-secreting tumors is unknown, all patients should be carefully monitored and, if evidence of tumor expansion develops, discontinuation of treatment and alternative procedures considered.
Parkinson's Disease
Safety during long-term use for more than 2 years at the doses required for parkinsonism has not been established.
As with any chronic therapy, periodic evaluation of hepatic, hematopoietic, cardiovascular, and renal function is recommended. Symptomatic hypotension can occur and, therefore, caution should be exercised when treating patients receiving antihypertensive drugs.
High doses of bromocriptine may be associated with confusion and mental disturbances. Since parkinsonian patients may manifest mild degrees of dementia, caution should be used when treating such patients.
Bromocriptine administered alone or concomitantly with levodopa may cause hallucinations (visual or auditory). Hallucinations usually resolve with dosage reduction; occasionally, discontinuation of bromocriptine is required. Rarely, after high doses, hallucinations have persisted for several weeks following discontinuation of bromocriptine.
As with levodopa, caution should be exercised when administering bromocriptine to patients with a history of myocardial infarction who have a residual atrial, nodal, or ventricular arrhythmia.
Retroperitoneal fibrosis has been reported in a few patients receiving long-term therapy (2 years to 10 years) with bromocriptine mesylate capsules in doses ranging from 30 mg to 140 mg daily.
Epidemiological studies have shown that patients with Parkinson's disease have a higher risk (2-approximately 6-fold higher) of developing melanoma than the general population. Whether the increased risk observed was due to Parkinson's disease or other factors, such as drugs used to treat Parkinson's disease, is unclear. For the reasons stated above, patients and providers are advised to monitor for melanomas frequently and on a regular basis when using bromocriptine for any indication. Ideally, periodic skin examinations should be performed by appropriately qualified individuals (e.g., dermatologists).
Discontinuation of bromocriptine should be undertaken gradually whenever possible, even if the patient is to remain on levodopa. A symptom complex resembling the neuroleptic malignant syndrome (characterized by elevated temperature, muscular rigidity, altered consciousness, and autonomic instability), with no other obvious etiology, has been reported in association with rapid dose reduction, withdrawal of, or changes in antiparkinsonian therapy.
Symptoms including apathy, anxiety, depression, fatigue, insomnia, sweating, and pain have been reported during taper or after discontinuation of dopamine agonists, including bromocriptine. These symptoms generally do not respond to levodopa. Prior to discontinuation of bromocriptine, patients should be informed about potential withdrawal symptoms, and closely monitored during and after discontinuation of bromocriptine. In case of severe withdrawal symptoms, re-administration of a dopamine agonist at the lowest effective dose may be considered.
Information for Patients
During clinical trials, dizziness, drowsiness, faintness, fainting, and syncope have been reported early in the course of bromocriptine mesylate capsule therapy. In postmarketing reports, bromocriptine has been associated with somnolence, and episodes of sudden sleep onset, particularly in patients with Parkinson's disease. Sudden onset of sleep during daily activities, in some cases without awareness or warning signs, has been reported very rarely. All patients receiving bromocriptine should be cautioned with regard to engaging in activities requiring rapid and precise responses, such as driving an automobile or operating machinery. Patients being treated with bromocriptine and presenting with somnolence and/or sudden sleep episodes must be advised not to drive or engage in activities where impaired alertness may put themselves or others at risk of serious injury or death (e.g., operating machines).
Patients receiving bromocriptine for hyperprolactinemic states associated with macroadenoma or those who have had previous transsphenoidal surgery should be told to report any persistent watery nasal discharge to their physician. Patients receiving bromocriptine for treatment of a macroadenoma should be told that discontinuation of drug may be associated with rapid regrowth of the tumor and recurrence of their original symptoms.
Patients and their caregivers should be alerted to the possibility that patients may experience intense urges to spend money uncontrollably, intense urges to gamble, increased sexual urges and other intense urges and the inability to control these urges while taking bromocriptine. Advise patients and their caregivers to inform their healthcare provider if they develop new or increased uncontrolled spending, gambling urges, sexual urges, or other urges while being treated with bromocriptine [see PRECAUTIONS].
Especially during the first days of treatment, hypotensive reactions may occasionally occur and result in reduced alertness, particular care should be exercised when driving a vehicle or operating machinery.
Advise patients to contact their healthcare provider if they wish to discontinue bromocriptine or decrease the dose of bromocriptine. Advise patients who have been prescribed a lower dose or who have been withdrawn from the drug to notify their healthcare provider if they have withdrawal symptoms such as fever, muscular rigidity, altered consciousness, apathy, anxiety, depression, fatigue, insomnia, sweating, or pain (see Precautions).
Drug Interactions
The risk of using bromocriptine in combination with other drugs has not been systematically evaluated, but alcohol may potentiate the side effects of bromocriptine. Bromocriptine may interact with dopamine antagonists, butyrophenones, and certain other agents. Compounds in these categories result in a decreased efficacy of bromocriptine: phenothiazines, haloperidol, metoclopramide and pimozide. Bromocriptine is a substrate of CYP3A4. Caution should therefore be used when coadministering drugs which are strong inhibitors of this enzyme (such as azole antimycotics, HIV protease inhibitors). The concomitant use of macrolide antibiotics such as erythromycin was shown to increase the plasma levels of bromocriptine (mean AUC and Cmax values increased 3.7-fold and 4.6-fold, respectively).1 The concomitant treatment of acromegalic patients with bromocriptine and octreotide led to increased plasma levels of bromocriptine (bromocriptine AUC increased about 38%).4Concomitant use of bromocriptine with other ergot alkaloids is not recommended. Dose adjustment may be necessary in those cases where high doses of bromocriptine are being used (such as Parkinson's disease indication).
Carcinogenesis, Mutagenesis, Impairment of Fertility
A 74-week study was conducted in mice using dietary levels of bromocriptine mesylate equivalent to oral doses of 10 and 50 mg/kg/day. A 100-week study in rats was conducted using dietary levels equivalent to oral doses of 1.7, 9.8, and 44 mg/kg/day. The highest doses tested in mice and rats were approximately 2.5 and 4.4 times, respectively, the maximum human dose administered in controlled clinical trials (100 mg/day) based on body surface area. Malignant uterine tumors, endometrial and myometrial, were found in rats as follows: 0/50 control females, 2/50 females given 1.7 mg/kg daily, 7/49 females given 9.8 mg/kg daily, and 9/50 females given 44 mg/kg daily. The occurrence of these neoplasms is probably attributable to the high estrogen/progesterone ratio which occurs in rats as a result of the prolactin-inhibiting action of bromocriptine mesylate. The endocrine mechanisms believed to be involved in the rats are not present in humans. There is no known correlation between uterine malignancies occurring in bromocriptine-treated rats and human risk. In contrast to the findings in rats, the uteri from mice killed after 74 weeks of treatment did not exhibit evidence of drug-related changes.
Bromocriptine mesylate was evaluated for mutagenic potential in the battery of tests that included Ames bacterial mutation assay, mutagenic activity in vitro on V79 Chinese hamster fibroblasts, cytogenetic analysis of Chinese hamster bone marrow cells following in vivo treatment, and an in vivo micronucleus test for mutagenic potential in mice.
No mutagenic effects were obtained in any of these tests.
Fertility and reproductive performance in female rats were not influenced adversely by treatment with bromocriptine beyond the predicted decrease in the weight of pups due to suppression of lactation. In males treated with 50 mg/kg of this drug, mating and fertility were within the normal range. Increased perinatal loss was produced in the subgroups of dams, sacrificed on day 21 postpartum (p.p.) after mating with males treated with the highest dose (50 mg/kg).
Pregnancy
Administration of 10 mg/kg to 30 mg/kg of bromocriptine to 2 strains of rats on days 6 to 15 postcoitum (p.c.) as well as a single dose of 10 mg/kg on day 5 p.c. interfered with nidation. Three mg/kg given on days 6 to 15 were without effect on nidation, and did not produce any anomalies. In animals treated from day 8 to 15 p.c., i.e., after implantation, 30 mg/kg produced increased prenatal mortality in the form of increased incidence of embryonic resorption. One anomaly, aplasia of spinal vertebrae and ribs, was found in the group of 262 fetuses derived from the dams treated with 30 mg/kg bromocriptine. No fetotoxic effects were found in offspring of dams treated during the peri- or postnatal period.
Two studies were conducted in rabbits (2 strains) to determine the potential to interfere with nidation. Dose levels of 100 mg/kg/day or 300 mg/kg/day from day 1 to day 6 p.c. did not adversely affect nidation. The high dose was approximately 63 times the maximum human dose administered in controlled clinical trials (100 mg/day), based on body surface area. In New Zealand white rabbits, some embryo mortality occurred at 300 mg/kg which was a reflection of overt maternal toxicity. Three studies were conducted in 2 strains of rabbits to determine the teratological potential of bromocriptine at dose levels of 3 mg/kg, 10 mg/kg, 30 mg/kg, 100 mg/kg, and 300 mg/kg given from day 6 to day 18 p.c. In 2 studies with the Yellow-silver strain, cleft palate was found in 3 and 2 fetuses at maternally toxic doses of 100 mg/kg and 300 mg/kg, respectively. One control fetus also exhibited this anomaly. In the third study conducted with New Zealand white rabbits using an identical protocol, no cleft palates were produced.
No teratological or embryotoxic effects of bromocriptine were produced in any of 6 offspring from 6 monkeys at a dose level of 2 mg/kg.
Information concerning 1,276 pregnancies in women taking bromocriptine has been collected. In the majority of cases, bromocriptine was discontinued within 8 weeks into pregnancy (mean 28.7 days), however, 8 patients received the drug continuously throughout pregnancy. The mean daily dose for all patients was 5.8 mg (range 1 mg to 40 mg).
Of these 1,276 pregnancies, there were 1,088 full-term deliveries (4 stillborn), 145 spontaneous abortions (11.4%), and 28 induced abortions (2.2%). Moreover, 12 extrauterine gravidities and 3 hydatidiform moles (twice in the same patient) caused early termination of pregnancy. These data compare favorably with the abortion rate (11% to 25%) cited for pregnancies induced by clomiphene citrate, menopausal gonadotropin, and chorionic gonadotropin.
Although spontaneous abortions often go unreported, especially prior to 20 weeks of gestation, their frequency has been estimated to be 15%.
The incidence of birth defects in the population at large ranges from 2% to 4.5%. The incidence in 1,109 live births from patients receiving bromocriptine is 3.3%.
There is no suggestion that bromocriptine contributed to the type or incidence of birth defects in this group of infants.
Nursing Mothers
Bromocriptine should not be used during lactation in postpartum women.
Pediatric Use
The safety and effectiveness of bromocriptine for the treatment of prolactin-secreting pituitary adenomas have been established in patients age 16 to adult. No data are available for bromocriptine use in pediatric patients under the age of 8 years. A single 8-year-old patient treated with bromocriptine for a prolactin-secreting pituitary macroadenoma has been reported without therapeutic response.
The use of bromocriptine for the treatment of prolactin-secreting adenomas in pediatric patients in the age group 11 to under 16 years is supported by evidence from well-controlled trials in adults, with additional data in a limited number (n=14) of children and adolescents 11 years to 15 years of age with prolactin-secreting pituitary macro- and microadenomas who have been treated with bromocriptine. Of the 14 reported patients, 9 had successful outcomes, 3 partial responses, and 2 failed to respond to bromocriptine treatment. Chronic hypopituitarism complicated macroadenoma treatment in 5 of the responders, both in patients receiving bromocriptine alone and in those who received bromocriptine in combination with surgical treatment and/or pituitary irradiation.
Safety and effectiveness of bromocriptine in pediatric patients have not been established for any other indication listed in the INDICATIONS AND USAGE section.
Geriatric Use
Clinical studies for bromocriptine did not include sufficient numbers of subjects aged 65 years and over to determine whether the elderly respond differently from younger subjects. However, other reported clinical experiences, including postmarketing reporting of adverse events, have not identified differences in response or tolerability between elderly and younger patients. Even though no variation in efficacy or adverse reaction profile in geriatric patients taking bromocriptine has been observed, greater sensitivity of some elderly individuals cannot be categorically ruled out. In general, dose selection for an elderly patient should be cautious, starting at the lower end of the dose range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy in this population.
-
ADVERSE REACTIONS
Adverse Reactions from Clinical Trials
Hyperprolactinemic Indications
The incidence of adverse effects is quite high (69%) but these are generally mild to moderate in degree. Therapy was discontinued in approximately 5% of patients because of adverse effects. These in decreasing order of frequency are: nausea (49%), headache (19%), dizziness (17%), fatigue (7%), lightheadedness (5%), vomiting (5%), abdominal cramps (4%), nasal congestion (3%), constipation (3%), diarrhea (3%) and drowsiness (3%).
A slight hypotensive effect may accompany bromocriptine mesylate treatment. The occurrence of adverse reactions may be lessened by temporarily reducing dosage to ½ a bromocriptine mesylate tablet 2 or 3 times daily. A few cases of cerebrospinal fluid rhinorrhea have been reported in patients receiving bromocriptine for treatment of large prolactinomas. This has occurred rarely, usually only in patients who have received previous transsphenoidal surgery, pituitary radiation, or both, and who were receiving bromocriptine for tumor recurrence. It may also occur in previously untreated patients whose tumor extends into the sphenoid sinus.
Acromegaly
The most frequent adverse reactions encountered in acromegalic patients treated with bromocriptine were: nausea (18%), constipation (14%), postural/orthostatic hypotension (6%), anorexia (4%), dry mouth/nasal stuffiness (4%), indigestion/dyspepsia (4%), digital vasospasm (3%), drowsiness/tiredness (3%) and vomiting (2%).
Less frequent adverse reactions (less than 2%) were: gastrointestinal bleeding, dizziness, exacerbation of Raynaud's syndrome, headache and syncope. Rarely (less than 1%) hair loss, alcohol potentiation, faintness, lightheadedness, arrhythmia, ventricular tachycardia, decreased sleep requirement, visual hallucinations, lassitude, shortness of breath, bradycardia, vertigo, paresthesia, sluggishness, vasovagal attack, delusional psychosis, paranoia, insomnia, heavy headedness, reduced tolerance to cold, tingling of ears, facial pallor and muscle cramps have been reported.
Parkinson's Disease
In clinical trials in which bromocriptine was administered with concomitant reduction in the dose of levodopa/carbidopa, the most common newly appearing adverse reactions were: nausea, abnormal involuntary movements, hallucinations, confusion, "on-off" phenomenon, dizziness, drowsiness, faintness/fainting, vomiting, asthenia, abdominal discomfort, visual disturbance, ataxia, insomnia, depression, hypotension, shortness of breath, constipation, and vertigo.
Less common adverse reactions which may be encountered include: anorexia, anxiety, blepharospasm, dry mouth, dysphagia, edema of the feet and ankles, erythromelalgia, epileptiform seizure, fatigue, headache, lethargy, mottling of skin, nasal stuffiness, nervousness, nightmares, paresthesia, skin rash, urinary frequency, urinary incontinence, urinary retention, and rarely, signs and symptoms of ergotism such as tingling of fingers, cold feet, numbness, muscle cramps of feet and legs or exacerbation of Raynaud's syndrome.
Abnormalities in laboratory tests may include elevations in blood urea nitrogen, SGOT, SGPT, GGPT, CPK, alkaline phosphatase and uric acid, which are usually transient and not of clinical significance.
Adverse Reactions from Postmarketing Experience
The following adverse reactions have been reported during postapproval use of bromocriptine (All Indications Combined). Because adverse reactions from spontaneous reports are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Psychiatric disorders
Confusion, psychomotor agitation/excitation, hallucinations, psychotic disorders, insomnia, libido increase, hypersexuality and impulse control/compulsive behaviors (including gambling, spending, and other intense urges).
Nervous system disorders
Headache, drowsiness, dizziness, dyskinaesia, somnolence, paraesthesia, excess daytime somnolence, sudden onset of sleep.
Eye disorders
Visual disturbance, vision blurred.
Ear and labyrinth disorders
Tinnitus.
Cardiac disorders
Pericardial effusion, constrictive pericarditis, tachycardia, bradycardia, arrhythmia, cardiac valve fibrosis.
Vascular disorders
Hypotension, orthostatic hypotension (very rarely leading to syncope), reversible pallor of fingers and toes induced by cold (especially in patients with history of Raynaud's phenomenon).
Respiratory, thoracic and mediastinal disorders
Nasal congestion, pleural effusion, pleural fibrosis, pleurisy, pulmonary fibrosis, dyspnoea.
Gastrointestinal disorders
Nausea, constipation, vomiting, dry mouth, diarrhea, abdominal pain, retroperitoneal fibrosis, gastrointestinal ulcer, gastrointestinal hemorrhage.
Skin and subcutaneous tissue disorders
Allergic skin reactions, hair loss.
Musculoskeletal and connective tissue disorders
Leg cramps.
General disorders and administration site conditions
Fatigue, peripheral oedema, a syndrome resembling Neuroleptic Malignant Syndrome on abrupt withdrawal of bromocriptine, withdrawal symptoms (including apathy, anxiety, depression, fatigue, insomnia, sweating, and pain) with taper or after discontinuation (see Precautions).
Adverse Events Observed in Other Conditions
Postpartum Patients (see above Warnings)
In postpartum studies with bromocriptine 23 percent of postpartum patients treated had at least 1 side effect, but they were generally mild to moderate in degree. Therapy was discontinued in approximately 3% of patients. The most frequently occurring adverse reactions were: headache (10%), dizziness (8%), nausea (7%), vomiting (3%), fatigue (1.0%), syncope (0.7%), diarrhea (0.4%) and cramps (0.4%). Decreases in blood pressure (≥ 20 mm Hg systolic and ≥ 10 mm Hg diastolic) occurred in 28% of patients at least once during the first 3 postpartum days; these were usually of a transient nature. Reports of fainting in the puerperium may possibly be related to this effect. In postmarketing experience in the U.S., serious adverse reactions reported include 72 cases of seizures (including 4 cases of status epilepticus), 30 cases of stroke, and 9 cases of myocardial infarction among postpartum patients. Seizure cases were not necessarily accompanied by the development of hypertension. An unremitting and often progressively severe headache, sometimes accompanied by visual disturbance, often preceded by hours to days many cases of seizure and/or stroke. Most patients had shown no evidence of any of the hypertensive disorders of pregnancy including eclampsia, preeclampsia or pregnancy-induced hypertension. One stroke case was associated with sagittal sinus thrombosis, and another was associated with cerebral and cerebellar vasculitis. One case of myocardial infarction was associated with unexplained disseminated intravascular coagulation and a second occurred in conjunction with use of another ergot alkaloid. The relationship of these adverse reactions to bromocriptine administration has not been established.
In rare cases serious adverse events, including hypertension, myocardial infarction, seizures, stroke, or psychic disorders have been reported in postpartum women treated with bromocriptine. In some patients the development of seizures or stroke was preceded by severe headache and/or transient visual disturbances. Although the causal relationship of these events to the drug is uncertain, periodic monitoring of blood pressure is advisable in postpartum women receiving bromocriptine. If hypertension, severe, progressive, or unremitting headache (with or without visual disturbances), or evidence of CNS toxicity develop, the administration of bromocriptine should be discontinued and the patient should be evaluated promptly.
Particular caution is required in patients who have recently been treated or are on concomitant therapy with drugs that can alter blood pressure, e.g., vasoconstrictors such as sympathomimetics or ergot alkaloids including ergometrine or methylergometrine and their concomitant use in the puerperium is not recommended.
To report SUSPECTED ADVERSE REACTIONS, contact Zydus Pharmaceuticals (USA) Inc. at 1-877-993-8779 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
OVERDOSAGE
The most commonly reported signs and symptoms associated with acute bromocriptine overdose are: nausea, vomiting, constipation, diaphoresis, dizziness, pallor, severe hypotension, malaise, confusion, lethargy, drowsiness, delusions, hallucinations, and repetitive yawning. The lethal dose has not been established and the drug has a very wide margin of safety. However, one death occurred in a patient who committed suicide with an unknown quantity of bromocriptine and chloroquine.
Treatment of overdose consists of removal of the drug by emesis (if conscious), gastric lavage, activated charcoal, or saline catharsis. Careful supervision and recording of fluid intake and output is essential. Hypotension should be treated by placing the patient in the Trendelenburg position and administering I.V. fluids. If satisfactory relief of hypotension cannot be achieved by using the above measures to their fullest extent, vasopressors should be considered.
There have been isolated reports of children who accidentally ingested bromocriptine. Vomiting, somnolence and fever were reported as adverse events. Patients recovered either spontaneously within a few hours or after appropriate management.
-
DOSAGE AND ADMINISTRATION
It is recommended that bromocriptine mesylate capsules be taken with food. Patients should be evaluated frequently during dose escalation to determine the lowest dosage that produces a therapeutic response.
Hyperprolactinemic Indications
The initial dosage of bromocriptine mesylate tablets in adults is one ½ to one 2½ mg scored tablet daily. An additional 2½ mg tablet may be added to the treatment regimen as tolerated every 2 to 7 days until an optimal therapeutic response is achieved. The therapeutic dosage ranged from 2.5 to 15 mg daily in adults studied clinically.
Based on limited data in children of age 11 to 15, (see Pediatric Use) the initial dose is one ½ to one 2½ mg scored tablet daily. Dosing may need to be increased as tolerated until a therapeutic response is achieved. The therapeutic dosage ranged from 2.5 to 10 mg daily in children with prolactin-secreting pituitary adenomas.
In order to reduce the likelihood of prolonged exposure to bromocriptine mesylate capsules should an unsuspected pregnancy occur, a mechanical contraceptive should be used in conjunction with bromocriptine mesylate capsule therapy until normal ovulatory menstrual cycles have been restored. Contraception may then be discontinued in patients desiring pregnancy.
Thereafter, if menstruation does not occur within 3 days of the expected date, bromocriptine mesylate capsule therapy should be discontinued and a pregnancy test performed.
Acromegaly
Virtually all acromegalic patients receiving therapeutic benefit from bromocriptine mesylate capsules also have reductions in circulating levels of growth hormone. Therefore, periodic assessment of circulating levels of growth hormone will, in most cases, serve as a guide in determining the therapeutic potential of bromocriptine. If, after a brief trial with bromocriptine mesylate capsule therapy, no significant reduction in growth hormone levels has taken place, careful assessment of the clinical features of the disease should be made, and if no change has occurred, dosage adjustment or discontinuation of therapy should be considered.
The initial recommended dosage is one ½ to one 2½ mg bromocriptine mesylate tablet on retiring (with food) for 3 days. An additional one ½ to 1 bromocriptine mesylate tablet should be added to the treatment regimen as tolerated every 3 to 7 days until the patient obtains optimal therapeutic benefit. Patients should be reevaluated monthly and the dosage adjusted based on reductions of growth hormone or clinical response. The usual optimal therapeutic dosage range of bromocriptine mesylate capsules varies from 20 mg/day to 30 mg/day in most patients. The maximal dosage should not exceed 100 mg/day.
Patients treated with pituitary irradiation should be withdrawn from bromocriptine mesylate capsule therapy on a yearly basis to assess both the clinical effects of radiation on the disease process as well as the effects of bromocriptine mesylate capsule therapy. Usually a 4 week to 8 week withdrawal period is adequate for this purpose. Recurrence of the signs/symptoms or increases in growth hormone indicate the disease process is still active and further courses of bromocriptine should be considered.
Parkinson's Disease
The basic principle of bromocriptine mesylate capsule therapy is to initiate treatment at a low dosage and, on an individual basis, increase the daily dosage slowly until a maximum therapeutic response is achieved. The dosage of levodopa during this introductory period should be maintained, if possible. The initial dose of bromocriptine is one ½ of a 2½ mg tablet twice daily with meals. Assessments are advised at 2-week intervals during dosage titration to ensure that the lowest dosage producing an optimal therapeutic response is not exceeded. If necessary, the dosage may be increased every 14 to 28 days by 2½ mg/day with meals. Should it be advisable to reduce the dosage of levodopa because of adverse reactions, the daily dosage of bromocriptine, if increased, should be accomplished gradually in small (2½ mg) increments.
The safety of bromocriptine mesylate capsules have not been demonstrated in dosages exceeding 100 mg/day.
-
HOW SUPPLIED
Bromocriptine Mesylate Capsules USP, 5 mg are white to off-white powder filled in size "3" empty Cellulose capsules with tan colored cap printed with "ZA 17" in black ink and white colored body printed with "5 mg" in black ink and are supplied as follows:
NDC 68382-110-06 in bottle of 30 capsules with child-resistant closure
NDC 68382-110-01 in bottle of 100 capsules
Storage:
Store at 20°C to 25°C (68°F to 77° F) [See USP Controlled Room Temperature]; in tight, light-resistant container.
Dispense in a tight, light-resistant container.
- SPL UNCLASSIFIED SECTION
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
BROMOCRIPTINE MESYLATE
bromocriptine mesylate capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68382-110 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BROMOCRIPTINE MESYLATE (UNII: FFP983J3OD) (BROMOCRIPTINE - UNII:3A64E3G5ZO) BROMOCRIPTINE 5 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MALEIC ACID (UNII: 91XW058U2C) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) CARRAGEENAN (UNII: 5C69YCD2YJ) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) FERRIC OXIDE RED (UNII: 1K09F3G675) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) FERROSOFERRIC OXIDE (UNII: XM0M87F357) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) WATER (UNII: 059QF0KO0R) SHELLAC (UNII: 46N107B71O) AMMONIA (UNII: 5138Q19F1X) Product Characteristics Color BROWN (TAN) , WHITE (WHITE) Score no score Shape CAPSULE (CAPSULE) Size 16mm Flavor Imprint Code ZA17;5mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68382-110-06 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/23/2009 2 NDC:68382-110-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 01/23/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078899 01/23/2009 Labeler - Zydus Pharmaceuticals USA Inc. (156861945) Registrant - Zydus Pharmaceuticals USA Inc. (156861945) Establishment Name Address ID/FEI Business Operations Zydus Lifesciences Limited 918596198 ANALYSIS(68382-110) , MANUFACTURE(68382-110)
