Label: BOSULIF- bosutinib tablet, film coated


- NDC Code(s): 63539-117-01, 63539-193-30
- Packager: U.S. Pharmaceuticals
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated October 28, 2021
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BOSULIF safely and effectively. See full prescribing information for BOSULIF.
BOSULIF® (bosutinib) tablets, for oral use
Initial U.S. Approval: 2012RECENT MAJOR CHANGES
Indications and Usage (1) 5/2021 Warnings and Precautions, Gastrointestinal Toxicity (5.1) 5/2021 Warnings and Precautions, Hepatic Toxicity (5.3) 5/2021 Warnings and Precautions, Cardiovascular Toxicity (5.4) 5/2021 Warnings and Precautions, Fluid Retention (5.5) 5/2021 Warnings and Precautions, Renal Toxicity (5.6) 5/2021 INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- Newly-diagnosed chronic phase Ph+ CML: 400 mg orally once daily with food. (2.1)
- Chronic, accelerated, or blast phase Ph+ CML with resistance or intolerance to prior therapy: 500 mg orally once daily with food. (2.1)
- Consider dose escalation by increments of 100 mg once daily to a maximum of 600 mg daily in patients who do not reach complete hematologic, cytogenetic, or molecular response and do not have Grade 3 or greater adverse reactions. (2.2)
- Adjust dosage for toxicity and organ impairment (2)
DOSAGE FORMS AND STRENGTHS
Tablets: 100 mg, 400 mg, and 500 mg. (3)
CONTRAINDICATIONS
Hypersensitivity to BOSULIF. (4)
WARNINGS AND PRECAUTIONS
- Gastrointestinal Toxicity: Monitor and manage as necessary. Withhold, dose reduce, or discontinue BOSULIF. (2.3, 5.1)
- Myelosuppression: Monitor blood counts and manage as necessary. (2.4, 5.2)
- Hepatic Toxicity: Monitor liver enzymes at least monthly for the first 3 months and as needed. Withhold, dose reduce, or discontinue BOSULIF. (2.3, 5.3)
- Cardiovascular Toxicity: Monitor and manage as necessary. (5.4)
- Fluid Retention: Monitor patients and manage using standard of care treatment. Withhold, dose reduce, or discontinue BOSULIF. (2.3, 5.5)
- Renal Toxicity: Monitor patients for renal function at baseline and during therapy with BOSULIF. (5.6)
- Embryo-Fetal Toxicity: BOSULIF can cause fetal harm. Advise female patients of reproductive potential of potential risk to a fetus and to use effective contraception. (5.7)
ADVERSE REACTIONS
- Most common adverse reactions (≥20%), in patients with CML are diarrhea, rash, nausea, abdominal pain, vomiting, fatigue, hepatic dysfunction, respiratory tract infection, pyrexia, and headache. The most common laboratory abnormalities (≥20%) are creatinine increased, hemoglobin decreased, lymphocyte count decreased, platelets decreased, ALT increased, calcium decreased, white blood cell count decreased, absolute neutrophil count decreased, AST increased, glucose increased, phosphorus decreased, urate increased, alkaline phosphatase increased, lipase increased, creatine kinase increased, and amylase increased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2021
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Dose Escalation
2.3 Dose Adjustments for Non-Hematologic Adverse Reactions
2.4 Dose Adjustments for Myelosuppression
2.5 Dose Adjustments for Renal Impairment or Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Toxicity
5.2 Myelosuppression
5.3 Hepatic Toxicity
5.4 Cardiovascular Toxicity
5.5 Fluid Retention
5.6 Renal Toxicity
5.7 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on BOSULIF
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Newly-Diagnosed CP Ph+ CML
14.2 Imatinib-Resistant or -Intolerant Ph+ CP, AP, and BP CML
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dose is taken orally once daily with food. The tablet is to be swallowed whole and should not be broken or cut. Continue treatment with BOSULIF until disease progression or intolerance to therapy.
If a dose is missed beyond 12 hours, the patient should skip the dose and take the usual prescribed dose on the following day.
2.2 Dose Escalation
In clinical studies of adult Ph+ CML patients, dose escalation by increments of 100 mg once daily to a maximum of 600 mg once daily was allowed in patients who did not achieve or maintain a hematologic, cytogenetic, or molecular response and who did not have Grade 3 or higher adverse reactions at the recommended starting dosage.
2.3 Dose Adjustments for Non-Hematologic Adverse Reactions
Elevated liver transaminases: If elevations in liver transaminases greater than 5×institutional upper limit of normal (ULN) occur, withhold BOSULIF until recovery to less than or equal to 2.5×ULN and resume at 400 mg once daily thereafter. If recovery takes longer than 4 weeks, discontinue BOSULIF. If transaminase elevations greater than or equal to 3×ULN occur concurrently with bilirubin elevations greater than 2×ULN and alkaline phosphatase less than 2×ULN (Hy's law case definition), discontinue BOSULIF [see Warnings and Precautions (5.3)].
Diarrhea: For National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) Grade 3–4 diarrhea (increase of greater than or equal to 7 stools/day over baseline/pretreatment), withhold BOSULIF until recovery to Grade less than or equal to 1. BOSULIF may be resumed at 400 mg once daily [see Warnings and Precautions (5.1)].
For other clinically significant, moderate or severe non-hematological toxicity, withhold BOSULIF until the toxicity has resolved, then consider resuming BOSULIF at a dose reduced by 100 mg taken once daily. If clinically appropriate, consider re-escalating the dose of BOSULIF to the starting dose taken once daily. Doses less than 300 mg/day have been used in patients; however, efficacy has not been established.
2.4 Dose Adjustments for Myelosuppression
Dose reductions for severe or persistent neutropenia and thrombocytopenia are described below (Table 1).
Table 1: Dose Adjustments for Neutropenia and Thrombocytopenia - *
- Absolute Neutrophil Count
ANC* less than 1000×106/L
or
Platelets less than 50,000×106/LWithhold BOSULIF until ANC greater than or equal to1000×106/L and platelets greater than or equal to 50,000×106/L.
Resume treatment with BOSULIF at the same dose if recovery occurs within 2 weeks. If blood counts remain low for greater than 2 weeks, upon recovery, reduce dose by 100 mg and resume treatment.
If cytopenia recurs, reduce dose by an additional 100 mg upon recovery and resume treatment.
Doses less than 300 mg/day have been used in patients; however, efficacy has not been established.2.5 Dose Adjustments for Renal Impairment or Hepatic Impairment
The recommended starting doses for patients with renal and hepatic impairment are described in Table 2 below.
Table 2: Dose Adjustments for Renal and Hepatic Impairment Recommended Starting Dosage Newly-diagnosed chronic phase Ph+ CML2 Chronic, accelerated, or blast phase Ph+ CML with resistance or intolerance to prior therapy [see Use in Specific Populations (8.6, 8.7) and Clinical Pharmacology (12.3)]. Abbreviations: CML=chronic myelogenous leukemia; Ph+=Philadelphia chromosome-positive. - *
- There are no clinical data for efficacy at the dose of 200 mg once daily in patients with CML.
Normal renal and hepatic function 400 mg daily 500 mg daily Renal impairment Creatinine clearance 30 to 50 mL/min 300 mg daily 400 mg daily Creatinine clearance less than 30 mL/min 200 mg daily 300 mg daily Hepatic impairment Mild (Child-Pugh A), Moderate (Child-Pugh B) or Severe (Child-Pugh C) 200 mg daily* 200 mg daily* -
3 DOSAGE FORMS AND STRENGTHS
- 100 mg tablets: yellow, oval, biconvex, film-coated tablets debossed with "Pfizer" on one side and "100" on the other.
- 400 mg tablets: orange, oval, biconvex, film-coated tablets debossed with "Pfizer" on one side and "400" on the other.
- 500 mg tablets: red, oval, biconvex, film-coated tablets debossed with "Pfizer" on one side and "500" on the other.
-
4 CONTRAINDICATIONS
BOSULIF is contraindicated in patients with a history of hypersensitivity to BOSULIF. Reactions have included anaphylaxis [see Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Toxicity
Diarrhea, nausea, vomiting, and abdominal pain occur with BOSULIF treatment. Monitor and manage patients using standards of care, including antidiarrheals, antiemetics, and fluid replacement.
In the randomized clinical trial in patients with newly-diagnosed Ph+ CML, the median time to onset for diarrhea (all grades) was 4 days and the median duration per event was 3 days.
Among 546 patients in a single-arm study in patients with CML who were resistant or intolerant to prior therapy, the median time to onset for diarrhea (all grades) was 2 days and the median duration per event was 2 days. Among the patients who experienced diarrhea, the median number of episodes of diarrhea per patient during treatment with BOSULIF was 3 (range 1–268).
To manage gastrointestinal toxicity, withhold, dose reduce, or discontinue BOSULIF as necessary [see Dosage and Administration (2.3) and Adverse Reactions (6)].
5.2 Myelosuppression
Thrombocytopenia, anemia and neutropenia occur with BOSULIF treatment. Perform complete blood counts weekly for the first month of therapy and then monthly thereafter, or as clinically indicated. To manage myelosuppression, withhold, dose reduce, or discontinue BOSULIF as necessary [see Dosage and Administration (2.4) and Adverse Reactions (6)].
5.3 Hepatic Toxicity
Bosutinib may cause elevations in serum transaminases (alanine aminotransferase [ALT], aspartate aminotransferase [AST]).
Two cases consistent with drug induced liver injury (defined as concurrent elevations in ALT or AST greater than or equal to 3×ULN with total bilirubin greater than 2×ULN and alkaline phosphatase less than 2×ULN) have occurred without alternative causes. This represented 2 out 1711 patients in BOSULIF clinical trials.
In the 268 patients from the safety population in the randomized clinical trial in patients with newly-diagnosed CML in the BOSULIF treatment group, the incidence of ALT elevation was 68.3% and AST elevation was 56.0%. Of patients who experienced transaminase elevations of any grade, 73% experienced their first event within the first 3 months. The median time to onset of increased ALT and AST was 29 and 56 days, respectively, and the median duration was 19 and 15 days, respectively.
Among the 546 patients in a single-arm study in patients with CML who were resistant or intolerant to prior therapy, the incidence of ALT elevation was 53.3% and AST elevation was 46.7%. Sixty percent of the patients experienced an increase in either ALT or AST. Most cases of transaminase elevations in this study occurred early in treatment; of patients who experienced transaminase elevations of any grade, more than 81% experienced their first event within the first 3 months. The median time to onset of increased ALT and AST was 22 and 29 days, respectively, and the median duration for each was 21 days.
Perform hepatic enzyme tests monthly for the first 3 months of BOSULIF treatment and as clinically indicated. In patients with transaminase elevations, monitor liver enzymes more frequently. Withhold, dose reduce, or discontinue BOSULIF as necessary [see Dosage and Administration (2.3) and Adverse Reactions (6)].
5.4 Cardiovascular Toxicity
BOSULIF can cause cardiovascular toxicity including cardiac failure, left ventricular dysfunction, and cardiac ischemic events. Cardiac failure events occurred more frequently in previously treated patients than in patients with newly diagnosed CML and were more frequent in patients with advanced age or risk factors, including previous medical history of cardiac failure. Cardiac ischemic events occurred in both previously treated patients and in patients with newly diagnosed CML and were more common in patients with coronary artery disease risk factors, including history of diabetes, body mass index greater than 30, hypertension, and vascular disorders.
In a randomized study with newly diagnosed CML, cardiac failure occurred in 1.9% of patients treated with BOSULIF compared to 0.8% of patients treated with imatinib. Cardiac ischemic events occurred in 4.9% of patients treated with BOSULIF compared to 0.8% of patients treated with imatinib.
In a single-arm study in patients with CML who were resistant or intolerant to prior therapy, cardiac failure was observed in 5.3% of patients and cardiac ischemic events were observed in 4.9% of patients treated with BOSULIF.
Monitor patients for signs and symptoms consistent with cardiac failure and cardiac ischemia and treat as clinically indicated. Interrupt, dose reduce, or discontinue BOSULIF as necessary [see Dosage and Administration (2.3)].
5.5 Fluid Retention
Fluid retention occurs with BOSULIF and may manifest as pericardial effusion, pleural effusion, pulmonary edema, and/or peripheral edema.
In the randomized clinical trial of 268 patients with newly-diagnosed CML in the bosutinib treatment group, 3 patients (1.1%) experienced severe fluid retention of Grade 3, 1 patient experienced Grade 3 pericardial effusion, and 2 patients experienced Grade 3 pleural effusion. Among 546 patients in a single-arm study in patients with Ph+ CML who were resistant or intolerant to prior therapy, Grade 3 or 4 fluid retention was reported in 30 patients (6%). Some patients experienced more than one fluid retention event. Specifically, 24 patients experienced Grade 3 or 4 pleural effusions, 9 patients experienced Grade 3 or Grade 4 pericardial effusions, and 6 patients experienced Grade 3 edema.
Monitor and manage patients using standards of care. Interrupt, dose reduce or discontinue BOSULIF as necessary [see Dosage and Administration (2.3) and Adverse Reactions (6)].
5.6 Renal Toxicity
An on-treatment decline in estimated glomerular filtration rate (eGFR) has occurred in patients treated with BOSULIF. Table 3 identifies the shift from baseline to lowest observed eGFR during BOSULIF therapy for patients in the pooled leukemia studies regardless of line of therapy. The median duration of therapy with BOSULIF was approximately 24 months (range, 0.03 to 155) for patients in these studies.
Table 3: Shift From Baseline to Lowest Observed eGFR Group During Treatment Safety Population in Clinical Studies (N=1372)* Baseline Follow-Up Renal Function Status N Normal
n (%)Mild
n (%)Mild to Moderate
n (%)Moderate to Severe
n (%)Severe
n (%)Kidney Failure
n (%)Abbreviations: eGFR=estimated glomerular filtration rate; N/n=number of patients. Notes: eGFR was calculated using Modification in Diet in Renal Disease method (MDRD). Notes: Grading is based on Kidney Disease Improving Global Outcomes (KDIGO) Classification by eGFR: Normal: greater than or equal to 90, Mild: 60 to less than 90, Mild to Moderate: 45 to less than 60, Moderate to Severe: 30 to less than 45, Severe: 15 to less than 30, Kidney Failure: less than 15 ml/min/1.73 m2. - *
- Among the 1372 patients, eGFR was missing in 7 patients at baseline or on-therapy. There were no patients with kidney failure at baseline.
Normal 527 115 (21.8) 330 (62.6) 50 (9.5) 23 (4.4) 3 (0.6) 5 (0.9) Mild 672 10 (1.5) 259 (38.5) 271 (40.3) 96 (14.3) 26 (3.9) 6 (0.9) Mild to Moderate 137 0 6 (4.4) 40 (29.2) 66 (48.2) 24 (17.5) 1 (0.7) Moderate to Severe 33 0 1 (3.0) 1 (3.0) 8 (24.2) 19 (57.6) 4 (12.1) Severe 1 0 0 0 0 0 1 (100) Total 1370 125 (9.1) 596 (43.5) 362 (26.4) 193 (14.1) 72 (5.2) 17 (1.2) Monitor renal function at baseline and during therapy with BOSULIF, with particular attention to those patients who have preexisting renal impairment or risk factors for renal dysfunction. Consider dose adjustment in patients with baseline and treatment emergent renal impairment [see Dosage and Administration (2.5)].
5.7 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, BOSULIF can cause fetal harm when administered to a pregnant woman. There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies conducted in rats and rabbits, oral administration of bosutinib during organogenesis caused adverse developmental outcomes, including structural abnormalities, embryo-fetal mortality, and alterations to growth at maternal exposures (AUC) as low as 1.2 times the human exposure at the dose of 500 mg/day. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for at least 2 weeks after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Gastrointestinal toxicity [see Warnings and Precautions (5.1)].
- Myelosuppression [see Warnings and Precautions (5.2)].
- Hepatic toxicity [see Warnings and Precautions (5.3)].
- Cardiovascular toxicity [see Warnings and Precautions (5.4)]
- Fluid retention [see Warnings and Precautions (5.5)].
- Renal toxicity [see Warnings and Precautions (5.6)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reactions, in ≥20% of patients with newly diagnosed CP Ph+ CML or CP, AP, or BP Ph+ CML with resistance or intolerance to prior therapy (N=814) were diarrhea (80%), rash (44%), nausea (44%), abdominal pain (43%), vomiting (33%), fatigue (33%), hepatic dysfunction (33%), respiratory tract infection (25%), pyrexia (24%), and headache (21%).
The most common laboratory abnormalities that worsened from baseline in ≥20% of patients were creatinine increased (93%), hemoglobin decreased (90%), lymphocyte count decreased (72%), platelets decreased (69%), ALT increased (58%), calcium decreased (53%), white blood cell count decreased (52%), absolute neutrophils count decreased (50%), AST increased (50%), glucose increased (46%), phosphorus decreased (44%), urate increased (41%), alkaline phosphatase increased (40%), lipase increased (36%), creatine kinase increased (29%), and amylase increased (24%).
Adverse Reactions in Patients With Newly-Diagnosed CP CML
The clinical trial randomized and treated 533 patients with newly-diagnosed CP CML to receive BOSULIF 400 mg daily or imatinib 400 mg daily as single agents (Newly-Diagnosed CP CML Study) [see Clinical Studies (14.1)]. The safety population (received at least 1 dose of BOSULIF) included:
- two hundred sixty-eight (268) patients with newly-diagnosed CP CML had a median duration of BOSULIF treatment of 55 months (range: 0.3 to 60 months) and a median dose intensity of 394 mg/day.
Serious adverse reactions occurred in 22% of patients with newly-diagnosed CP CML who received bosutinib. Serious adverse reactions reported in >2% of patients included hepatic dysfunction (4.1%), pneumonia (3.4%), coronary artery disease (3.4%), and gastroenteritis (2.2%). Fatal adverse reactions occurred in 3 patients (1.1%) due to coronary artery disease (0.4%), cardiac failure acute (0.4%), and renal failure (0.4%).
Permanent discontinuation of bosutinib due to an adverse reaction occurred in 20% of patients with newly-diagnosed CP CML who received bosutinib. Adverse reactions which resulted in permanent discontinuation in > 2% of patients included hepatic dysfunction (9%).
Dose modifications (dose interruption or reductions) of bosutinib due to an adverse reaction occurred in 68% of patients with newly-diagnosed CP CML. Adverse reactions which required dose interruptions or reductions in >5% of patients included hepatic dysfunction (27%), thrombocytopenia (16%), diarrhea (16%), lipase increased (10%), neutropenia (7%), abdominal pain (6%), rash (5%).
The most common adverse reactions, in >20% of bosutinib-treated patients with newly-diagnosed CML (N=268) were diarrhea (75%), hepatic dysfunction (45%), rash (40%), abdominal pain (39%), nausea (37%), fatigue (33%), respiratory tract infection (27%), headache (22%), and vomiting (21%).
The most common laboratory abnormalities that worsened from baseline in ≥20% of patients were creatinine increased (94%), hemoglobin decreased (89%), lymphocyte count decreased (84%), ALT increased (68%), platelet count decreased (68%), glucose increased (57%), AST increased (56%), calcium decreased (55%), phosphorus decreased (54%), lipase increased (53%), white blood cell count decreased (50%), absolute neutrophil count decreased (42%), alkaline phosphatase increased (41%), creatine kinase increased (36%), and amylase increased (32%).
Table 4 identifies adverse reactions greater than or equal to 10% for All Grades and Grades 3 or 4 (3/4) for the Phase 3 CP CML safety population.
Table 4: Adverse Reactions (10% or Greater) in Patients With Newly-Diagnosed CML in Bosutinib 400 mg Study* System Organ Class Preferred Term Bosutinib 400 mg
Chronic Phase CML
(N=268)Imatinib 400 mg
Chronic Phase CML
(N=265)All Grades
%Grade 3/4
%All Grades
%Grade 3/4
%Adverse drug reactions are based on all-causality treatment-emergent adverse events. The commonality stratification is based on 'All Grades' under Total column. 'Grade 3', 'Grade 4' columns indicate maximum toxicity. - *
- Based on a Minimum of 57 Months of Follow-up.
- †
- Abdominal pain includes the following preferred terms: Abdominal discomfort, Abdominal pain, Abdominal pain lower, Abdominal pain upper, Abdominal tenderness, Dyspepsia, Epigastric discomfort, Gastrointestinal pain.
- ‡
- Hepatic dysfunction includes the preferred terms: Alanine aminotransferase increased, Aspartate aminotransferase, Aspartate aminotransferase increased, Bilirubin conjugated increased, Blood alkaline phosphatase increased, Blood bilirubin increased, Drug-induced liver injury, Gamma-glutamyltransferase increased, Hepatic enzyme increased, Hepatic steatosis, Hepatitis, Hepatitis toxic, Hepatocellular injury, Hepatotoxicity, Hyperbilirubinemia, Jaundice, Liver disorder, Liver function test increased, Ocular icterus, Transaminases increased.
- §
- Rash includes the following preferred terms: Acne, Blister, Dermatitis, Dermatitis acneiform, Dermatitis bullous, Dermatitis exfoliative generalized, Drug reaction with eosinophilia and systemic symptoms, Dyshidrotic eczema, Eczema, Eczema asteatotic, Erythema, Erythema nodosum, Genital rash, Lichen planus, Perivascular dermatitis, Photosensitivity reaction, Psoriasis, Rash, Rash erythematous, Rash macular, Rash maculo-papular, Rash papular, Rash pruritic, Rash pustular, Rash vesicular, Seborrhoeic keratosis, Skin discoloration, Skin exfoliation, Skin hypopigmentation, Skin irritation, Skin lesion, Stasis dermatitis.
- ¶
- Fatigue includes the following preferred terms: Asthenia, Fatigue, Malaise.
- #
- Edema includes the following preferred terms: Eye edema, Eyelid edema, Face edema, Edema, Edema peripheral, Orbital edema, Periorbital edema, Periorbital swelling, Peripheral swelling, Swelling, Swelling face, Swelling of eyelid, Swollen tongue.
- Þ
- Respiratory tract infection includes the following preferred terms: Nasopharyngitis, Respiratory tract congestion, Respiratory tract infection, Respiratory tract infection viral, Upper respiratory tract infection.
- ß
- Hypertension* includes the preferred terms: Blood pressure systolic increased, Hypertension, Hypertensive crisis, Hypertensive heart disease, Retinopathy hypertensive.
Gastrointestinal disorders Diarrhea 75 9 40 1 Abdominal pain† 39 2 27 1 Nausea 37 0 42 0 Vomiting 21 1 20 0 Constipation 13 0 6 0 Hepatobiliary disorders Hepatic dysfunction‡ 45 27 15 4 Skin and subcutaneous tissue disorders Rash§ 40 2 30 2 Pruritus 11 <1 4 0 General disorders and administration-site conditions Fatigue¶ 33 1 30 <1 Pyrexia 17 1 11 0 Edema# 15 0 46 2 Infections and infestations Respiratory tract infectionÞ 27 1 25 <1 Nervous system disorders Headache 22 1 15 1 Musculoskeletal and connective tissue disorders Arthralgia 18 1 18 <1 Back pain 12 <1 9 <1 Respiratory, thoracic, and mediastinal disorders Cough 11 0 10 0 Dyspnea 11 1 6 1 Metabolism and nutrition disorders Decreased appetite 11 <1 6 0 Vascular disorders Hypertensionß 10 5 11 5 In the randomized study in patients with newly-diagnosed CP CML, one patient in the group treated with BOSULIF experienced a Grade 3 QTcF prolongation (>500 msec). Patients with uncontrolled or significant cardiovascular disease including QT interval prolongation were excluded by protocol.
Table 5 identifies the clinically relevant or severe Grade 3/4 laboratory test abnormalities for the Phase 3 newly-diagnosed CML safety population.
Table 5: Select Laboratory Abnormalities (>20%) That Worsened From Baseline in Patients with Newly-Diagnosed CML in Bosutinib 400 mg Study* Bosutinib
N=268
%Imatinib
N=265
%All Grade Grade 3–4 All Grade Grade 3–4 Abbreviations: ALT=alanine aminotransferase; AST=aspartate aminotransferase; CML=chronic myelogenous leukemia; SGPT=serum glutamic-pyruvic transaminase; SGOT=serum glutamic-oxaloacetic transaminase; N/n=number of patients; ULN=upper limit of normal. Graded using CTCAE v 4.03 - *
- Based on a Minimum of 57 Months of Follow-up.
Hematology Parameters Platelet Count decreased 68 14 60 6 Absolute Neutrophil Count decreased 42 9 65 20 Hemoglobin decreased 89 9 90 7 White Blood Cell Count decreased 50 6 70 8 Lymphocyte Count decreased 84 12 82 14 Biochemistry Parameters SGPT/ALT increased 68 26 28 3 SGOT/AST increased 56 13 29 3.4 Lipase increased 53 19 35 8 Phosphorus decreased 54 9 69 21 Amylase increased 32 3.4 18 2.3 Alkaline Phosphatase increased 41 0 43 0.4 Calcium decreased 55 1.5 57 1.1 Glucose increased 57 3 65 3.4 Creatine Kinase increased 36 3 65 5 Creatinine increased 94 1.1 98 0.8 Adverse Reactions in Patients With Imatinib-Resistant or -Intolerant Ph+ CP, AP, and BP CML
The single-arm clinical trial enrolled patients with Ph+ CP, AP, or BP CML and with resistance or intolerance to prior therapy [see Clinical Studies (14.2)]. The safety population (received at least 1 dose of BOSULIF) included 546 CML patients:
- two hundred eighty-four (284) patients with CP CML previously treated with imatinib only who had a median duration of BOSULIF treatment of 26 months (range: 0.2 to 155 months), and a median dose intensity of 437 mg/day.
- one hundred nineteen (119) patients with CP CML previously treated with both imatinib and at least 1 additional tyrosine kinase inhibitor (TKI) who had a median duration of BOSULIF treatment of 9 months (range: 0.2 to 148 months) and a median dose intensity of 427 mg/day.
- one hundred forty-three (143) patients with advanced phase (AdvP) CML including 79 patients with AP CML and 64 patients with BP CML. In the patients with AP CML and BP CML, the median duration of BOSULIF treatment was 10 months (range: 0.1 to 140 months) and 3 months (range: 0.03 to 71 months), respectively. The median dose intensity was 406 mg/day, and 456 mg/day, in the AP CML and BP CML cohorts, respectively.
Serious adverse reactions occurred in 30% of patients in the safety population of the single-arm trial in patients with CML (N=546) who were resistant or intolerant to prior therapy. Serious adverse reactions reported in >2% of patients included pneumonia (7%), pleural effusion (6%), pyrexia (3.7%), coronary artery disease (3.5%), dyspnea (2.6%), rash (2.2%), thrombocytopenia (2%), abdominal pain (2%), and diarrhea (2%).
Fatal adverse reactions occurred in 12 patients (2.2%) due to coronary artery disease (0.9%), pneumonia (0.4%), respiratory failure (0.4%), gastrointestinal hemorrhage (0.2%), acute kidney injury (0.2%), and acute pulmonary edema (0.2%).
Permanent discontinuation of bosutinib due to an adverse reaction occurred in 22% of patients with CML who were resistant or intolerant to prior therapy. Adverse reactions which resulted in permanent discontinuation in >2% of patients included thrombocytopenia (6%), hepatic dysfunction (3.3%), and neutropenia (2%).
Dose modifications (dose interruption or reductions) of bosutinib due to an adverse reaction occurred in 66% of patients with CML who were resistant or intolerant to prior therapy. Adverse reactions which required dose interruptions or reductions in >5% of patients included thrombocytopenia (24%), diarrhea (14%), rash (13%), hepatic dysfunction (10%), neutropenia (9%), pleural effusion (8%), vomiting (7%), anemia (6%), and abdominal pain (6%).
The most common adverse reactions, in ≥20% of patients in the safety population of the single-arm trial in patients with CML (N=546) who were resistant or intolerant to prior therapy were diarrhea (83%), nausea (47%), rash (46%), abdominal pain (45%), vomiting (39%), fatigue (33%), pyrexia (28%), hepatic dysfunction (27%), respiratory tract infection (24%), cough (23%), and headache (21%).
The most common laboratory abnormalities that worsened from baseline in ≥20% were creatinine increased (93%), hemoglobin decreased (91%), lymphocyte decreased (80%), platelets decreased (69%), absolute neutrophil count (54%), ALT increased (53%), calcium decreased (53%), white blood cell count decreased (52%), urate increased (48%), AST increased (47%), phosphorus decreased (39%), alkaline phosphatase increased (39%), lipase increased (28%), magnesium increased (25%), potassium decreased (24%), potassium increased (23%). See Table 7 for Grade 3/4 laboratory abnormalities.
Table 6 identifies adverse reactions greater than or equal to 10% for All Grades and Grades 3 or 4 for the Phase 1/2 CML safety population based on long-term follow-up.
Table 6: Adverse Reactions (10% or Greater) in Patients With CML Who Were Resistant or Intolerant to Prior Therapy in Single-Arm Trial* System Organ Class Preferred Term CP CML
(N=403)AdvP CML
(N=143)All Grades
%Grade 3/4
%All Grades
%Grade 3/4
%ADR Definition Adverse drug reactions are based on all-causality treatment-emergent adverse events. The commonality stratification is based on 'All Grades' under Total column. 'Grade 3', 'Grade 4' columns indicate maximum toxicity. - *
- Based on a Minimum of 105 Months of Follow-up.
- †
- Abdominal pain includes the following preferred terms: Abdominal discomfort, Abdominal pain, Abdominal pain lower, Abdominal pain upper, Abdominal tenderness, Dyspepsia, Epigastric discomfort, Gastrointestinal pain, Hepatic pain.
- ‡
- Rash includes the following preferred terms: Acarodermatitis, Acne, Angular cheilitis, Blister, Dermatitis, Dermatitis acneiform, Dermatitis psoriasiform, Drug eruption, Eczema, Eczema asteatotic, Erythema, Erythema annulare, Exfoliative rash, Lichenoid keratosis, Palmar erythema, Photosensitivity reaction, Pigmentation disorder, Psoriasis, Pyoderma gangrenosum, Pyogenic granuloma, Rash, Rash erythematous, Rash generalised, Rash macular, Rash maculo-papular, Rash pruritic, Rash pustular, Seborrhoeic dermatitis, Seborrhoeic keratosis, Skin depigmentation, Skin discoloration, Skin disorder, Skin exfoliation, Skin hyperpigmentation, Skin hypopigmentation, Skin irritation, Skin lesion, Skin plaque, Skin toxicity, Stasis dermatitis.
- §
- Edema includes the following preferred terms: Eye edema, Eyelid edema, Face edema, Generalized edema, Localized edema, Edema, Edema peripheral, Penile edema, Periorbital edema, Periorbital swelling, Peripheral swelling, Scrotal edema, Scrotal swelling, Swelling, Swelling face, Swelling of eyelid, Testicular edema, Tongue edema.
- ¶
- Chest pain includes the following preferred terms: Chest discomfort, Chest pain.
- #
- Hepatic dysfunction includes the following preferred terms: Alanine aminotransferase increased, Aspartate aminotransferase increased, Bilirubin conjugated increased, Blood alkaline phosphatase increased, Blood bilirubin increased, Blood bilirubin unconjugated increased, Gamma-glutamyltransferase increased, Hepatic enzyme increased, Hepatic function abnormal, Hepatic steatosis, Hepatitis toxic, Hepatomegaly, Hepatotoxicity, Hyperbilirubinemia, Liver disorder, Liver function test abnormal, Liver function test increased, Transaminases increased.
- Þ
- Respiratory tract infection includes the following preferred terms: Nasopharyngitis, Respiratory tract congestion, Respiratory tract infection, Respiratory tract infection viral, Upper respiratory tract infection, Viral upper respiratory tract infection.
- ß
- Influenza includes the following preferred terms: H1N1 influenza, Influenza.
- à
- Pneumonia includes the following preferred terms: Atypical pneumonia, Lower respiratory tract congestion, Lower respiratory tract infection, Pneumonia, Pneumonia aspiration, Pneumonia bacterial, Pneumonia fungal, Pneumonia necrotising, Pneumonia streptococcal.
- è
- Hypertension* includes the following preferred terms: Blood pressure increased, Blood pressure systolic increased, Essential hypertension, Hypertension, Hypertensive crisis, Retinopathy hypertensive.
* ADR identified post-marketing.
Gastrointestinal disorders Diarrhea 85 10 76 4 Abdominal pain† 49 2 36 7 Nausea 47 1 48 2 Vomiting 38 3 43 3 Constipation 15 <1 17 1 Skin and subcutaneous tissue disorders Rash‡ 48 9 42 5 Pruritus 12 1 7 0 General disorders and administration-site conditions Fatigue 35 3 27 6 Pyrexia 25 1 37 3 Edema§ 19 <1 17 1 Chest pain¶ 8 1 12 1 Hepatobiliary disorders Hepatic dysfunction# 29 11 21 10 Infections and infestations Respiratory tract infectionÞ 27 <1 17 0 Influenzaß 11 1 3 0 Pneumoniaà 10 4 18 12 Respiratory, thoracic, and mediastinal disorders Cough 24 0 22 0 Pleural effusion 14 4 9 4 Dyspnea 12 2 20 6 Nervous system disorders Headache 21 1 18 4 Dizziness 11 0 14 1 Musculoskeletal and connective tissue disorders Arthralgia 19 1 15 0 Back pain 14 1 8 1 Metabolism and nutrition disorders Decreased appetite 14 1 14 0 Vascular disorders Hypertensionè 11 3 8 3 In the single-arm study in patients with CML who were resistant or intolerant to prior therapy, 2 patients (0.4%) experienced QTcF interval of greater than 500 milliseconds. Patients with uncontrolled or significant cardiovascular disease including QT interval prolongation were excluded by protocol.
Table 7 identifies the clinically relevant or severe Grade 3/4 laboratory test abnormalities for the safety population of the study in patients with CML who were resistant or intolerant to prior therapy based on long-term follow-up.
Table 7: Number (%) of Patients With Clinically Relevant All Grade or Grade 3/4 Laboratory Test Abnormalities in the Safety Population of the Study of Patients With CML Who Were Resistant or Intolerant to Prior Therapy* CP CML
N=403
%AdvP CML
N=143
%All grade Grade 3/4 All grade Grade 3/4 Abbreviations: AdvP=advanced phase; ALT=alanine aminotransferase; AST=aspartate aminotransferase; CML=chronic myelogenous leukemia; CP=chronic phase; N/n=number of patients; SGPT=serum glutamate-pyruvate transaminase; SGOT=serum glutamate-oxaloacetate aminotransferase; ULN=upper limit of normal. - *
- Based on a Minimum of 105 Months of Follow-up.
Hematology Parameters Platelet Count decreased 66 26 80 57 Absolute Neutrophil Count decreased 50 16 66 39 Hemoglobin decreased 89 13 97 38 Lymphocyte decreased 79 14 82 21 White Blood Cell Count decreased 51 7 57 27 Biochemistry Parameters SGPT/ALT increased 58 11 39 6 SGOT/AST increased 50 5 37 3.5 Lipase increased 32 12 19 6 Phosphorus decreased 41 8 33 7 Total Bilirubin increased 16 0.7 22 2.8 Creatinine increased 95 3 87 1.4 Alkaline Phosphatase increased 39 0 39 1.4 Glucose increased 42 2.7 39 6 Sodium increased 23 0.5 11 0 Sodium decreased 18 2.2 27 6 Calcium decreased 55 4.7 45 3.5 Urate increased 49 6 43 6 Magnesium increased 27 7 18 4.9 Potassium decreased 22 1.7 29 4.9 Potassium increased 25 2.7 19 2.1 Additional Adverse Reactions From Multiple Clinical Trials
The following adverse reactions were reported in patients in clinical trials with BOSULIF (less than 10% of BOSULIF-treated patients). They represent an evaluation of the adverse reaction data from all 1372 patients with leukemia who received at least 1 dose of single-agent BOSULIF. These adverse reactions are presented by system organ class and are ranked by frequency. These adverse reactions are included based on clinical relevance and ranked in order of decreasing seriousness within each category.
Blood and Lymphatic System Disorders: 0.1% and less than 1% - Febrile neutropenia
Cardiac Disorders: 1% and less than 10% - Pericardial effusion; 0.1% and less than 1% - Pericarditis
Ear and Labyrinth Disorders: 1% and less than 10% - Tinnitus
Endocrine Disorders: 1% and less than 10% - Hypothyroidism; 0.1% and less than 1% - Hyperthyroidism
Gastrointestinal Disorders: 1% and less than 10% - Gastritis, Pancreatitis (includes Edematous pancreatitis, Pancreatic enzymes increased, Pancreatitis, Pancreatitis acute, Pancreatitis chronic), Gastrointestinal hemorrhage (includes Anal hemorrhage, Gastric hemorrhage, Gastrointestinal hemorrhage, Intestinal hemorrhage, Lower gastrointestinal hemorrhage, Rectal hemorrhage, Upper gastrointestinal hemorrhage)
General Disorders and Administrative Site Conditions: 1% and less than 10% - Pain
Immune System Disorders: 1% and less than 10% - Drug hypersensitivity; 0.1% and less than 1% - Anaphylactic shock
Infections and Infestations: 1% and less than 10% - Bronchitis
Investigations: 1% and less than 10% - Electrocardiogram QT prolonged (includes Electrocardiogram QT prolonged, Long QT syndrome)
Metabolism and Nutrition Disorders: 1% and less than 10% - Dehydration
Musculoskeletal and Connective Tissue Disorders: 1% and less than 10% - Myalgia
Nervous System Disorders: 1% and less than 10% - Dysgeusia
Renal and Urinary Disorders: 1% and less than 10% - Acute kidney injury, Renal impairment, Renal failure
Respiratory, Thoracic and Mediastinal Disorders: 1% and less than 10% - Pulmonary hypertension (includes Pulmonary hypertension, Pulmonary arterial hypertension, Pulmonary arterial pressure increased); 0.1% and less than 1% - Acute pulmonary edema (includes Acute pulmonary edema, Pulmonary edema), Respiratory failure
Skin and Subcutaneous Disorders: 0.1% and less than 1% - Erythema multiforme
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during post-approval use of BOSULIF. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: Thrombotic microangiopathy
Skin and Subcutaneous Tissue Disorders: Stevens-Johnson syndrome
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on BOSULIF
Strong or Moderate CYP3A Inhibitors
Concomitant use with a strong or moderate CYP3A inhibitor increased bosutinib Cmax and AUC compared to BOSULIF alone [see Clinical Pharmacology (12.3)] which may increase the risk of toxicities. Avoid the concomitant use of strong or moderate CYP3A inhibitors with BOSULIF.
Strong CYP3A Inducers
Concomitant use with a strong CYP3A inducer decreased bosutinib Cmax and AUC compared to BOSULIF alone [see Clinical Pharmacology (12.3)] which may reduce BOSULIF efficacy. Avoid the concomitant use of strong CYP3A inducers with BOSULIF.
Proton Pump Inhibitors (PPI)
Concomitant use with a PPI decreased bosutinib Cmax and AUC compared to BOSULIF alone [see Clinical Pharmacology (12.3)] which may reduce BOSULIF efficacy. As an alternative to PPIs, use short-acting antacids or H2 blockers and separate dosing by more than 2 hours from BOSULIF dosing.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, BOSULIF can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)].
There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies conducted in rats and rabbits, oral administration of bosutinib during organogenesis caused adverse developmental outcomes, including structural abnormalities, embryo-fetal mortality, and alterations to growth at maternal exposures (AUC) as low as 1.2 times the human exposure at the dose of 500 mg/day (see Data). Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2–4% and 15–20%, respectively.
Data
Animal Data
In a rat fertility and early embryonic development study, bosutinib was administered orally to female rats for approximately 3 to 6 weeks, depending on day of mating (2 weeks prior to cohabitation with untreated breeder males until gestation day [GD] 7). Increased embryonic resorptions occurred at greater than or equal to 10 mg/kg/day of bosutinib (1.6 and 1.2 times the human exposure at the recommended doses of 400 or 500 mg/day, respectively), and decreased implantations and reduced number of viable embryos at 30 mg/kg/day of bosutinib (3.4 and 2.5 times the human exposure at the recommended doses of 400 or 500 mg/day, respectively).
In an embryo-fetal development study conducted in rabbits, bosutinib was administered orally to pregnant animals during the period of organogenesis at doses of 3, 10, and 30 mg/kg/day. At the maternally-toxic dose of 30 mg/kg/day of bosutinib, there were fetal anomalies (fused sternebrae, and 2 fetuses had various visceral observations), and an approximate 6% decrease in fetal body weight. The dose of 30 mg/kg/day resulted in exposures (AUC) approximately 5.1 and 3.8 times the human exposures at the recommended doses of 400 and 500 mg/day, respectively.
Fetal exposure to bosutinib-derived radioactivity during pregnancy was demonstrated in a placental-transfer study in pregnant rats. In a rat pre- and postnatal development study, bosutinib was administered orally to pregnant animals during the period of organogenesis through lactation day 20 at doses of 10, 30, and 70 mg/kg/day. Reduced number of pups born occurred at greater than or equal to 30 mg/kg/day bosutinib (3.4 and 2.5 times the human exposure at the recommended doses of 400 or 500 mg/day, respectively), and increased incidence of total litter loss and decreased growth of offspring after birth occurred at 70 mg/kg/day bosutinib (6.9 and 5.1 times the human exposure at the recommended doses of 400 or 500 mg/day, respectively).
8.2 Lactation
Risk Summary
No data are available regarding the presence of bosutinib or its metabolites in human milk or its effects on a breastfed child or on milk production. However, bosutinib is present in the milk of lactating rats. Because of the potential for serious adverse reactions in a nursing child, breastfeeding is not recommended during treatment with BOSULIF and for at least 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy
Based on findings from animal studies, BOSULIF can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Females of reproductive potential should have a pregnancy test prior to starting treatment with BOSULIF.
Contraception
Females
Based on findings from animal studies, BOSULIF can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception (methods that result in less than 1% pregnancy rates) during treatment with BOSULIF and for at least 2 weeks after the last dose.
Infertility
The risk of infertility in females or males of reproductive potential has not been studied in humans. Based on findings from animal studies, BOSULIF may cause reduced fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of BOSULIF in patients less than 18 years of age have not been established.
8.5 Geriatric Use
In the single-arm study in patients with CML who were resistant or intolerant to prior therapy of BOSULIF in patients with Ph+ CML, 20% were age 65 and over, 4% were 75 and over. Of the 268 patients who received bosutinib in the study for newly diagnosed CML, 20% were age 65 and over, 5% were 75 and over. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
Reduce the BOSULIF starting dose in patients with moderate (creatinine clearance [CLcr] 30 to 50 mL/min, estimated by Cockcroft-Gault (C-G)) and severe (CLcr less than 30 mL/min, C-G) renal impairment at baseline. For patients who have declining renal function while on BOSULIF who cannot tolerate the starting dose, follow dose adjustment recommendations for toxicity [see Dosage and Administration (2.3, 2.5) and Clinical Pharmacology (12.3)]. BOSULIF has not been studied in patients undergoing hemodialysis.
8.7 Hepatic Impairment
Reduce the BOSULIF dosage in patients with hepatic impairment (Child-Pugh A, B, or C) [see Dosage and Administration (2.3, 2.5) and Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
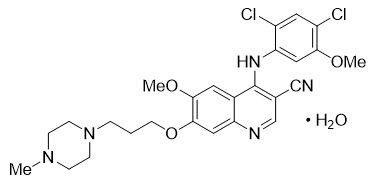
Bosutinib is a kinase inhibitor. The chemical name for bosutinib monohydrate is 3-Quinolinecarbonitrile, 4-[(2,4-dichloro-5-methoxyphenyl)amino]-6-methoxy-7-[3-(4-methyl-1-piperazinyl) propoxy]-, hydrate (1:1). Its chemical formula is C26H29Cl2N5O3∙H2O (monohydrate); its molecular weight is 548.46 (monohydrate), equivalent to 530.46 (anhydrous). Bosutinib monohydrate has the following chemical structure:
Bosutinib monohydrate is a white to yellowish-tan powder. Bosutinib monohydrate has a pH dependent solubility across the physiological pH range. At or below pH 5, bosutinib monohydrate behaves as a highly soluble compound. Above pH 5, the solubility of bosutinib monohydrate reduces rapidly.
BOSULIF® (bosutinib) tablets are supplied for oral administration in 3 strengths: a 100 mg yellow, oval, biconvex, film-coated tablet debossed with "Pfizer" on one side and "100" on the other; a 400 mg orange, oval, biconvex, film-coated tablet debossed with "Pfizer" on one side and "400" on the other; and a 500 mg red, oval, biconvex, film-coated tablet debossed with "Pfizer" on one side and "500" on the other.
Each 100 mg BOSULIF tablet contains 103.40 mg of bosutinib monohydrate, equivalent to 100 mg of bosutinib; each 400 mg BOSULIF tablet contains 413.60 mg of bosutinib monohydrate, equivalent to 400 mg of bosutinib; each 500 mg BOSULIF tablet contains 516.98 mg of bosutinib monohydrate, equivalent to 500 mg of bosutinib. The following inactive ingredients are included in the tablets: microcrystalline cellulose, croscarmellose sodium, poloxamer, povidone, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow (for 100 mg, and 400 mg tablet) and iron oxide red (for 400 mg, and 500 mg tablet).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bosutinib is a TKI. Bosutinib inhibits the BCR-ABL kinase that promotes CML; it is also an inhibitor of Src-family kinases including Src, Lyn, and Hck. Bosutinib inhibited 16 of 18 imatinib-resistant forms of BCR-ABL kinase expressed in murine myeloid cell lines. Bosutinib did not inhibit the T315I and V299L mutant cells.
12.2 Pharmacodynamics
Based on the exposure response analyses for efficacy, a relationship between drug exposure and a greater likelihood of response was observed in clinical studies. Based on the exposure response analyses for safety, a relationship between drug exposure and a greater likelihood of safety events was observed in clinical studies.
12.3 Pharmacokinetics
The bosutinib pharmacokinetics following oral dosing was assessed with food, unless otherwise specified.
Bosutinib exhibits dose proportional increases in AUC and Cmax over the oral dose range of 200 to 800 mg (0.33 to 1.3 times the maximum approved recommended dosage of 600 mg). The mean (standard deviation [SD]) Cmax was 146 (20) ng/mL and the mean (SD) AUC was 2720 (442) ng∙h/mL following multiple oral doses of BOSULIF 400 mg in patients with CML; Cmax was 200 (12) ng/mL and AUC was 3650 (425) ng∙h/mL following multiple oral doses of BOSULIF 500 mg in patients with CML.
Absorption
Following administration of a single oral dose of BOSULIF 500 mg with food in patients with CML, the median (minimum, maximum) time-to-peak concentration (tmax) was 6.0 (6.0, 6.0) hours. The absolute bioavailability was 34% in healthy subjects.
Distribution
Following a single intravenous dose of bosutinib 120 mg (0.2 times the maximum approved recommended oral dosage of 600 mg) in healthy subjects, bosutinib had a mean (SD) volume of distribution of 2441 (796) L. The mean (SD) apparent volume of distribution after an oral dose of 500 mg of BOSULIF to patients with CML was 6080 (1230) L. Protein binding of bosutinib is 94% in vitro and 96% ex vivo, and is independent of concentration.
Elimination
Following a single intravenous dose of bosutinib 120 mg (0.2 times the maximum approved recommended oral dosage of 600 mg), the mean (SD) terminal phase elimination half-life (t½) was 35.5 (8.5) hours, and the mean (SD) clearance (Cl) was 63.6 (14.1) L/h. Following a single oral dose of BOSULIF in patients with CML, the mean (SD) t½ was 22.5 (1.7) hours, and the mean (SD) Cl was 189 (48) L/h.
Specific Populations
Patients with Renal Impairment
Following a single oral dose of BOSULIF 200 mg (0.33 times the maximum approved recommended dosage of 600 mg), bosutinib AUC increased 1.4-fold in subjects with moderate renal impairment (CLcr: 30 to 50 mL/min, estimated by Cockcroft-Gault (C-G)) and increased 1.6-fold in subjects with severe renal impairment (CLcr less than 30 mL/min) compared to subjects with normal renal function (CLcr > 80 mL/min, C-G). No clinically significant difference in the pharmacokinetics of bosutinib was observed in subjects with mild renal impairment (CLcr: 51 to 80 mL/min, C-G).
Patients with Hepatic Impairment
Following a single oral dose of BOSULIF 200 mg (0.33 times the maximum approved recommended dosage of 600 mg), bosutinib Cmax increased 2.4-fold, 2-fold, and 1.5-fold, and AUC increased 2.3-fold, 2-fold, and 1.9-fold in hepatic impairment Child-Pugh A, B, and C, respectively.
Drug Interaction Studies
Clinical Studies
The following interactions were evaluated in crossover studies of healthy subjects, unless otherwise specified.
Strong and Moderate CYP3A Inhibitors
A single oral dose of BOSULIF 100 mg (0.17 times the maximum approved recommended dosage) was administered alone or following multiple daily doses of 400 mg ketoconazole (a strong CYP3A inhibitor) without food. Ketoconazole increased bosutinib Cmax and AUC 5.2-fold and 8.6-fold, respectively.
A single oral dose of BOSULIF 500 mg was administered alone or in combination with 125 mg aprepitant (a moderate CYP3A inhibitor) with food. Aprepitant increased bosutinib Cmax 1.5-fold and AUC 2.0-fold.
Strong CYP3A Inducers
A single dose of BOSULIF 500 mg was administered alone or following multiple daily doses of 600 mg rifampin with food. Rifampin decreased bosutinib Cmax by 86% and AUC by 94%.
Proton Pump Inhibitors
BOSULIF displays pH-dependent aqueous solubility, in vitro. A single oral dose of BOSULIF 400 mg was administered alone or following multiple oral doses of lansoprazole 60 mg without food. Lansoprazole decreased bosutinib Cmax by 46% and AUC by 26%.
P-gp Substrates
A single oral dose of 500 mg BOSULIF was administered in combination with a single oral dose of 150 mg dabigatran etexilate mesylate (a P-glycoprotein (P-gp) substrate). No clinically significant difference in the pharmacokinetics of dabigatran was observed following bosutinib administration.
In Vitro Studies
Bosutinib Effect on Transporters
Bosutinib may have the potential to inhibit breast cancer resistance protein (BCRP) in the gastrointestinal tract but has a low potential to inhibit BCRP, systemically, or organic anion transporting polypeptide (OATP)1B1, OATP1B3, organic anion transporter (OAT)1, OAT3, organic cation transporter (OCT)1, and OCT2 at clinically relevant concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Bosutinib was not carcinogenic in rats or transgenic mice. The rat 2-year carcinogenicity study was conducted at bosutinib oral doses up to 25 mg/kg in males and 15 mg/kg in females. Exposures at these doses were approximately 1.5 times (males) and 3.1 times (females) the human exposure at the 400 mg dose and 1.2 times (males) and 2.4 times (females) exposure in humans at the 500 mg dose. The 6-month RasH2 transgenic mouse carcinogenicity study was conducted at bosutinib oral doses up to 60 mg/kg.
Bosutinib was not mutagenic or clastogenic in a battery of tests, including the bacteria reverse mutation assay (Ames Test), the in vitro assay using human peripheral blood lymphocytes and the micronucleus test in orally treated male mice.
In a rat fertility study, drug-treated males were mated with untreated females, or untreated males were mated with drug-treated females. Females were administered the drug from pre-mating through early embryonic development. The dose of 70 mg/kg/day of bosutinib resulted in reduced fertility in males as demonstrated by 16% reduction in the number of pregnancies. There were no lesions in the male reproductive organs at this dose. This dose of 70 mg/kg/day resulted in exposure (AUC) in male rats approximately 1.5 times and equal to the human exposure at the recommended doses of 400 and 500 mg/day, respectively. Fertility (number of pregnancies) was not affected when female rats were treated with bosutinib. However, there were increased embryonic resorptions at greater than or equal to 10 mg/kg/day of bosutinib (1.6 and 1.2 times the human exposure at the recommended doses of 400 and 500 mg/day, respectively), and decreased implantations and reduced number of viable embryos at 30 mg/kg/day of bosutinib (3.4 and 2.5 times the human exposure at the recommended doses of 400 or 500 mg/day, respectively).
-
14 CLINICAL STUDIES
14.1 Newly-Diagnosed CP Ph+ CML
The efficacy of BOSULIF in patients with newly-diagnosed chronic phase Ph+ CML was evaluated in the Bosutinib trial in First-line chrOnic myelogenous leukemia tREatment (BFORE) Trial: "A Multicenter Phase 3, Open-Label Study of Bosutinib Versus Imatinib in Adult Patients With Newly Diagnosed Chronic Phase Chronic Myelogenous Leukemia" [NCT02130557].
The BFORE Trial is a 2-arm, open-label, randomized, multicenter trial conducted to investigate the efficacy and safety of BOSULIF 400 mg once daily alone compared with imatinib 400 mg once daily alone in adult patients with newly-diagnosed CP Ph+ CML. The trial randomized 536 patients (268 in each arm) with Ph+ or Ph- newly-diagnosed CP CML (intent-to-treat [ITT] population) including 487 patients with Ph+ CML harboring b2a2 and/or b3a2 transcripts at baseline and baseline BCR-ABL copies >0 (modified intent-to-treat [mITT] population). Randomization was stratified by Sokal score and geographical region. All patients are being treated and/or followed for up to 5 years (240 weeks). Efficacy was evaluated in the mITT population. The major efficacy outcome measure was major molecular response (MMR) at 12 months (48 weeks) defined as ≤0.1% BCR-ABL ratio on international scale (corresponding to ≥3 log reduction from standardized baseline) with a minimum of 3000 ABL transcripts as assessed by the central laboratory. Additional efficacy outcomes included CCyR by 12 months, defined as the absence of Ph+ metaphases in chromosome banding analysis of ≥20 metaphases derived from bone marrow aspirate or MMR if an adequate cytogenetic assessment was unavailable and MMR by 18 months (72 weeks).
In the mITT population in this study, 57% of patients were males, 78% were Caucasian, and 19% were 65 years or older. The median age was 53 years. At baseline, the distribution of Sokal risk scores was similar in bosutinib and imatinib-treated patients (low risk: 35% and 39%; intermediate risk: 44% and 38%; high risk: 22% and 22%, respectively). After a minimum of 12 months follow-up, 78% of the 246 bosutinib-treated patients and 72% of the 239 imatinib-treated patients were still receiving treatment and with a minimum of 60 months of follow-up, 60% and 60% of patients, respectively, were still receiving treatment. The median treatment duration was 55.1 months for BOSULIF and 55.0 months for imatinib.
The efficacy results from the BFORE trial are summarized in Table 8.
Table 8: Summary of Major Molecular Response (MMR) and Complete Cytogenetic Response (CCyR), by Treatment Group in the Modified Intent-to-Treat (mITT) Population Response Bosutinib
N=246
n (%)Imatinib
N=241
n (%)2-sided p-value Abbreviations: CCyR=complete cytogenetic response; CI=confidence interval; CMH=Cochran-Mantel-Haenszel; MMR=major molecular response; N/n=number of patients. - *
- Derived from CMH test stratified by Geographical region and Sokal score at randomization.
MMR at Month 12 (Week 48) MMR (%) 116 (47) 89 (37) 0.0200* (95% CI) (41, 53) (31, 43) CCyR by Month 12 (Week 48) CCyR (%) 190 (77) 160 (66) (95% CI) (72, 83) (60, 72) 0.0075* MMR by Month 18 (Week 72) MMR (%) 150 (61) 127 (53) (95% CI) (55, 67) (46, 59) 0.0606* The MMR rate at Month 12 for all randomized patients (ITT population) was consistent with the mITT population (47% [95% CI: 41, 53] in the bosutinib treatment group and 36% [95% CI: 30, 42] in the imatinib treatment group; odds ratio of 1.57 [95% CI: 1.10, 2.22]). MMR by Month 60 (Week 240) in the mITT population was 74% (95% CI: 69, 80) in the bosutinib treatment group and 66% (95% CI: 60, 72) in the imatinib treatment group; odds ratio of 1.52 (95% CI: 1.02, 2.25). MMR by Month 60 in the ITT population was also consistent with the mITT population (1.57 [95% CI: 1.08, 2.28]).
After 60 months of follow-up, the median time to MMR in responders was 9.0 months for bosutinib and 11.9 months for imatinib.
By 60 months, the MMR rates in each Sokal risk group for the bosutinib and imatinib-treated patients, respectively, were 78% and 72% for low risk, 74% and 67% for intermediate risk and 68% and 52% for high risk.
After 60 months of follow-up, 6 (2%) bosutinib patients and 7 (3%) imatinib patients transformed to AP CML or BP CML while on treatment.
At 60 months, the estimated overall survival rate was 95% (95% CI: 91, 97) in the bosutinib group and 94% (95% CI: 90, 96) in the imatinib group.
14.2 Imatinib-Resistant or -Intolerant Ph+ CP, AP, and BP CML
Study 200 (NCT00261846), a single-arm, open-label, multicenter study in patients with CML who were resistant or intolerant to prior therapy was conducted to evaluate the efficacy and safety of BOSULIF 500 mg once daily in patients with imatinib-resistant or -intolerant CML with separate cohorts for CP, AP, and BP disease previously treated with 1 prior TKI (imatinib) or more than 1 TKI (imatinib followed by dasatinib and/or nilotinib). The definition of imatinib resistance included (1) failure to achieve or maintain any hematologic improvement within 4 weeks; (2) failure to achieve a CHR by 3 months, cytogenetic response by 6 months or major cytogenetic response (MCyR) by 12 months; (3) progression of disease after a previous cytogenetic or hematologic response; or (4) presence of a genetic mutation in the BCR-ABL gene associated with imatinib resistance. Imatinib intolerance was defined as inability to tolerate imatinib due to toxicity, or progression on imatinib and inability to receive a higher dose due to toxicity. The definitions of resistance and intolerance to both dasatinib and nilotinib were similar to those for imatinib. The protocol was amended to exclude patients with a known history of the T315I mutation after 396 patients were enrolled in the trial.
The efficacy endpoints for patients with CP CML previously treated with 1 prior TKI (imatinib) were the rate of attaining MCyR by Week 24 and the duration of MCyR. The efficacy endpoints for patients with CP CML previously treated with both imatinib and at least 1 additional TKI were the cumulative rate of attaining MCyR by Week 24 and the duration of MCyR. The efficacy endpoints for patients with previously treated AP and BP CML were confirmed CHR and overall hematologic response (OHR).
The study enrolled 546 patients with CP, AP or BP CML. Of the total patient population 73% were imatinib resistant and 27% were imatinib intolerant. In this trial, 53% of patients were males, 65% were Caucasian, and 20% were 65 years old or older. Of the 546 treated patients, 506 were considered evaluable for cytogenetic or hematologic efficacy assessment. Patients were evaluable for efficacy if they had received at least 1 dose of BOSULIF and had a valid baseline efficacy assessment. Among evaluable patients, there were 262 patients with CP CML previously treated with 1 prior TKI (imatinib), 112 patients with CP CML previously treated with both imatinib and at least 1 additional TKI, and 132 patients with advanced phase CML previously treated with at least 1 TKI.
Median duration of BOSULIF treatment was 26 months in patients with CP CML previously treated with 1 TKI (imatinib), 9 months in patients with CP CML previously treated with imatinib and at least 1 additional TKI, 10 months in patients with AP CML previously treated with at least imatinib, and 3 months in patients with BP CML previously treated with at least imatinib.
The 24 week efficacy and MCyR at any time results are summarized in Table 9.
Table 9: Efficacy Results in Patients with Ph+ CP CML With Resistance to or Intolerance to Imatinib Prior Treatment With Imatinib Only
(N=262 evaluable)
n (%)Prior Treatment With Imatinib and Dasatinib or Nilotinib
(N=112 evaluable)
n (%)Abbreviations: CI=confidence interval; CML=chronic myelogenous leukemia; CP=chronic phase; MCyR=major cytogenetic response; N/n=number of patients; Ph+=Philadelphia chromosome positive. By Week 24 MCyR 105 (40.1) 29 (25.9) (95% CI) (34.1, 46.3) (18.1, 35.0) MCyR any time 156 (59.5) 45 (40.2) (53.3, 65.5) (31.0, 49.9) The long term follow-up data analysis was based on a minimum of 60 months for patients with CP CML treated with 1 prior TKI (imatinib) and a minimum of 48 months for patients with CP CML treated with imatinib and at least 1 additional TKI. For the 59.5% of patients with CP CML treated with 1 prior TKI (imatinib) who achieved a MCyR at any time, the median duration of MCyR was not reached. Among these patients, 65.4% and 42.9% had a MCyR lasting at least 18 and 54 months, respectively. For the 40.2% of patients with CP CML treated with imatinib and at least 1 additional TKI who achieved a MCyR at any time, the median duration of MCyR was not reached. Among these patients, 64.4% and 35.6% had a MCyR lasting at least 9 and 42 months, respectively. Of the 403 treated patients with CP CML, 20 patients had confirmed disease transformation to AP or BP while on treatment with BOSULIF.
The 48 week efficacy results in patients with accelerated and blast phases CML previously treated with at least imatinib are summarized in Table 10.
Table 10: Efficacy Results in Patients With Accelerated Phase and Blast Phase CML Previously Treated With at Least Imatinib AP CML
(N=72 evaluable)
n (%)BP CML
(N=60 evaluable)
n (%)Abbreviations: AP=accelerated phase; BP=blast phase; CHR=complete hematologic response; CI=confidence interval; CML=chronic myelogenous leukemia; CI=confidence interval, OHR=overall hematologic response, CHR=complete hematologic response, N/n=number of patients - *
- Overall hematologic response (OHR) = major hematologic response (complete hematologic response + no evidence of leukemia) or return to chronic phase (RCP). All responses were confirmed after 4 weeks. Complete hematologic response (CHR) for AP and BP CML: WBC less than or equal to institutional ULN, platelets greater than or equal to 100,000/mm3 and less than 450,000/mm3, absolute neutrophil count (ANC) greater than or equal to 1.0×109 /L, no blasts or promyelocytes in peripheral blood, less than 5% myelocytes + metamyelocytes in bone marrow, less than 20% basophils in peripheral blood, and no extramedullary involvement. No evidence of leukemia (NEL): Meets all other criteria for CHR except may have thrombocytopenia (platelets greater than or equal to 20,000/mm3 and less than 100,000/mm3) and/or neutropenia (ANC greater than or equal to 0.5×109 /L and less than 1.0×109 /L). Return to chronic phase (RCP) = disappearance of features defining accelerated or blast phases but still in chronic phase.
CHR* by Week 48 22 (30.6) 10 (16.7) (95% CI) (20.2, 42.5) (8.3, 28.5) OHR* by Week 48 41 (56.9) 17 (28.3) (95% CI) (44.7, 68.6) (17.5, 41.4) The long term follow-up data analysis was based on a minimum of 48 months for patients with AP CML and BP CML. Of the 79 treated patients with AP CML, 3 patients had confirmed disease transformation to BP while on BOSULIF treatment.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
BOSULIF (bosutinib) tablets are supplied for oral administration in 3 strengths: a 100 mg yellow, oval, biconvex, film-coated tablet debossed with "Pfizer" on one side and "100" on the other; a 400 mg orange, oval, biconvex, film coated tablet debossed with "Pfizer" on one side and "400" on the other; and a 500 mg red, oval, biconvex, film-coated tablet debossed with "Pfizer" on one side and "500" on the other. BOSULIF (bosutinib) tablets are available in the following packaging configurations (Table 11):
Table 11: Tablet Presentations BOSULIF Tablets Package Configuration Tablet Strength
(mg)NDC Tablet Description Abbreviation: NDC=National drug code. 120 tablets per bottle 100 mg 0069-0135-01 Yellow, oval, biconvex, film-coated tablets, debossed "Pfizer" on one side and "100" on the other. 30 tablets per bottle 400 mg 0069-0193-01 Orange, oval, biconvex, film-coated tablet debossed with "Pfizer" on one side and "400" on the other. 30 tablets per bottle 500 mg 0069-0136-01 Red, oval, biconvex, film-coated tablets, debossed "Pfizer" on one side and "500" on the other. -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
• Dosage and Administration
Instruct patients to take BOSULIF exactly as prescribed, not to change their dose or to stop taking BOSULIF unless they are told to do so by their doctor. If patients miss a dose beyond 12 hours, they should be advised to take the next scheduled dose at its regular time. A double dose should not be taken to make up for any missed dose. Advise patients to take BOSULIF with food. Patients should be advised: "Do not crush, break, or cut tablet. Do not touch or handle crushed or broken tablets."
• Gastrointestinal Toxicity
Advise patients that they may experience diarrhea, nausea, vomiting, abdominal pain, or blood in their stools with BOSULIF and to seek medical attention promptly for these symptoms [see Warnings and Precautions (5.1)].
• Myelosuppression
Advise patients of the possibility of developing low blood cell counts and to immediately report fever, any suggestion of infection, or signs or symptoms suggestive of bleeding or easy bruising [see Warnings and Precautions (5.2)].
• Hepatic Toxicity
Advise patients of the possibility of developing liver function abnormalities and to immediately report jaundice [see Warnings and Precautions (5.3)].
• Cardiovascular Toxicity
Advise patients that cardiac failure, left ventricular dysfunction, and cardiac ischemic events have been reported. Advise patients to seek immediate medical attention if any symptoms suggestive of cardiac failure and cardiac ischemia occur, such as shortness of breath, weight gain, or fluid retention [see Warnings and Precautions (5.4)].
• Fluid Retention
Advise patients of the possibility of developing fluid retention (swelling, weight gain, or shortness of breath) and to seek medical attention promptly if these symptoms arise [see Warnings and Precautions (5.5)].
• Renal Toxicity
Advise patients of the possibility of developing renal problems and to immediately report frequent urination, polyuria or oliguria [see Warnings and Precautions (5.6)].
• Adverse Reactions
Advise patients that they may experience other adverse reactions such as respiratory tract infections, rash, fatigue, loss of appetite, headache, dizziness, back pain, arthralgia, or pruritus with BOSULIF and to seek medical attention if symptoms are significant. There is a possibility of anaphylactic shock [see Contraindications (4) and Adverse Reactions (6)].
• Embryo-Fetal Toxicity
Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1)].
Advise females of reproductive potential, to use effective contraception during treatment and for at least 2 weeks after receiving the last dose of BOSULIF [see Warnings and Precautions (5.7) and Use in Specific Populations (8.1, 8.3)].
Advise lactating women not to breastfeed during treatment with BOSULIF and for at least 2 weeks after the last dose [see Use in Specific Populations (8.2)].
• Drug Interactions
Advise patients that BOSULIF and certain other medicines, including over the counter medications or herbal supplements (such as St. John's wort) can interact with each other and may alter the effects of BOSULIF [see Drug Interactions (7)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
BOSULIF® (BAH-su-lif)
(bosutinib)
tabletsThis Patient Information has been approved by the U.S. Food and Drug Administration. Revised: May 2021 What is BOSULIF?
BOSULIF is a prescription medicine used to treat adults who have a certain type of leukemia called Philadelphia chromosome-positive chronic myelogenous leukemia (Ph+ CML) who are newly-diagnosed or who no longer benefit from or did not tolerate other treatment.
It is not known if BOSULIF is safe and effective in children less than 18 years of age.Do not take BOSULIF if you are allergic to bosutinib or any of the ingredients in BOSULIF. See the end of this leaflet for a complete list of ingredients of BOSULIF. Before taking BOSULIF, tell your doctor about all of your medical conditions, including if you: - have liver problems
- have heart problems
- have kidney problems
- have high blood pressure
- have diabetes
- are pregnant or plan to become pregnant. BOSULIF can harm your unborn baby. Females who are able to become pregnant should have a pregnancy test before starting treatment with BOSULIF. Tell your doctor right away if you become pregnant during treatment with BOSULIF.
- Females who are able to become pregnant should use effective birth control (contraception) during treatment with BOSULIF and for at least 2 weeks after the last dose. Talk to your doctor about birth control methods that may be right for you.
- are breastfeeding or plan to breastfeed. It is not known if BOSULIF passes into your breast milk or if it can harm your baby. Do not breastfeed during treatment with BOSULIF and for at least 2 weeks after the last dose.
Know the medicines you take. Keep a list of your medicines and show it to your doctor and pharmacist when you get a new medicine.How should I take BOSULIF? - Take BOSULIF exactly as prescribed by your doctor.
- Do not change your dose or stop taking BOSULIF without first talking with your doctor.
- Take BOSULIF with food.
- Swallow BOSULIF tablets whole. Do not crush, break, or cut BOSULIF tablets. Do not touch or handle crushed or broken BOSULIF tablets.
- If you take an antacid or H2 blocker medicine, take it at least 2 hours before or 2 hours after BOSULIF. If you take a Proton Pump Inhibitor (PPI) medicine, talk to your doctor or pharmacist.
- You should avoid grapefruit, grapefruit juice, and supplements that contain grapefruit extract during treatment with BOSULIF. Grapefruit products increase the amount of BOSULIF in your body.
- If you miss a dose of BOSULIF, take it as soon as you remember. If you miss a dose by more than 12 hours, skip that dose and take your next dose at your regular time. Do not take 2 doses at the same time.
- If you take too much BOSULIF, call your doctor or go to the nearest hospital emergency room right away.
What are the possible side effects of BOSULIF?
BOSULIF may cause serious side effects, including:- Stomach problems. BOSULIF may cause stomach (abdomen) pain, nausea, diarrhea, vomiting, or blood in your stools. Get medical help right away for any stomach problems.
- Low blood cell counts. BOSULIF may cause low platelet counts (thrombocytopenia), low red blood cell counts (anemia) and low white blood cell counts (neutropenia). Your doctor should do blood tests to check your blood cell counts regularly during your treatment with BOSULIF. Call your doctor right away if you have unexpected bleeding or bruising, blood in your urine or stools, fever, or any signs of an infection.
- Liver problems. Your doctor should do blood tests to check your liver function regularly during your treatment with BOSULIF. Call your doctor right away if your skin or the white part of your eyes turns yellow (jaundice) or you have dark "tea color" urine.
- Heart problems. BOSULIF may cause heart problems, including heart failure and decreased blood flow to the heart which can lead to heart attack. Get medical help right away if you get shortness of breath, weight gain, chest pain, or swelling in your hands, ankles or feet.
- Your body may hold too much fluid (fluid retention). Fluid may build up in the lining of your lungs, the sac around your heart, or your stomach cavity. Get medical help right away if you get any of the following symptoms during your treatment with BOSULIF:
- shortness of breath and cough
- chest pain
- swelling in your hands, ankles, or feet
- swelling all over your body
- weight gain
-
Kidney problems. Your doctor should do tests to check your kidney function when you start treatment with BOSULIF and during your treatment. Call your doctor right away if you get any of the following symptoms during your treatment with BOSULIF:
- you urinate more often than normal
- you urinate less often than normal
- you make a much larger amount of urine than normal
- you make a much smaller amount of urine than normal
- diarrhea
- rash
- nausea
- stomach (abdomen) pain
- vomiting
- tiredness
- liver problems
- respiratory tract infections (infections in nose, throat or lungs)
- fever
- headache
- changes in certain blood tests. Your doctor may do blood tests during treatment with BOSULIF to check for changes
Tell your doctor or get medical help right away if you get respiratory tract infections, loss of appetite, headache, dizziness, back pain, joint pain, rash or itching while taking BOSULIF. These may be symptoms of a severe allergic reaction.
Your doctor may change your dose, temporarily stop, or permanently stop treatment with BOSULIF if you have certain side effects.
BOSULIF may cause fertility problems in females and males. This may affect your ability to have a child. Talk to your doctor if this is a concern for you.
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects of BOSULIF.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store BOSULIF? - Store BOSULIF at room temperature between 68°F to 77°F (20°C to 25°C).
- Ask your doctor or pharmacist about the right way to throw away outdated or unused BOSULIF.
General information about the safe and effective use of BOSULIF.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use BOSULIF for a condition for which it is not prescribed. Do not give BOSULIF to other people even if they have the same symptoms you have. It may harm them. You can ask your doctor or pharmacist for information about BOSULIF that is written for health professionals.What are the ingredients in BOSULIF?
Active ingredient: bosutinib.
Inactive ingredients: microcrystalline cellulose, croscarmellose sodium, poloxamer, povidone, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow (for 100 mg and 400 mg tablets) and iron oxide red (for 400 mg and 500 mg tablets).
LAB-0639-11.0
For more information, go to www.Bosulif.com or www.pfizermedicalinformation.com or call 1-800-438-1985. - PRINCIPAL DISPLAY PANEL - 100 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 400 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
BOSULIF
bosutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63539-117 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BOSUTINIB MONOHYDRATE (UNII: 844ZJE6I55) (BOSUTINIB - UNII:5018V4AEZ0) BOSUTINIB 100 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) POVIDONE K25 (UNII: K0KQV10C35) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) POLOXAMER 188 (UNII: LQA7B6G8JG) Product Characteristics Color YELLOW Score no score Shape OVAL (biconvex) Size 11mm Flavor Imprint Code Pfizer;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63539-117-01 120 in 1 BOTTLE; Type 0: Not a Combination Product 09/04/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203341 09/04/2012 BOSULIF
bosutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63539-193 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BOSUTINIB MONOHYDRATE (UNII: 844ZJE6I55) (BOSUTINIB - UNII:5018V4AEZ0) BOSUTINIB 400 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) POVIDONE K25 (UNII: K0KQV10C35) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) POLOXAMER 188 (UNII: LQA7B6G8JG) Product Characteristics Color ORANGE Score no score Shape OVAL (biconvex) Size 18mm Flavor Imprint Code Pfizer;400 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63539-193-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 02/21/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203341 09/04/2012 Labeler - U.S. Pharmaceuticals (829076905) Establishment Name Address ID/FEI Business Operations Pfizer Manufacturing Deutschland GmbH 341970073 ANALYSIS(63539-117, 63539-193) , MANUFACTURE(63539-117, 63539-193) , PACK(63539-117, 63539-193) , LABEL(63539-117, 63539-193) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals 985052076 ANALYSIS(63539-117, 63539-193) , API MANUFACTURE(63539-117, 63539-193)