Label: AMMONUL- sodium phenylacetate and sodium benzoate injection, solution, concentrate


-
Contains inactivated NDC Code(s)
NDC Code(s): 62592-720-50 - Packager: Ucyclyd Pharma Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated January 12, 2018
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AMMONUL ® safely and effectively. See full prescribing information for AMMONUL ®
AMMONUL ® (sodium phenylacetate and sodium benzoate) injection, for
intravenous use
Initial U.S. Approval: 1987INDICATIONS AND USAGE
AMMONUL is a nitrogen binding agent indicated as adjunctive therapy for the treatment of acute hyperammonemia and associated encephalopathy in patients with deficiencies in enzymes of the urea cycle. ( 1)
DOSAGE AND ADMINISTRATION
- AMMONUL must be diluted with sterile 10% Dextrose Injection (D10W) before administration. Administration must be through a central venous catheter. Administration through a peripheral line may cause burns. ( 2)
- AMMONUL is administered intravenously as a loading dose infusion administered over 90 to 120 minutes, followed by an equivalent maintenance dose infusion administered over 24 hours. ( 2)
DOSAGE FORMS AND STRENGTHS
Injection: 10% per 10% sterile, concentrated, aqueous solution of sodium phenylacetate and sodium benzoate. ( 3)
CONTRAINDICATIONS
None ( 4)
WARNINGS AND PRECAUTIONS
- Management of Acute Hyperammonemia: Monitor plasma ammonia levels during treatment. Prolonged exposure to elevated plasma ammonia levels can rapidly result in injury to the brain or death. Prompt use of all therapies necessary to reduce plasma ammonia levels is essential. ( 5.1)
- Decreased Potassium Levels: Plasma potassium levels should be carefully monitored and appropriate treatment given when necessary. ( 5.2)
- Conditions Associated with Fluid Overload: AMMONUL contains 30.5 mg of sodium per mL of undiluted product. Caution should be used if AMMONUL is administered to patients with congestive heart failure, severe renal insufficiency, or with conditions in which there is sodium retention with edema. ( 5.3)
- Extravasation: Extravasation of AMMONUL into the perivenous tissues during high flow bolus infusion may lead to skin necrosis, especially in infants. The infusion site must be monitored closely for possible tissue infiltration during drug administration. ( 5.4)
- Neurotoxicity of Phenylacetate: Because of prolonged plasma levels achieved by phenylacetate in pharmacokinetic studies, repeat loading doses should not be administered. Additionally, neurotoxicity related to phenylacetate has been reported in cancer patients. Monitor patients for symptoms of acute neurotoxicity.( 5.5)
- Hyperventilation and Metabolic Acidosis: AMMONUL may cause side effects typically associated with salicylate overdose, such as hyperventilation and metabolic acidosis. Monitor patient blood chemistry profiles and perform frequent blood pH and pCO 2 measurements. ( 5.6)
ADVERSE REACTIONS
The most frequently reported adverse reactions (incidence ≥ 6%) are vomiting, hyperglycemia, hypokalemia, convulsions, and mental impairment. ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact Ucyclyd Pharma, Inc. at 1-800-321-4576 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Some antibiotics such as penicillin may affect the overall disposition of the infused drug. ( 7)
- Probenecid may affect renal excretion of phenylacetylglutamine and hippurate. ( 7)
- Valproic acid given to patients with urea cycle disorders may exacerbate their condition and antagonize the efficacy of AMMONUL through inhibition of the synthesis of N-acetylglutamate, a co-factor for carbamyl phosphate synthetase. ( 7)
- Use of corticosteroids may cause the breakdown of body protein and potentially increase plasma ammonia levels in patients with impaired ability to form urea ( 7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2013
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Hyperammonemia
5.2 Decreased Potassium Levels
5.3 Conditions Associated with Fluid Overload
5.4 Extravasation
5.5 Neurotoxicity of Phenylacetate
5.6 Hyperventilation and Metabolic Acidosis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Gender
8.7 Hepatic Insufficiency
8.8 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
AMMONUL is indicated as adjunctive therapy in pediatric and adult patients for the treatment of acute hyperammonemia and associated encephalopathy in patients with deficiencies in enzymes of the urea cycle. During acute hyperammonemic episodes, arginine supplementation, caloric supplementation, dietary protein restriction, hemodialysis, and other ammonia lowering therapies should be considered [ see Warnings and Precautions (5)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
AMMONUL must be diluted with sterile 10% Dextrose Injection (D10W) before administration. The dilution and dosage of AMMONUL are determined by weight for neonates, infants and young children, and by body surface area for larger patients, including older children, adolescents, and adults (Table 1).
Table 1 Dosage and Administration Patient Population Components of Infusion Solution
AMMONUL must be diluted with sterile 10% Dextrose Injection at ≥ 25 mL/Kg before administration.Dosage Provided AMMONUL Arginine HCl Injection, 10% Sodium Phenylacetate Sodium Benzoate Arginine HCl Patients 0 to 20 kg: CPS and OTC Deficiency Dose Loading: over 90 to 120 minutes 2.5 mL/kg 2 mL/kg 250 mg/kg 250 mg/kg 200 mg/kg Maintenance: over 24 hours ASS and ASL Deficiency Dose Loading: over 90 to 120 minutes 2.5 mL/kg 6 mL/kg 250 mg/kg 250 mg/kg 600 mg/kg Maintenance: over 24 hours Patients > 20 kg: CPS and OTC Deficiency Dose Loading: over 90 to 120 minutes 55 mL/m 2 2 mL/kg 5.5 g/m 2 5.5 g/m 2 200 mg/kg Maintenance: over 24 hours ASS and ASL Deficiency Dose Loading: over 90 to 120 minutes 55 mL/m 2 6 mL/kg 5.5 g/m 2 5.5 g/m 2 600 mg/kg Maintenance: over 24 hours 2.2 Administration
AMMONUL is a concentrated solution and must be diluted before intravenous administration via a central venous catheter. Administration through a peripheral intravenous catheter may cause burns. AMMONUL may not be administered by any other route.
AMMONUL should be administered as a loading dose infusion over 90 to 120 minutes, followed by the same dose repeated as a maintenance infusion administered over 24 hours. Because of prolonged plasma levels achieved by phenylacetate in pharmacokinetic studies, repeat loading doses of AMMONUL should not be administered. Maintenance infusions may be continued until elevated plasma ammonia levels have been normalized or the patient can tolerate oral nutrition and medications. An antiemetic may be administered during AMMONUL infusion to aid control of infusion-associated nausea and vomiting. Administration of analogous oral drugs, such as sodium phenylbutyrate, should be terminated prior to AMMONUL infusion.
AMMONUL infusion should be started as soon as the diagnosis of hyperammonemia is made. Treatment of hyperammonemia also requires caloric supplementation and restriction of dietary protein. Non-protein calories should be supplied principally as glucose (8–10 mg/kg/min) with an intravenous fat emulsion added. Attempts should be made to maintain a caloric intake of greater than 80 kcal/kg/day. During and after infusion of AMMONUL, ongoing monitoring of the following clinical laboratory values is crucial: plasma ammonia, glutamine, quantitative plasma amino acids, blood glucose, electrolytes, venous or arterial blood gases, AST and ALT. On-going monitoring of the following clinical responses is also crucial to assess patient response to treatment: neurological status, Glasgow Coma Scale, tachypnea, CT or MRI scan or fundoscopic evidence of cerebral edema, and/or of gray matter and white matter damage. Hemodialysis should be considered in patients with severe hyperammonemia or who are not responsive to AMMONUL administration [ see Warnings and Precautions (5.1)]. In the non-neonatal study patient population treated with AMMONUL, dialysis was required in 13% of hyperammonemic episodes. Standard hemodialysis was the most frequently used dialysis method. High levels of ammonia can be reduced quickly when AMMONUL is used with hemodialysis, as the ammonia-scavenging of AMMONUL suppresses the production of ammonia from catabolism of endogenous protein and hemodialysis eliminates the ammonia and ammonia conjugates.
AMMONUL solutions are physically and chemically stable for up to 24 hours at room temperature and room lighting conditions. No compatibility information is presently available for AMMONUL infusion solutions except for Arginine HCl Injection, 10%, which may be mixed in the same container as AMMONUL. Other infusion solutions and drug products should not be administered together with AMMONUL infusion solution. AMMONUL solutions may be prepared in glass and PVC containers.
Arginine Administration
Intravenous arginine is an essential component of therapy for patients with carbamyl phosphate synthetase (CPS), ornithine transcarbamylase (OTC), argininosuccinate synthetase (ASS), or argininosuccinate lyase (ASL) deficiency. Because hyperchloremic acidosis may develop after high-dose arginine hydrochloride administration, chloride and bicarbonate levels should be monitored and appropriate amounts of bicarbonate administered.
In hyperammonemic infants with suspected, but unconfirmed urea cycle disorders, intravenous arginine should be given (6 mL/kg of Arginine HCl Injection 10%, over 90 minutes followed by the same dose given as a maintenance infusion over 24 hours). If deficiencies of ASS or ASL are excluded as diagnostic possibilities, the intravenous dose of arginine HCl should be reduced to 2 mL/kg/day Arginine HCl Injection 10%.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Hyperammonemia
Any episode of acute symptomatic hyperammonemia should be treated as a life-threatening emergency. Uncontrolled hyperammonemia can rapidly result in brain damage or death, and prompt use of all therapies necessary, including hemodialysis, to reduce ammonia levels is essential.
Hyperammonemic coma (regardless of cause) in the newborn infant should be aggressively treated while the specific diagnosis is pursued. Hemodialysis should be promptly initiated in all newborn patients. A blood flow rate of 150 mL/min/m 2 should be targeted (ammonia clearance [mL/min] is similar to the blood flow rate [mL/min] through the dialyzer). Clearance of ammonia is approximately ten times greater by hemodialysis than by peritoneal dialysis or hemofiltration. Exchange transfusion is ineffective in the management of hyperammonemia. Hemodialysis may be repeated until the plasma ammonia level is stable at normal or near normal levels.
Hyperammonemia due to urea cycle disorders should be managed in coordination with medical personnel experienced in metabolic disorders. Ongoing monitoring of plasma ammonia levels, neurological status, laboratory tests, and clinical response in patients receiving AMMONUL is crucial to assess patient response to treatment.
5.2 Decreased Potassium Levels
Because urine potassium loss is enhanced by the excretion of the non-reabsorbable anions, phenylacetylglutamine and hippurate, plasma potassium levels should be carefully monitored and appropriate treatment given when necessary.
5.3 Conditions Associated with Fluid Overload
AMMONUL contains 30.5 mg of sodium per mL of undiluted product. Thus, AMMONUL should be used with great care, if at all, in patients with congestive heart failure or severe renal insufficiency, and in clinical states in which there is sodium retention with edema. Discontinue administration of AMMONUL, evaluate the patient, and institute appropriate therapeutic countermeasures if an adverse event occurs.
5.4 Extravasation
Administration must be through a central line. Administration through a peripheral line may cause burns. Bolus infusion flow rates are relatively high, especially for infants [ see Dosage and Administration (2)]. Extravasation of AMMONUL into the perivenous tissues may lead to skin necrosis. If extravasation is suspected, discontinue the infusion and resume at a different infusion site, if necessary. The infusion site must be monitored closely for possible infiltration during drug administration. Do not administer undiluted product.
5.5 Neurotoxicity of Phenylacetate
Because of prolonged plasma levels achieved by phenylacetate in pharmacokinetic studies, repeat loading doses of AMMONUL should not be administered. Additionally, neurotoxicity was reported in cancer patients receiving intravenous phenylacetate, 250–300 mg/kg/day for 14 days, repeated at 4-week intervals. Manifestations were predominantly somnolence, fatigue, and lightheadedness, with less frequent headaches, dysgeusia, hypoacusis, disorientation, impaired memory, and exacerbation of a pre-existing neuropathy. The acute onset of symptoms upon initiation of treatment and reversibility of symptoms when the phenylacetate was discontinued suggest a drug effect. [See Animal Toxicology and/or Pharmacology (13.2)]
5.6 Hyperventilation and Metabolic Acidosis
Due to structural similarities between phenylacetate and benzoate to salicylate, AMMONUL may cause side effects typically associated with salicylate overdose, such as hyperventilation and metabolic acidosis. Monitoring of blood chemistry profiles, blood pH and pCO 2 should be performed.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety data were obtained from 316 patients who received AMMONUL as emergency (rescue) or prospective treatment for hyperammonemia as part of an uncontrolled, open-label study. The study population included patients between the ages of 0 to 53 years with a mean (SD) of 6.2 (8.54) years; 51% were male and 49% were female who had the following diagnoses: OTC (46%), ASS (22%), CPS (12%), ASL (2%), ARG (< 1%), THN (< 1%), and other (18%).
Table 2. Adverse Reactions Occurring in ≥ 3% of Patients Treated with AMMONUL Patients
N=316Number of patients with any adverse event 163 (52%) Blood and lymphatic system disorders 35 (11%) Anemia 12 (4%) Disseminated intravascular coagulation 11 (3%) Cardiac disorders 28 (9%) Gastrointestinal disorders 42 (13%) Diarrhea 10 (3%) Nausea 9 (3%) Vomiting 29 (9%) General disorders and administration-site conditions 45 (14%) Injection-site reaction 11 (3%) Pyrexia 17 (5%) Infections 39 (12%) Urinary tract infection 9 (3%) Injury, poisoning and procedural complications 12 (4%) Investigations 32 (10%) Metabolism and nutrition disorders 67 (21%) Acidosis 8 (3%) Hyperammonemia 17 (5%) Hyperglycemia 22 (7%) Hypocalcemia 8 (3%) Hypokalemia 23 (7%) Metabolic acidosis 13 (4%) Nervous system disorders 71 (22%) Brain edema 17 (5%) Coma 10 (3%) Convulsions 19 (6%) Mental impairment 18 (6%) Psychiatric disorders 16 (5%) Agitation 8 (3%) Renal and urinary disorders 14 (4%) Respiratory, thoracic and mediastinal disorders 47 (15%) Respiratory distress 9 (3%) Skin and subcutaneous tissue disorders 19 (6%) Vascular disorders 19 (6%) Hypotension 14 (4%) Adverse reactions were reported with similar frequency in patients with OTC, ASS, CPS, and diagnoses categorized as "other." Nervous system disorders were more frequent in patients with OTC and CPS, compared with patients with ASS and patients with "other" diagnoses. Convulsions and mental impairment were reported in patients with OTC and CPS. These observations are consistent with literature reports that patients with enzyme deficiencies occurring earlier in the urea cycle (i.e., OTC and CPS) tend to be more severely affected.
Adverse reactions profiles differed by age group. Patients ≤ 30 days of age had more blood and lymphatic system disorders and vascular disorders (specifically hypotension), while patients > 30 days of age had more gastrointestinal disorders (specifically nausea, vomiting and diarrhea).
Less common adverse reactions (< 3% of patients) that are characterized as severe are listed below by body system.
BLOOD AND LYMPHATIC SYSTEM DISORDERS: coagulopathy, pancytopenia, thrombocytopenia
CARDIAC DISORDERS: atrial rupture, bradycardia, cardiac or cardiopulmonary arrest/failure, cardiogenic shock, cardiomyopathy, pericardial effusion
EYE DISORDERS: blindness
GASTROINTESTINAL DISORDERS: abdominal distension, gastrointestinal hemorrhage
GENERAL DISORDERS AND ADMINISTRATION-SITE CONDITIONS: asthenia, brain death, chest pain, multiorgan failure, edema
HEPATOBILIARY DISORDERS: cholestasis, hepatic artery stenosis, hepatic failure/hepatotoxicity, jaundice
INFECTIONS AND INFESTATIONS: sepsis/septic shock
INJURY, POISONING AND PROCEDURAL COMPLICATIONS: brain herniation, subdural hematoma, overdose
INVESTIGATIONS: blood carbon dioxide changes, blood glucose changes, blood pH increased, cardiac output decreased, pCO 2 changes, respiratory rate increased
METABOLISM AND NUTRITION DISORDERS: alkalosis, dehydration, fluid overload/retention, hypoglycemia, hyperkalemia, hypernatremia, alkalosis, tetany
NEOPLASMS BENIGN, MALIGNANT AND UNSPECIFIED: hemangioma acquired
NERVOUS SYSTEM DISORDERS: areflexia, ataxia, brain infarction, brain hemorrhage, cerebral atrophy, clonus, depressed level of consciousness, encephalopathy, nerve paralysis, intracranial pressure increased, subdural hematoma, tremor
PSYCHIATRIC DISORDERS: acute psychosis, aggression, confusional state, hallucinations
RENAL AND URINARY DISORDERS: anuria, renal failure, urinary retention
RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS: acute respiratory distress syndrome, dyspnea, hypercapnia, hyperventilation, Kussmaul respiration, pneumonia aspiration, pneumothorax, pulmonary hemorrhage, pulmonary edema, respiratory acidosis or alkalosis, respiratory arrest/failure
SKIN AND SUBCUTANEOUS TISSUE DISORDERS: alopecia, blister, pruritis generalized, rash, urticaria
VASCULAR DISORDERS: flushing, hemorrhage, hypertension, phlebothrombosis/thrombosis
-
7 DRUG INTERACTIONS
Formal drug interaction studies have not been performed with AMMONUL.
Some antibiotics such as penicillin may compete with phenylacetylglutamine and hippurate for active secretion by renal tubules, which may affect the overall disposition of the infused drug.
Probenecid is known to inhibit the renal transport of many organic compounds, including aminohippuric acid, and may affect renal excretion of phenylacetylglutamine and hippurate.
There have been reports that valproic acid can induce hyperammonemia through inhibition of the synthesis of N-acetylglutamate, a co-factor for carbamyl phosphate synthetase. Therefore, administration of valproic acid to patients with urea cycle disorders may exacerbate their condition and antagonize the efficacy of AMMONUL.
Use of corticosteroids may cause a protein catabolic state and, thereby, potentially increase plasma ammonia levels in patients with impaired ability to form urea.
-
8 USE IN SPECIFIC POPULATIONS
8.3 Nursing Mothers
It is not known whether sodium phenylacetate, sodium benzoate, or their conjugation products are excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when AMMONUL is administered to a nursing woman.
8.4 Pediatric Use
AMMONUL has been used as a treatment for acute hyperammonemia in pediatric patients including patients in the early neonatal period [ see Dosage and Administration (2)].
8.5 Geriatric Use
Clinical studies of AMMONUL did not include any patients aged 65 and over to determine whether they respond differently from younger patients. Urea cycle disorders are presently diseases of the pediatric and younger adult populations. No pharmacokinetic studies of AMMONUL have been performed in geriatric patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and concomitant disease or other drug therapy in this patient population.
8.6 Gender
Pharmacokinetic parameters of AMMONUL were compared in healthy males and females. Bioavailability of both benzoate and phenylacetate was slightly higher in females than in males. However, conclusions cannot be drawn due to the limited number of subjects in this study.
8.7 Hepatic Insufficiency
Limited information is available on the metabolism and excretion of sodium phenylacetate and sodium benzoate in patients with impaired hepatic function. However, metabolic conjugation of sodium phenylacetate and sodium benzoate is known to take place in the liver and kidney. Therefore, caution should be used in administering AMMONUL to patients with hepatic insufficiency.
-
10 OVERDOSAGE
Overdosage has been reported during AMMONUL treatment in urea cycle-deficient patients. All patients in the uncontrolled open-label study were to be treated with the same dose of AMMONUL. However, some patients received more than the dose level specified in the protocol. In 16 of the 64 deaths, the patient received a known overdose of AMMONUL. Causes of death in these patients included cardiorespiratory failure/arrest (6 patients), hyperammonemia (3 patients), increased intracranial pressure (2 patients), pneumonitis with septic shock and coagulopathy (1 patient), error in dialysis procedure (1 patient), respiratory failure (1 patient), intractable hypotension and probable sepsis (1 patient), and unknown (1 patient). Additionally, other signs of intoxication may include obtundation (in the absence of hyperammonemia), hyperventilation, a severe compensated metabolic acidosis, perhaps with a respiratory component, large anion gap, hypernatremia and hyperosmolarity, progressive encephalopathy, cardiovascular collapse, and death.
In case of overdose of AMMONUL, discontinue the drug and institute appropriate emergency medical monitoring and procedures. In severe cases, the latter may include hemodialysis (procedure of choice) or peritoneal dialysis (when hemodialysis is unavailable).
-
11 DESCRIPTION
AMMONUL (sodium phenylacetate and sodium benzoate) Injection 10% per 10% (a nitrogen binding agent), is a sterile, concentrated, aqueous solution of sodium phenylacetate and sodium benzoate. The pH of the solution is between 6 and 8. Sodium phenylacetate is a crystalline, white to off-white powder with a strong, offensive odor. It is soluble in water. Sodium benzoate is a white and odorless, crystalline powder that is readily soluble in water.
Figure 1
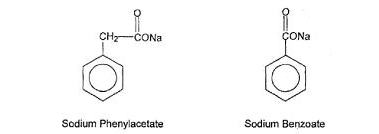
Sodium phenylacetate has a molecular weight of 158.13 and the molecular formula C 8H 7NaO 2. Sodium benzoate has a molecular weight of 144.11 and the molecular formula C 7H 5NaO 2.
Each mL of AMMONUL contains 100 mg of sodium phenylacetate and 100 mg of sodium benzoate, and Water for Injection. Sodium hydroxide and/or hydrochloric acid may have been used for pH adjustment.
AMMONUL injection is a sterile, concentrated solution intended for intravenous administration via a central line only after dilution [ see Dosage and Administration (2)].
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Urea cycle disorders can result from decreased activity of any of the following enzymes: N-acetylglutamate synthetase (NAGS), carbamyl phosphate synthetase (CPS), argininosuccinate synthetase (ASS), ornithine transcarbamylase (OTC), argininosuccinate lyase (ASL), or arginase (ARG).
Sodium phenylacetate and sodium benzoate are metabolically active compounds that can serve as alternatives to urea for the excretion of waste nitrogen. Figure 2 is a schematic illustrating how the components of AMMONUL, phenylacetate and benzoate, provide an alternative pathway for nitrogen disposal in patients without a fully functioning urea cycle. Phenylacetate conjugates with glutamine in the liver and kidneys to form phenylacetylglutamine, via acetylation. Phenylacetylglutamine is excreted by the kidneys via glomerular filtration and tubular secretion. The nitrogen content of phenylacetylglutamine per mole is identical to that of urea (both contain two moles of nitrogen). Two moles of nitrogen are removed per mole of phenylacetate when it is conjugated with glutamine. Similarly, preceded by acylation, benzoate conjugates with glycine to form hippuric acid, which is rapidly excreted by the kidneys by glomerular filtration and tubular secretion. One mole of hippuric acid contains one mole of waste nitrogen. Thus, one mole of nitrogen is removed per mole of benzoate when it is conjugated with glycine
Figure 2
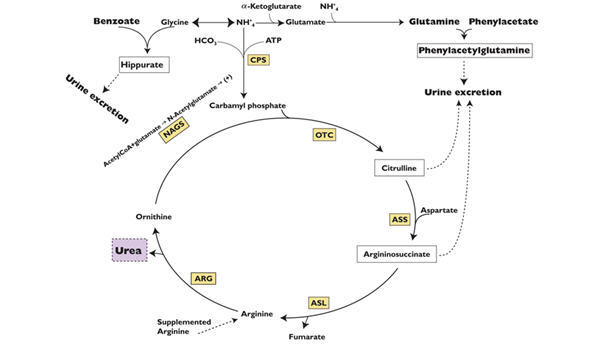
CPS = carbamyl phosphate synthetase;
OTC = ornithine transcarbamylase;
ASS = argininosuccinate synthetase;
ASL = argininosuccinate lyase;
ARG = arginase;
NAGS = N-acetylglutamate synthetase12.2 Pharmacodynamics
In patients with hyperammonemia due to deficiencies in enzymes of the urea cycle, AMMONUL has been shown to decrease elevated plasma ammonia levels. These effects are considered to be the result of reduction in nitrogen overload through glutamine and glycine scavenging by AMMONUL in combination with appropriate dietary and other supportive measures.
12.3 Pharmacokinetics
The pharmacokinetics of intravenously administered AMMONUL was characterized in healthy adult volunteers. Both benzoate and phenylacetate exhibited nonlinear kinetics. Following 90 minute intravenous infusion mean AUClast for benzoate was 20.3, 114.9, 564.6, 562.8, and 1599.1 mcg/mL following doses of 1, 2, 3.75, 4, and 5.5 g/m 2, respectively. The total clearance decreased from 5.19 to 3.62 L/h/m 2 at the 3.75 and 5.5 g/m 2 doses, respectively.
Similarly, phenylacetate exhibited nonlinear kinetics following the priming dose regimens. AUClast was 175.6, 713.8, 2040.6, 2181.6, and 3829.2 mcg∙h/mL following doses of 1, 2, 3.75, 4, and 5.5 g/m 2, respectively. The total clearance decreased from 1.82 to 0.89 mcg∙h/mL with increasing dose (3.75 and 4 g/m 2, respectively).
During the sequence of 90 minute priming infusion followed by a 24 hour maintenance infusion, phenylacetate was detected in the plasma at the end of infusion (Tmax of 2 hr at 3.75 g/m 2) whereas, benzoate concentrations declined rapidly (Tmax of 1.5 hr at 3.75 g/m 2) and were undetectable at 14 and 26 hours following the 3.75 and 4 g/m 2 dose, respectively.
A difference in the metabolic rates for phenylacetate and benzoate was noted. The formation of hippurate from benzoate occurred more rapidly than that of phenylacetylglutamine from phenylacetate, and the rate of elimination for hippurate appeared to be more rapid than that for phenylacetylglutamine.
Pharmacokinetic observations have also been reported from twelve episodes of hyperammonemic encephalopathy in seven children diagnosed (age 3 to 26 months) with urea cycle disorders who had been administered AMMONUL intravenously. These data showed peak plasma levels of phenylacetate and benzoate at approximately the same times as were observed in healthy adults. As in healthy adults, the plasma levels of phenylacetate were higher than benzoate and were present for a longer time.
The pharmacokinetics of intravenous phenylacetate have been reported following administration to adult patients with advanced solid tumors. The decline in serum phenylacetate concentrations following a loading infusion of 150 mg/kg was consistent with saturable enzyme kinetics. Ninety-nine percent of administered phenylacetate was excreted as phenylacetylglutamine [2,3].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals have not been performed to evaluate the carcinogenic potential of AMMONUL. Studies to evaluate the possible impairment of fertility or mutagenic potential of AMMONUL have not been performed. Results indicate that sodium benzoate is not mutagenic or carcinogenic, and does not impair fertility.
13.2 Animal Toxicology and/or Pharmacology
In animal studies, subcutaneous administration to rat pups of 190–474 mg/kg of phenylacetate caused decreased proliferation and increased loss of neurons, and reduced central nervous system (CNS) myelin. Cerebral synapse maturation was retarded, and the number of functioning nerve terminals in the cerebrum was reduced, which resulted in impaired brain growth. Pregnant rats were given phenylacetate at 3.5 µmol/g/day subcutaneously from gestation day 7 through normal delivery. Prenatal exposure of rat pups to phenylacetate produced lesions in layer 5 cortical pyramidal cells; dendritic spines were longer and thinner than normal and reduced in number.
-
14 CLINICAL STUDIES
The efficacy of AMMONUL in improving patient survival of acute hyperammonemic episodes was demonstrated in an analysis of 316 patients (1045 episodes of hospitalization) treated between 1981 and 2003.
The demographic characteristics and diagnoses of the patient population are shown in Table 3.
Table 3 Baseline Characteristics and Diagnoses of Study Population Patients *
N=316OTC = ornithine transcarbamylase deficiency; ASS = argininosuccinate synthetase deficiency; CPS = carbamyl phosphate synthetase deficiency; ASL = argininosuccinate lyase deficiency; ARG = arginase deficiency; THN = transient hyperammonemia of the newborn - *
- For the summary at the patient level, data obtained at first episode used.
- †
- Diagnosis unknown or pending (33 episodes), acidemia (14 episodes), HHH syndrome (6 episodes), carnitine translocase deficiency (4 episodes), liver disease (3 episodes), HMG CoA lyase deficiency (1 episode), non-ketotic hyperglycinemia (1 episode), suspected fatty acid oxidation deficiency (1 episode), and valproic-acid-induced hyperammonemia (1 episode).
Gender Male 158 (51%) Female 150 (49%) Age (years) N 310 Mean (SD) 6.2 (8.54) Min–Max 0.0–53.0 Age groups 0–30 days 104 (34%) 31 days–2 years 55 (18%) > 2–12 years 90 (29%) > 12–16 years 30 (10%) > 16 years 31 (10%) Enzyme OTC 146 (46%) deficiency ASS 71 (22%) CPS 38 (12%) ASL 7 (2%) ARG 2 (< 1%) THN 2 (< 1%) Other † 56 (18%) On admission to the hospital, patients with hyperammonemia and a suspected or confirmed urea cycle disorder (UCD) diagnosis were treated with a bolus dose of 0.25 g/kg (or 5.5 g/m 2) sodium phenylacetate + 0.25 g/kg (or 5.5 g/m 2) sodium benzoate over a period of 90 minutes to 6 hours, depending on the specific UCD. Infusions also contained arginine; the dose of arginine depended on the specific UCD. After completion of the bolus dose, maintenance infusions of the same dose over 24 hours were continued until the patient was no longer hyperammonemic or oral therapy could be tolerated. The mean (SD) duration of treatment was 4.6 (6.45) days per episode, and ranged from 1 to 72 days.
Survival was substantially improved after AMMONUL treatment compared with historical values (estimated 14% 1-year survival rate with dietary therapy alone) and with dialysis (estimated 43% survival of acute hyperammonemia) .
Eighty percent of patients (252 of 316) survived their last episode. Of the 64 patients who died, 53 (83%) died during their first hyperammonemic episode. Of the 104 neonates (< 30d) treated with AMMONUL, 34 (33%) died during the first hyperammonemic episode.
Ammonia levels decreased from very high levels (> 4 times the upper limit of normal [ULN]) to lower levels in 91% of episodes after treatment. In patients responding to therapy, mean ammonia concentrations decreased from 200.9 umol/L at hour zero to 101.6 umol/L within four hours of initiation of AMMONUL therapy and were maintained. Hemodialysis is recommended for those patients who's plasma ammonia levels fail to fall below 150 umol/L or by more than 40% within 4 to 8 hours after receiving AMMONUL. A shift from high (≤ 4 times ULN) to very high (> 4 times ULN) levels was observed in only 4% of the episodes.
Overall, investigators rated neurological status as improved, much improved, or the same in 93% of episodes, and overall status in response to treatment as improved, much improved, or the same in 97% of episodes. Recovery from coma was observed in 97% of episodes where coma was present at admission (111 of 114 episodes).
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Physicians should advise patients and caregivers about the following for safe use of AMMONUL:
- When plasma ammonia levels have normalized, dietary protein intake can usually be increased with the goal of unrestricted protein intake.
- Caution should be exercised when AMMONUL is administered to a nursing woman.
- The most common adverse reactions are vomiting, hyperglycemia, hypokalemia, convulsions, and mental impairment.
- Generally BUPHENYL is stopped during the time AMMONUL is used.
- SPL UNCLASSIFIED SECTION
-
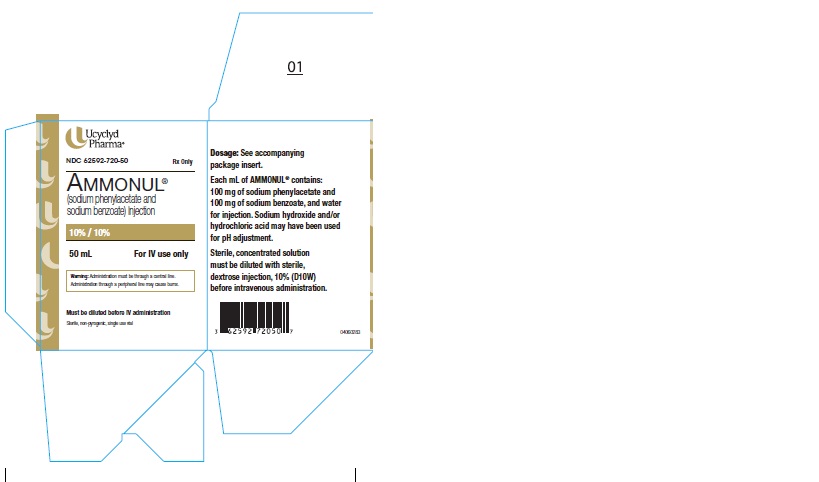
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 50 mL Vial Carton
Ucyclyd
Pharma ®NDC 62592-720-50
Rx OnlyAMMONUL®
(sodium phenylacetate and
sodium benzoate) Injection10% / 10%
50 mL
For IV use onlyWarning: Administration must be through a central line.
Administration through a peripheral line may cause burns.Must be diluted before IV administration
Sterile, non-pyrogenic, single use vial

-
INGREDIENTS AND APPEARANCE
AMMONUL
sodium phenylacetate and sodium benzoate injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:62592-720 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SODIUM PHENYLACETATE (UNII: 48N6U1781G) (PHENYLACETIC ACID - UNII:ER5I1W795A) SODIUM PHENYLACETATE 100 mg in 1 mL SODIUM BENZOATE (UNII: OJ245FE5EU) (BENZOIC ACID - UNII:8SKN0B0MIM) SODIUM BENZOATE 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:62592-720-50 1 in 1 CARTON 02/17/2005 1 50 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020645 02/17/2005 Labeler - Ucyclyd Pharma Inc. (150393221) Establishment Name Address ID/FEI Business Operations Cangene bioPharma Inc 050783398 manufacture(62592-720)