Label: PRASTERA- prasterone and ibuprofen kit



-
Contains inactivated NDC Code(s)
NDC Code(s): 55607-400-10 - Packager: Health Science Funding, LLC
- This is a repackaged label.
- Source NDC Code(s): 25000-121
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: unapproved drug other
DISCLAIMER: This drug has not been found by FDA to be safe and effective, and this labeling has not been approved by FDA. For further information about unapproved drugs, click here.
Drug Label Information
Updated February 25, 2014
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PRASTERA® safely and effectively. See full prescribing information for PRASTERA®.
PRASTERA® prasterone oral softgels 200mg are for oral use only.
Initial U.S. ApprovalINDICATIONS AND USAGE
PRASTERA® prasterone 200 mg oral softgels is indicated in female patients with mild to moderate, active (SLEDAI ≥2) systemic lupus erythematosus (SLE) to restore serum 5-dehydroandrosterone sulfate to levels typical of women without SLE. In Phase III clinical trials in female patients with mild to moderate active SLE, prasterone 200 mg was associated with reduced risk of auto-immune flare, reduced risk of breast cancer and reduced risk of death from any cause. (§§1, 6.2, 6.4, 6.5)
DOSAGE AND ADMINISTRATION
The recommended dose is one (1) softgel daily. (§2)
DOSAGE FORMS AND STRENGTHS
200mg oral softgel capsules supplied in a convenience package with ibuprofen oral tablets 400mg. (§3)
CONTRAINDICATIONS
- Known hypersensitivity to any of its ingredients. (§4)
- Undiagnosed abnormal genital bleeding. (§4)
- Known, suspected or history of breast cancer. (§4, §6.5)
- History of, or known, deep vein thrombosis, pulmonary embolism, arterial thromboembolic disease (e.g., stroke, myocardial infarction). (§4)
- Hypercholesterolemia or ischemic heart disease. (§4, §7.2.4)
- Hepatic or renal impairment (pharmacokinetic data lacking). (§4, §7.2)
- Breast-feeding or known or suspected pregnancy. (§4)
- History of psychiatric disorder. (§4, §8)
WARNINGS AND PRECAUTIONS
- PRASTERA® is not intended for use in nursing or pregnant women, children nor males (safety data is lacking). (§§4, 9)
- PRASTERA® use may be prohibited by certain athletic anti-doping regulations. (§9.5)
- PRASTERA® may in certain patients elevate serum levels of 5-dehydroepiandrosterone, testosterone or estrogen above the normal range for healthy, non-afflicted women of similar age. Periodic measurement of serum hormones is prudent. (§7.2.1)
- Hypertension may occur with prasterone treatment. Monitor blood pressure closely. (§6.3.2)
ADVERSE REACTIONS
The most common adverse event with PRASTERA® is acne. This is generally treatable with topical anti-acne medication. (§6.1)
Another common adverse reaction is hirsutism. Both acne and hirsutism are reversible on cessation of prasterone. (§6.1)
To report SUSPECTED ADVERSE REACTIONS, contact 1 (855) FLARE-FREE or FDA (1-800-FDA-1088 or www.fda.gov/medwatch) or your doctor.
DRUG INTERACTIONS
- PRASTERA® may interact with certain psychiatric drugs. (§8)
- Concomitant administration of PRASTERA® with endogenous testosterone or estrogens, or dietary supplements containing DHEA or dehydroepiandrosterone, is not generally recommended unless serum sex hormone levels are monitored, because of the potential to elevate levels above the ranges considered normal in healthy individuals. (§§7.1, 7.2.1, 8, 10, 16.3)
USE IN SPECIFIC POPULATIONS
- Not recommended for use in nursing nor pregnant women, pediatric patients, or men (safety data is lacking). (§9)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 8/2013
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Instructions
2.2 Special Precautions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Allergy Warning
5 WARNINGS AND PRECAUTIONS
6 ADVERSE REACTIONS
6.1 Increased Risk of Acne and Hirsutism
6.2 Reduced Risk of Myalgia and Other Flare Symptoms
6.3 Other Common Adverse Events
6.4 Reduced Risk of Death
6.5 Reduced Risk of Breast Cancer
6.6 Other Serious Adverse Events
7 SAFETY
7.1 Relationship Of Dose To Safety
7.2 Clinical Laboratory Evaluation
7.3 Post-Marketing Experience
8 DRUG INTERACTIONS
9 USE IN SPECIFIC POPULATIONS
9.1 Males
9.2 Patients with Active SLE Disease
9.3 Pregnancy
9.4 Pediatric Use
9.5 Athletic Anti-Doping
10 OVERDOSE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Pharmacodynamics
12.2 Pharmacokinetics
13 PRE-CLINICAL TOXICOLOGY
14 CLINICAL STUDIES
14.1 Reduction In Risk Of Flare
14.2 Reduction In Risk of Death
15 HOW SUPPLIED / DOSAGE AND HANDLING
16 PATIENT COUNSELING INFORMATION
16.1 Patient / Caregiver Instructions
16.2 Benefits
16.3 Other Medications
16.4 Adverse Reactions
16.5 Pregnancy
17 MEDICATION GUIDE
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Oral prasterone (200mg per day) in female patients with active systemic lupus erythematosus (SLE) has in several blinded, placebo-controlled randomized clinical studies been associated with a reduced risk of auto-immune flare, §§6.2, 14.1.1, a reduced risk of breast cancer and a reduced risk of death from any cause, §§6.4, 14.2.
Patients with SLE may have depressed serum levels of 5-dehydroepiandrosterone sulfate (5-DHEAS). Oral prasterone has been shown to restore SLE patients' serum 5-DHEAS levels. Prastera® oral softgels are intended for use in patients for whom medical evaluation shows a depressed serum level of DHEA and thus a distinctive need for exogenous DHEA. Prastera® oral prasterone softgels are intended to be used under medical supervision, for a patient receiving active and ongoing medical supervision, wherein the patient obtains medical care on a recurring basis for, among other things, instructions on the use of this product. Prastera® oral prasterone softgels are intended for the dietary management of SLE by meeting the distinctive nutritional requirement of women with mild-to-moderate active SLE. Prastera® oral prasterone softgels are intended for oral intake only.
Prastera® does not cure, treat, mitigate or prevent SLE. To the contrary, patients taking Prastera® will continue to have SLE, and thus may continue to require other appropriate therapy.
- 2 DOSAGE AND ADMINISTRATION
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Prastera® oral softgels should not be used in patients with any of the following conditions:
- Known hypersensitivity to prasterone, DHEA, testosterone, estrogens or any component of Prastera® oral prasterone softgels. §4.1.
- Undiagnosed abnormal genital bleeding.
- Known, suspected, or history of breast cancer. §6.5.
- Active deep vein thrombosis, pulmonary embolism or history of these conditions.
- Active arterial thromboembolic disease (for example, stroke and myocardial infarction), or a history of these conditions. §6.3.2.
- Hypercholesterolemia, §7.2.4, or ischemic heart disease.
- Liver disease or renal impairment (pharmacokinetic data lacking).
- Known or suspected pregnancy; breast-feeding (safety data lacking).
- History of psychiatric disorders (risk of exacerbation). The risk of mania may be increased during concomitant use with antidepressants (tricyclic or SSRIs) and/or alcohol, or with high prasterone doses, or in patients with a history of mood disorders.
-
5 WARNINGS AND PRECAUTIONS
PRASTERA® is not intended for use in children, nor males, nor women who are breastfeeding, pregnant, or who expect to become pregnant. §9. Monitoring of blood pressure, serum lipids, serum sex hormones is prudent. See Clinical Laboratory Evaluation, §§7.2.1, 7.2.4.
-
6 ADVERSE REACTIONS
The most-frequent adverse reactions observed in placebo-controlled, blinded clinical studies GL94-01, GL95-01, GL95-02 and GLB96-01 are as follows:
6.1 Increased Risk of Acne and Hirsutism
Acne was the most frequently reported adverse event. Acne was reported less frequently by African American patients, in approximately 26%, compared to approximately 36% in Caucasian patients.
The second most-frequent adverse event was hirsutism. In study GLB96-01 involving patients of Chinese extraction, hirsutism was not reported; this may indicate a decreased racial susceptibility to hirsutism.
Acne and hirsutism were both reversible on cessation of prasterone therapy. In addition, both were more likely to be reported early in treatment; patients who had not developed these within the first 6 months of exposure are less likely to develop them later.
6.2 Reduced Risk of Myalgia and Other Flare Symptoms
Placebo-treated patients had higher incidences of myalgia, joint disorder, anorexia, nasal ulcers and LE skin rash than did prasterone-treated patients. These differences may be due to the decreased risk of flare observed in prasterone-treated patients. §14.1.
6.3 Other Common Adverse Events
No adverse events increase in frequency with longer duration of treatment. The Table displays all adverse events reported in a frequency of 10% or greater from either the 200 mg dose group or the placebo group for the pooled double-blind phases of Studies GL94-01 and GL95-02. Because the number of patients who received prasterone 100 mg was substantially fewer, adverse events for this group are only presented for those adverse events which were reported in ≥ 10% of either placebo or prasterone 200 mg patients.
ADVERSE EVENTS WITH FREQUENCY ≥10%* (pooled GL94-01 and GL95-02 results) *Frequency > 10% in either prasterone 200 mg or placebo patients.
** P< 0.05, Placebo vs. prasterone 200 mg.
COSTART TERM Placebo
N=256200mg
N=253Rash 77 (30.1%) 93 (36.8%) Acne 39 (15.2%) 91 (36.0%) ** Arthralgia 95 (37.1%) 88 (34.8%) Asthenia 70 (27.3%) 68 (26.9%) Headache 76 (29.7%) 60 (23.7%) Arthritis 58 (22.7%) 57 (22.5%) Myalgia 79 (30.9%) 55 (21.7%) ** Pain Abdomen 34 (13.3%) 41 (16.2%) Flu Syndrome 46 (18.0%) 40 (15.8%) Stomatitis Ulcer 50 (19.5%) 38 (15.0%) Hirsutism 6 (2.3%) 36 (14.2%) ** Fever 39 (15.2%) 36 (14.2%) Depression 33 (12.9%) 35 (13.8%) Alopecia 48 (18.8%) 35 (13.8%) Infection 37 (14.5%) 26 (10.3%) Sinusitis 33 (12.9%) 22 (8.7%) Pain Chest 27 (10.5%) 22 (8.7%) For adverse events occurring in < 10% of patients, the following showed an absolute difference of at least 3% between placebo or prasterone 200 mg, or, if less than 3% difference, the difference was significant (p < 0.05):
ADVERSE EVENTS WITH FREQUENCY <10% AND AT LEAST A 3% OR A SIGNIFICANT DIFFERENCE (pooled GL94-01 and GL95-02 results) ** P< 0.05, Placebo vs. prasterone 200 mg.
COSTART TERM Placebo
N=256prasterone 200 mg
N=253Less Frequent in Prasterone Anorexia 10 (3.9%) 2 (0.8%) ** Nasal Septum Disorder (nasal ulcers) 14 (5.5%) 5 (2.0%) ** Rash Lupus Erythematosus 13 (5.1%) 4 (1.6%) ** Joint Disorder 14 (5.5%) 4 (1.6%) ** More Frequent in Prasterone Creatinine Increase 0 (0.0%) 6 (2.4%)** Hypertension 7 (2.7%) 20 (7.9%) ** Hematuria 1 (0.4%) 9 (3.6%) ** Insignificant Difference Back Pain 16 (6.3%) 24 (9.5%) Pharyngitis 14 (5.5%) 6 (2.4%) Dyspnea 22 (8.6%) 11 (4.3%) Lymphadenopathy 21 (8.2%) 12 (4.7%) The pattern of adverse events in clinical study GLB96-01 showed a similar, but not identical pattern. Acne was the most common adverse event; this may reflect the fact that almost all patients were also receiving corticosteroids. This may also reflect racial differences in sensitivity to prasterone. By contrast, hirsutism was not reported at all in GLB96-01.
ADVERSE EVENTS REPORTED BY AT LEAST 10% OF EITHER TREATMENT GROUP (GBL96-01) *P-value<0.05, Treatment vs. Placebo, chi-square test
Placebo
N= 59Treatment
N= 61Arthralgia 37 ( 62.7%) 39 ( 63.9%) Acne * 17 ( 28.8%) 36 ( 59.0%)* Pharyngitis 32 ( 54.2%) 34 ( 55.7%) Myalgia 24 ( 40.7%) 28 ( 45.9%) Headache * 37 ( 62.7%) 26 ( 42.6%)* Pain Abdomen 25 ( 42.4%) 23 ( 37.7%) Asthenia 19 ( 32.2%) 18 ( 29.5%) Cough Increase 18 ( 30.5%) 18 ( 29.5%) Dizziness 19 ( 32.2%) 15 ( 24.6%) Pain Chest 11 ( 18.6%) 14 ( 23.0%) Dyspnea 8 ( 13.6%) 14 ( 23.0%) Rash 16 (27.1%) 14 ( 23.0%) Fever 17 ( 28.8%) 13 ( 21.3%) Alopecia 8 ( 13.6%) 13 ( 21.3%) Pain 8 ( 13.6%) 11 ( 18.0%) Diarrhea 11 ( 18.6%) 11 ( 18.0%) Rhinitis 13 ( 22.0%) 11 ( 18.0%) Stomatitis Ulcer 17 ( 28.8%) 10 ( 16.4%) Pain Back 10 ( 16.9%) 9 ( 14.8%) Edema 6 ( 10.2%) 9 ( 14.8%) Injury Accident 6 ( 10.2%) 8 ( 13.1%) Insomnia 7 ( 11.9%) 8 ( 13.1%) Pruritus 7 ( 11.9%) 8 ( 13.1%) Infection* 15 ( 25.4%) 6 ( 9.8%)* Dry Eye 10 ( 16.9%) 6 ( 9.8%) Vomit 8 ( 13.6%) 5 ( 8.2%) Peripheral Edema 8 ( 13.6%) 5 ( 8.2%) Rash Lupus Erythematosus 7 ( 11.9%) 5 ( 8.2%) Conjunctivitis 7 ( 11.9%) 5 ( 8.2%) Nausea 9 ( 15.3%) 4 ( 6.6%) Of adverse events reported by at least 10% of patients, acne was the only event significantly more frequent in the treatment group. Headache and infection were more frequent in the placebo group. Of the adverse events reported with an incidence of less than 10%, the only statistically significant difference was for seborrhea (0 placebo vs. 5 treatment patients).
6.3.1 Hypertension
Hypertension was reported as an adverse event more frequently in the prasterone 200 mg group than placebo. When measures of increased (changed) blood pressure were included, there appeared to be no difference between the groups. Whether prasterone increases hypertension is thus not clear.
6.4 Reduced Risk of Death
In the GL94-01, GL95-01 and GL95-02 placebo-controlled clinical studies (pooled data), the placebo group experienced 6 deaths in 77 patients, a risk of death of 7.8%. In contrast, the prasterone group (pooled treatment and cross-over) experienced 8 deaths in 495 patients, a risk of death of 1.6%. The risk of death from any cause was therefore five times higher in the placebo group.
6.5 Reduced Risk of Breast Cancer
In the GL94-01, GL95-01 and GL95-02 placebo-controlled clinical studies (pooled data), incidence of breast cancer was 1 in 336 patient-years (0.29%) for placebo and 3 in 1573 patient-years (0.19%) for prasterone (pooled treatment and cross-over). Prasterone was thus associated with reducing the risk of breast cancer by one third.
For patients over 44 years of age, the difference between placebo and prasterone groups was more pronounced. For women at least 45 years of age, the rate of breast cancer was 1 in 24 patient-years (4.2%) for placebo and 3 in 206 patient-years (1.5%) for prasterone treatment and cross-over patients. Prasterone was thus associated with reducing the risk of breast cancer by two thirds.
6.6 Other Serious Adverse Events
Adverse events that were assessed as “severe” occurred in similar frequencies in both treatment and placebo groups, with asthenia being the most common adverse event reported as severe in both placebo and treated patients. Although the patient numbers are small, abdominal pain reported as a severe adverse event occurred in 6 treated 200 mg patients, 2 treated 100mg patients, and no placebo patients.
Serious adverse events occurred in 39 200 mg, 7 100 mg, and 47 placebo patients participating in GL94-01 and GL95-02. However, only 3 serious adverse events were considered possibly related, 2 in the placebo group (one suicide and one patient with menomettrorhagia) and one in 200 mg (a patient with an acute psychosis).
In the Taiwan study, serious adverse events were reported in a significantly higher proportion of patients in the placebo group than in the treatment group. In most cases, the types of serious adverse events reported were consistent with SLE flares or hospitalization for manifestations of SLE, rather than adverse effects of the study drug.
SEVERE ADVERSE EVENTS OCCURRING IN AT LEAST 2 PATIENTS* (GL94-01 and GL95-02) *Frequency at least 2 patients in either prasterone 200 mg or placebo
COSTART TERM Placebo
N=256prasterone 200 mg
N=253Asthenia 22 (8.6%) 22 (8.7%) Headache 11 (4.3%) 8( 3.2%) Arthralgia 6 (2.3%) 6 (2.4%) Pain Abdomen 0 (0%) 6 (2.4%) Rash 5 (2.0%) 6 (2.4%) Arthritis 2 (0.8%) 5 (2.0%) Dyspnea 1 (0.4%) 4 (1.6%) Depression 4 (1.6%) 2 (0.8%) Diabetes Mellitus 0 (0%) 2 (0.8%) Emotional Lability 0 (0%) 2 (0.8%) Infection 0 (0%) 2 (0.8%) Myalgia 5 (2.0%) 2 (0.8%) Pain 1 (0.4%) 2 (0.8%) Pain Chest 4 (1.6%) 2 (0.8%) Paresthesia 1 (0.4%) 2 (0.8%) Pleural Disorder 1 (0.4%) 2 (0.8%) Vasculitis 0 (0%) 2 (0.8%) Joint Disorder 2 (0.8%) 1 (0.4%) Peripheral Edema 2 (0.8%) 0 (0%) Sepsis 2 (0.8%) 0 (0%) Cyst 2 (0.8%) 0 (0%) Thinking Abnormal 2 (0.8%) 0 (0%) -
7 SAFETY
7.1 Relationship Of Dose To Safety
Neither adverse events nor laboratory values showed a dose relationship.
7.2 Clinical Laboratory Evaluation
In the placebo-controlled trials, testosterone was increased in a dose related manner in SLE patients receiving prasterone.
Prasterone caused an increased number of patients (in comparison to placebo) to have a decrease in serum complement (C3). The decrease in C3 was not associated with renal dysfunction. The medical literature suggests that a decline in serum complement may be a direct effect of suppression of complement production by androgens.
The clinical studies suggest that administration of prasterone causes an early, but not progressive, decrease in serum lipids, primarily HDL-C and triglycerides. It would be prudent to follow NCEP guidelines while monitoring lipids in patients receiving prasterone.
There were no changes of potential significance in liver function tests (ALT, AST, alkaline phosphatase, or total bilirubin) within and between the treatment groups. Equally, serum calcium, phosphorus, uric acid, total protein and albumin showed no clinically relevant differences.
Mean 24-hour urine protein excretion was slightly higher in the prasterone group viz placebo.
There does not appear to be a difference between placebo and prasterone for new hematuria accompanied by SLE renal involvement, as manifested by changes in urinary protein excretion, increased creatinine, or new therapy for renal SLE.
7.3 Post-Marketing Experience
The following adverse reactions have been reported with unregulated dietary supplements containing DHEA. The identity, purity and strength of the product used was not always known. It is therefore not possible to establish a causal relation to prasterone exposure.
Cardiovascular Effects: Benign premature atrial contractions and occasional premature ventricular contractions occurred in a 55-year-old man after administration of DHEA; DHEA was discontinued and arrhythmias controlled by beta-blockers. (Sahelian 1998)
One case of hepatitis has been reported in a patient with high pre-treatment anti-nuclear antibody (ANA) titers; causality is uncertain. (Buster 1992)
Manic reactions during DHEA use have been described. (Dean 2000; Pies 2000) Risk factors for development of mania / psychosis are considered to be higher doses, combined use with antidepressants, alcohol or benzodiazepines, young patients (20-30 years, with peak endogenous DHEA levels), and cytochrome P450 polymorphisms (poor metabolizers).
-
8 DRUG INTERACTIONS
There is no known pharmacokinetic effect (bioavailability, pharmacokinetics, or pharmacodynamics) of prasterone on prednisone or hydroxychloroquine, with the possible exception of increasing the magnitude of a decrease in triglycerides seen with hydroxyquinoline.
Prasterone may theoretically interact with one or more of the following drugs: carbamazepine, phenothiazines (e.g., acetophenazine, chlorpromazine, chlorprothixene, ethopropazine, fluphenazine, mesordiazine, methdilazine, perazine, perphenazine, pipotiazine, prochlorperazine, promazine, promethazine, propiomazine, thioridazine, trifluoperazine, triflupromazine), citalopram; escitalopram; clozapine, conjugated estrogens; estherified estrogens; estradiol; estradiol cypionate; estropipate; ethinyl estradiol, fluoxetine, fluvoxamine; haloperidol, lithium, loxapine, molindone, olanzapine, paroxetine, quetiapine, risperodone, sertraline, testosterone, triazolam and valproic acid. For further information on potential interactions with these drugs, please see The Cochrane Review monograph for prasterone.
-
9 USE IN SPECIFIC POPULATIONS
9.1 Males
The placebo-controlled, double-blind clinical studies involved women. Use in men at this time is not recommended because data is lacking.
9.2 Patients with Active SLE Disease
The GL94-01 and GL95-02 clinical studies showed a difference in responder rates between placebo and prasterone increased with increasing baseline SLEDAI.
9.3 Pregnancy
Safety and effectiveness in nursing and pregnant women has not been established. Use is not recommended.
- 10 OVERDOSE
-
11 DESCRIPTION
Prastera® prasterone oral softgels are oblong blue soft gelatin capsules. Each PRASTERA® prasterone oral softgel contains 200mg prasterone (>98% pure), in a lipophilic vehicle.
Prasterone is chemically identical to the naturally-occurring pro-hormone 5-dehydroepiandrosterone, secreted by the adrenal cortex, gonads and brain tissue. It is designated chemically as (3S,8R,9S,10R,13S,14S)-3-hydroxy-10,13-dimethyl-3,4,7,8,9,10,11,12,13,14,15,16 –dodecahydro-1H-cyclopenta[a]phenanthren-17(2H)-one. Its molecular weight is 288.424 g/mol. Its molecular formula is C19H28O2.
-
12 CLINICAL PHARMACOLOGY
Prastera® softgels are an oral dosage form of pharmaceutical-grade prasterone, chemically identical to prasterone of native human origin, in a lipophilic vehicle.
12.1 Pharmacodynamics
Oral prasterone has been shown to increase serum levels of 5-DHEAS. The precise mechanism by which normal serum levels of 5-DHEAS may act to reduce the risk of auto-immune flare, breast cancer and death is not known.
12.2 Pharmacokinetics
12.2.1 Time To Peak Concentration
Oral prasterone reaches a peak serum concentration at 1.5 to 2 hours after administration. In healthy young women (mean age, 30 years) receiving prasterone 200 mg daily (given with prednisone), mean peak plasma levels on day 29 of prasterone and 5-DHEAS were 1.3 mcg/dL (13 ng/mL) and 942 mcg/dL (9.4 mcg/mL), and occurred in 2 hours and 2.4 hours, respectively, after administration. In elderly women (mean, 69 years) and elderly men (mean, 69 years), mean peak plasma concentrations (times to peak levels) after a single 200-mg micronized oral dose were 27 ng/mL (1.4 hours) and 22 ng/mL (1.3 hours), respectively. After a single 200 mg dose, mean serum levels of 5-DHEAS increased 5-fold in men (to 7 mcg/mL) and 21-fold in women (to 7.5 mcg/mL) relative to baseline healthy levels.
12.2.2 Absorption
During two weeks of daily administration (200 mg), plasma levels (and times to peak levels) of both 5-dehydroepiandrosterone and its sulfated metabolite did not change significantly in either women or men, indicating a lack of accumulation.
12.2.3 Distribution
Approximately 10% to 20% of prasterone is bound to serum protein; approximately 80% to 90% of 5-DHEAS is bound to protein. 5-DHEAS penetrates the blood-brain barrier; cerebro-spinal fluid levels of 5-DHEAS range from 0.2% to 5% of corresponding plasma levels.
12.2.4 Metabolism
Oral prasterone is sulfated to 5-DHEAS ester in the intestine and liver by sulfotransferases. Prasterone and 5-DHEAS are converted in peripheral tissues to androstenedione, androsterone sulfate, estradiol, estriol and estrone, dihydrotestosterone, 7-oxo-prasterone, and testosterone.
12.2.5 Excretion
Prasterone (200mg dose) elimination half-life: young women, 11 hours; elderly men, 7 hours. In elderly women, the elimination half-life progressively declined, from about 12 hours (day 1), to 9 hours (day 8), to 7 hours (day 15). 5-DHEAS half-life: young women, 12 hours; elderly men, 20-25 hours; elderly women, 24-27 hours.
12.2.6 Special Populations
The pharmacokinetics of oral prasterone has not been assessed in low body weight or obese patients. There is insufficient information available from placebo-controlled clinical trials to compare prasterone pharmacokinetics in different racial groups, nor for patients with renal or hepatic impairment.
-
13 PRE-CLINICAL TOXICOLOGY
The non-clinical literature indicates that prasterone may be either chemo-protective or carcinogenic, depending on the model. Prasterone may thus be inhibitory or stimulatory to hormone-senentive tumors. The literature, however, suggests that prasterone is less potent than its androgenic and estrogenic metabolites. Similarly to androgenic and extrogenic compounds, it is expected to be difficult to define the carcinogenic potential of prasterone.
-
14 CLINICAL STUDIES
The placebo-controlled studies (GL94-01, GL95-01, GL95-02 and GLB96-01) had very different study designs and efficacy endpoints. Pooling of efficacy data is thus not meaningful. Consequently, results are presented by individual study.
14.1 Reduction In Risk Of Flare
14.1.1 Clinical Study GLB96-01
GLB96-01 was a six month, multi-center, randomized, parallel group, double-blind, placebo-controlled study of prasterone (200mg daily) in Asian women (mean age = 32 years); 97% had baseline SLEDAI score >2. The treatment group (n = 60) had a somewhat higher baseline SLEDAI than did the placebo group (n = 59).
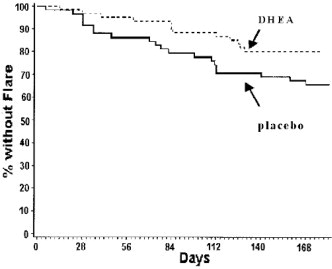
The treatment group significantly had fewer patients with at least one definite flare. The number of patients with definite flares in the treatment group was 46.0% less than in the placebo group (18.3% vs. 33.9%, p = 0.044 based on survival analysis using Cox model).
14.1.2 Clinical Study GL95-02
Study GL95-02 was a 12 month, multi-center, randomized, parallel group, double-blind, placebo-controlled study of prasterone (200mg daily) in women (n = 346) with active SLE (SLAM score ≥7 excluding ESR, SLEDAI score >2) receiving ≤10 mg/day prednisone (or its equivalent of other cortico-steroids). A secondary efficacy variable (flare) was defined as a modified SELENA definition flare.
Three hundred eighty one (381) patients were randomized, of whom 346 were in the per-protocol population. For all patients, 47/176 (27%) of placebo and 37/170 (22%) of prasterone patients experienced a definite flare.
For the subset of patients with a baseline SLEDAI>2, treated patients had a 23.7% decrease in the risk of experiencing at least one definite flare, compared to placebo. During the study period, 41/133 (31%) of placebo patients and 31/132 (23.5%) of prasterone patients experienced a definite flare. See bar chart.
For patients with a baseline SLEDAI>2, treated patients had a 23.7% decrease in the risk of experiencing at least one definite flare, compared to placebo (p=0.201, log-rank test for time to first definite flare).
14.2 Reduction In Risk of Death
In the GL95-02 study (discussed above), five placebo patients died, and no prasterone-treated patients died. These data show a statistically significant and clinically meaningful reduction in risk of death by any cause. In the GL94-01, GL95-01 and GL95-02 clinical trials (pooled data), the prasterone treated group (including prasterone-treated crossover patients) experienced 8 deaths among 495 patients, or a risk of death of 1.62%. In contrast, the placebo group experienced 6 deaths among 77 patients, or a risk of death of 7.80%.
14.2.1 Clinical Study GL94-01
Study GL94-01 compared the proportion of patients achieving sustained reduction of daily corticosteroid dose, without worsening of signs and symptoms of SLE (“Responders”), in placebo (n = 64) and prasterone 200 mg (n = 64) groups for about 7 months.
For all randomized patients, 26/64 (41%) placebo and 35/64 (55%) prasterone 200 mg patients responded: a strong trend in favor of prasterone (p = 0.110). For patients with baseline SLEDAI >2, 13/45 (29%) placebo and 23/45 (51%) prasterone 200mg patients responded (p = 0.031).
-
15 HOW SUPPLIED / DOSAGE AND HANDLING
Prastera® softgels 200mg are oblong blue soft, liquid filled gelatin capsules.
Store at not more than 25° C (77° F). Excursions are permitted to 15° C to 30° C (59° F to 86° F). See United States Pharmacopoeia, Controlled Room Temperature. Protect from excessive moisture or humidity. Dispense in a tight, light-resistant container as defined in USP/NF, using a child-resistant closure system, accompanied by a Patient Insert and in a convenience pack together with a blister strip of ibuprofen 400mg oral tablets.
NDC 55607-400-10
Keep out of reach of children.
-
Manufactured for:
Health Science Funding, LLC
55 Madison Avenue, 4th floor
Morristown, NJ 07960
info@healthsciencefunding.com
-
16 PATIENT COUNSELING INFORMATION
See Medication Guide (§17) for specific patient instructions.
16.1 Patient / Caregiver Instructions
Inform patients of the following information before initiating therapy with PRASTERA® and periodically during the course of on-going therapy. Encourage patients to read the Medication Guide that accompanies each prescription dispensed, prior to using PRASTERA®.
16.2 Benefits
Oral prasterone 200 mg / day reduced the risk of auto-immune flare, §§6.2, 14.1, and significantly reduced the risk of death, §§6.4, 14.2, in placebo-controlled, randomized, blinded clinical studies (GL94-01, GL95-01, GL95-02 and GLB96-01) in female patients with Systemic Lupus Erythematosus. Results observed in clinical trials may not, however, reflect the rates observed in practice.
PRASTERA® does not cure, mitigate, treat or prevent the patient's underlying SLE. To the contrary, the patient will continue to have SLE. The patient therefore should continue to be monitored by a physician and should continue other therapy (e.g., prednisone, NSAID) as believed appropriate.
Oral prasterone reduced the risk of auto-immune flare and death. See above. PRASTERA® may not, however, make the patient feel significantly different on a day-to-day basis. This does not mean PRASTERA® is not working; it may take at least six months of continuous therapy to achieve a statistically-significant reduction in risk of flare and death. §14.
16.3 Other Medications
PRASTERA® is a synthetic form of 5-dehydroepiandrosterone. PRASTERA® should not be combined with dietary supplements containing “DHEA” or “dehydroepiandrosterone.”
PRASTERA® is a precursor to testosterone and estrogens. If PRASTERA® is used in conjunction with testosterone or estrogens, levels of serum testosterone and estrogens should be monitored closely to assure levels do not exceed the range seen in healthy women of similar age.
16.4 Adverse Reactions
PRASTERA® may cause acne. This acne is reversible on cessation of prasterone therapy, and may be treateable by a topical anti-acne drug. You may request a Prastera® convenience kit which includes a topical anti-acne drug.
PRASTERA® may cause hirsutism, reversible on cessation of prasterone therapy.
PRASTERA® may cause hypertension, §6.3.2, and/or changes in serum lipids, §7.2.4, or serum hormone levels, §7.2.1. These should be monitored, and are reversible on cessation of prasterone therapy.
- 17 MEDICATION GUIDE
-
MEDICATION GUIDE
Prasterone is a naturally-occurring pre-hormone secreted by the adrenal cortex. SLE attacks the adrenal cortex. Women with SLE thus often have depressed levels of this pre-hormone.
Your doctor has prescribed PRASTERA® to supplement your body's level of this pre-hormone. Use PRASTERA® exactly as your doctor tells you.
You likely will not feel much different taking PRASTERA®. This does not mean that it is not working, so keep taking it unless your doctor tells you not to, or unless you feel you are having an adverse reaction to it.
In clinical studies, oral prasterone reduced lupus patients' risk of auto-immune flare and death. PRASTERA® does not, however, cure, mitigate, treat or prevent your SLE. Thus, even while taking your PRASTERA®, you will continue to have SLE. You therefore should continue to see your doctor and should continue other therapy they believe appropriate.
PRASTERA® may cause acne. If this happens, you can ask your doctor to give you a prescription for a PRASTERA® kit which includes a mild topical anti-acne treatment. If that does not work, talk with your health care provider because acne should go away simply by stopping PRASTERA®.
PRASTERA® may cause hirsutism (unwanted body hair). This is reversible by stopping taking PRASTERA®.
PRASTERA® may cause abdominal pain, hypertension, and/or changes in serum lipids or serum hormone levels; your doctor may want to monitor these.
Tell your doctor what other drugs or dietary supplements you are taking. If you are taking PRASTERA®, you should not use any dietary supplement containing “DHEA” or “dehydroepiandrosterone.”
If you are pregnant, intending to become pregnant, or are nursing, do not use PRASTERA®.
Store PRASTERA® at not more than 25° C (77° F), and protect from excessive moisture or humidity. Keep out of reach of children.
For medical advice about suspected side effects, call your doctor. You may report suspected side effects to your doctor, or the FDA at 1 (800) 332-1088, or to your pharmacist at 1 (855) FLARE-FREE.
-
Manufactured for:
Health Science Funding, LLC
55 Madison Avenue, 4th floor
Morristown, NJ 07960
info@healthsciencefunding.com
© 2012 Health Science Funding, LLC. All rights reserved. Patent pending. The PRASTERA name, PRASTERA device, product color scheme and package configuration are each trademarks of Health Science Funding, LLC.
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
BOXED WARNING
Cardiovascular Risk
- NSAIDs may cause an increased risk of serious cardiovascular thrombotic events, myocardial infarction, and stroke, which can be fatal. This risk may increase with duration of use. Patients with cardiovascular disease or risk factors for cardiovascular disease may be at greater risk (see WARNINGS).
- Ibuprofen tablets are contraindicated for treatment of peri-operative pain in the setting of coronary artery bypass graft (CABG) surgery (see WARNINGS).
Gastrointestinal Risk
- NSAIDS cause an increased risk of serious gastrointestinal adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Elderly patients are at greater risk for serious gastrointestinal events (see WARNINGS).
-
DESCRIPTION
Ibuprofen tablets contain the active ingredient ibuprofen, which is (±) - 2 - (p - isobutylphenyl) propionic acid. Ibuprofen is a white powder with a melting point of 74-77° C and is very slightly soluble in water (< 1 mg/mL) and readily soluble in organic solvents such as ethanol and acetone .
The structural formula is represented below:
Ibuprofen tablets, a nonsteroidal anti-inflammatory drug ( NSAID ), are available in 400 mg, 600 mg, and 800 mg tablets for oral administration. Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, pregelatinized starch, talc and titanium dioxide. -
CLINICAL PHARMACOLOGY
Ibuprofen tablets contain ibuprofen which possesses analgesic and antipyretic activities. Its mode of action, like that of other NSAIDs, is not completely understood, but may be related to prostaglandin synthetase inhibition.
In clinical studies in patients with rheumatoid arthritis and osteoarthritis , ibuprofen tablets have been shown to be comparable to aspirin in controlling pain and inflammation and to be associated with a statistically significant reduction in the milder gastrointestinal side effects (see ADVERSE REACTIONS ). Ibuprofen tablets may be well tolerated in some patients who have had gastrointestinal side effects with aspirin, but these patients when treated with ibuprofen tablets should be carefully followed for signs and symptoms of gastrointestinal ulceration and bleeding. Although it is not definitely known whether ibuprofen tablets causes less peptic ulceration than aspirin, in one study involving 885 patients with rheumatoid arthritis treated for up to one year, there were no reports of gastric ulceration with ibuprofen tablets whereas frank ulceration was reported in 13 patients in the aspirin group (statistically significant p< .001).
Gastroscopic studies at varying doses show an increased tendency toward gastric irritation at higher doses. However, at comparable doses, gastric irritation is approximately half that seen with aspirin. Studies using 51Cr-tagged red cells indicate that fecal blood loss associated with ibuprofen tablets in doses up to 2400 mg daily did not exceed the normal range, and was significantly less than that seen in aspirin-treated patients.
In clinical studies in patients with rheumatoid arthritis, ibuprofen tablets have been shown to be comparable to indomethacin in controlling the signs and symptoms of disease activity and to be associated with a statistically significant reduction of the milder gastrointestinal (see ADVERSE REACTIONS ) and CNS side effects.
Ibuprofen tablets may be used in combination with gold salts and/or corticosteroids.
Controlled studies have demonstrated that ibuprofen tablets are a more effective analgesic than propoxyphene for the relief of episiotomy pain, pain following dental extraction procedures, and for the relief of the symptoms of primary dysmenorrhea.
In patients with primary dysmenorrhea, ibuprofen tablets have been shown to reduce elevated levels of prostaglandin activity in the menstrual fluid and to reduce resting and active intrauterine pressure, as well as the frequency of uterine contractions. The probable mechanism of action is to inhibit prostaglandin synthesis rather than simply to provide analgesia .
The ibuprofen in ibuprofen tablets is rapidly absorbed. Peak serum ibuprofen levels are generally attained one to two hours after administration. With single doses up to 800 mg, a linear relationship exists between amount of drug administered and the integrated area under the serum drug concentration vs time curve. Above 800 mg, however, the area under the curve increases less than proportional to increases in dose. There is no evidence of drug accumulation or enzyme induction.
The administration of ibuprofen tablets either under fasting conditions or immediately before meals yields quite similar serum ibuprofen concentration-time profiles. When ibuprofen tablets are administered immediately after a meal, there is a reduction in the rate of absorption but no appreciable decrease in the extent of absorption. The bioavailability of the drug is minimally altered by the presence of food.
A bioavailability study has shown that there was no interference with the absorption of ibuprofen when ibuprofen tablets were given in conjunction with an antacid containing both aluminum hydroxide and magnesium hydroxide.
Ibuprofen is rapidly metabolized and eliminated in the urine . The excretion of ibuprofen is virtually complete 24 hours after the last dose. The serum half-life is 1.8 to 2.0 hours.
Studies have shown that following ingestion of the drug, 45%to 79%of the dose was recovered in the urine within 24 hours as metabolite A (25%), (+)-2-[p-(2hydroxymethyl-propyl) phenyl] propionic acid and metabolite B (37%), (+)-2-[p-(2carboxypropyl)phenyl] propionic acid; the percentages of free and conjugated ibuprofen were approximately 1%and 14%, respectively.
-
INDICATIONS AND USAGE
Carefully consider the potential benefits and risks of ibuprofen tablets and other treatment options before deciding to use ibuprofen tablets. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS ).
Ibuprofen tablets are indicated for relief of the signs and symptoms of rheumatoid arthritis and osteoarthritis .
Ibuprofen tablets are indicated for relief of mild to moderate pain.
Ibuprofen tablets are also indicated for the treatment of primary dysmenorrhea.
Controlled clinical trials to establish the safety and effectiveness of ibuprofen tablets in children have not been conducted.
-
CONTRAINDICATIONS
Ibuprofen tablets are contraindicated in patients with known hypersensitivity to Ibuprofen.
Ibuprofen tablets should not be given to patients who have experienced asthma, urticaria , or allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal, anaphylactic-like reactions to NSAIDs have been reported in such patients (see WARNINGS , Anaphylactoid Reactions, and PRECAUTIONS , Preexisting Asthma).
Ibuprofen tablets are contraindicated for the treatment of peri-operative pain in the setting of coronary artery bypass graft ( CABG ) surgery (see WARNINGS).
-
WARNINGS
Cardiovascular Effects
Cardiovascular Thrombotic Events
Clinical trials of several COX-2 selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, myocardial infarction, and stroke , which can be fatal. All NSAIDs, both COX-2 selective and nonselective, may have a similar risk. Patients with known CV disease or risk factors for CV disease may be at greater risk. To minimize the potential risk for an adverse CV event in patients treated with an NSAID , the lowest effective dose should be used for the shortest duration possible. Physicians and patients should remain alert for the development of such events, even in the absence of previous CV symptoms. Patients should be informed about the signs and/or symptoms of serious CV events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID does increase the risk of serious GI events (see WARNINGS, Gastrointestinal Effects-Risk of Ulceration, Bleeding, and Perforation).
Two large, controlled clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10-14 days following CABG surgery found an increased incidence of myocardial infarction and stroke (see CONTRAINDICATIONS).
Hypertension
NSAIDs including ibuprofen tablets, can lead to onset of new hypertension or worsening of pre-existing hypertension, either of which may contribute to the increased incidence of CV events. Patients taking thiazides or loop diuretics may have impaired response to these therapies when taking NSAIDs. NSAIDs, including ibuprofen tablets, should be used with caution in patients with hypertension. Blood pressure (BP) should be monitored closely during the initiation of NSAID treatment and throughout the course of therapy.
Gastrointestinal Effects-Risk of Ulceration, Bleeding, and Perforation
NSAIDs, including ibuprofen tablets, can cause serious gastrointestinal (GI) adverse events including inflammation, bleeding, ulceration, and perforation of the stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with NSAIDs. Only one in five patients, who develop a serious upper GI adverse event on NSAID therapy, is symptomatic. Upper GI ulcers, gross bleeding, or perforation caused by NSAIDs occur in approximately 1%of patients treated for 3-6 months, and in about 2-4%of patients treated for one year. These trends continue with longer duration of use, increasing the likelihood of developing a serious GI event at some time during the course of therapy. However, even short-term therapy is not without risk. NSAIDs should be prescribed with extreme caution in those with a prior history of ulcer disease or gastrointestinal bleeding. Patients with a prior history of peptic ulcer disease and/or gastrointestinal bleeding who use NSAIDs have a greater than 10-fold increased risk for developing a GI bleed compared to patients treated with neither of these risk factors. Other factors that increase the risk of GI bleeding in patients treated with NSAIDs include concomitant use of oral corticosteroids or anticoagulants, longer duration of NSAID therapy, smoking, use of alcohol, older age, and poor general health status. Most spontaneous reports of fatal GI events are in elderly or debilitated patients and therefore, special care should be taken in treating this population.
To minimize the potential risk for an adverse GI event in patients treated with an NSAID, the lowest effective dose should be used for the shortest possible duration. Patients and physicians should remain alert for signs and symptoms of GI ulcerations and bleeding during NSAID therapy and promptly initiate additional evaluation and treatment if a serious GI event is suspected. This should include discontinuation of the NSAID until a serious GI adverse event is ruled out. For high-risk patients, alternate therapies that do not involve NSAIDs should be considered.
Renal Effects
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of a NSAID may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
Advanced Renal Disease
No information is available from controlled clinical studies regarding the use of ibuprofen tablets in patients with advanced renal disease. Therefore, treatment with ibuprofen tablets is not recommended in these patients with advanced renal disease. If ibuprofen tablet therapy must be initiated, close monitoring of the patients renal function is advisable.
Anaphylactoid Reactions
As with other NSAIDs, anaphylactoid reactions may occur in patients without known prior exposure to ibuprofen tablets. Ibuprofen tablets should not be given to patients with the aspirin triad. This symptom complex typically occurs in asthmatic patients who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs (see CONTRAINDICATIONSand PRECAUTIONS, Preexisting Asthma). Emergency help should be sought in cases where an anaphylactoid reaction occurs.
Skin Reactions
NSAIDs, including ibuprofen tablets, can cause serious skin adverse events such as exfoliative dermatitis , Stevens-Johnson Syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. These serious events may occur without warning. Patients should be informed about the signs and symptoms of serious skin manifestations and use of the drug should be discontinued at the first appearance of skin rash or any other sign of hypersensitivity.
-
PRECAUTIONS
General Precautions
Ibuprofen tablets cannot be expected to substitute for corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to disease exacerbation. Patients on prolonged corticosteroid therapy should have their therapy tapered slowly if a decision is made to discontinue corticosteroids.
The pharmacological activity of ibuprofen tablets in reducing fever and inflammation may diminish the utility of these diagnostic signs in detecting complications of presumed noninfectious, painful conditions.
Information for Patients
Patients should be informed of the following information before initiating therapy with an NSAID and periodically during the course of ongoing therapy. Patients should also be encouraged to read the NSAID Medication Guide that accompanies each prescription dispensed.
- Ibuprofen tablets like other NSAIDs, may cause serious CV side effects, such as MI or stroke, which may result in hospitalization and even death. Although serious CV events can occur without warning symptoms, patients should be alert for the signs and symptoms of chest pain , shortness of breath, weakness, slurring of speech, and should ask for medical advice when observing any indicative sign or symptoms. Patients should be apprised of the importance of this follow-up (see WARNINGS, Cardiovascular Effects).
- Ibuprofen tablets, like other NSAIDs, can cause GI discomfort and, rarely, serious GI side effects, such as ulcers and bleeding, which may result in hospitalization and even death. Although serious GI tract ulcerations and bleeding can occur without warning symptoms, patients should be alert for the signs and symptoms of ulcerations and bleeding, and should ask for medical advice when observing any indicative signs or symptoms including epigastric pain, dyspepsia , melena , and hematemesis . Patients should be apprised of the importance of this follow-up (see WARNINGS, Gastrointestinal Effects- Risk of Ulceration, Bleeding and Perforation).
- Ibuprofen tablets, like other NSAIDs, can cause serious skin side effects such as exfoliative dermatitis, SJS and TEN, which may result in hospitalization and even death. Although serious skin reactions may occur without warning, patients should be alert for the signs and symptoms of skin rash and blisters, fever, or other signs hypersensitivity such as itching, and should ask for medical advice when observing any indicative sign or symptoms. Patients should be advised to stop the drug immediately if they develop any type of rash and contact their physicians as soon as possible.
- Patients should promptly report signs or symptoms of unexplained weight gain or edema to their physicians.
- Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness and "flu-like" symptoms). If these occur, patients should be instructed to stop therapy and seek immediate medical therapy.
- Patients should be informed of the signs of an anaphylactoid reaction (e.g. difficulty breathing, swelling of the face or throat). If these occur, patients should be instructed to seek immediate emergency help (see WARNINGS).
- In late pregnancy, as with other NSAIDs, ibuprofen tablets should be avoided because it may cause premature closure of the ductus arteriosus.
Laboratory Tests
Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs or symptoms of GI bleeding. Patients on long-term treatment with NSAIDs should have their CBC and chemistry profile checked periodically. If clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash etc.), or abnormal liver tests persist or worsen, ibuprofen tablets should be discontinued.
Drug Interactions
ACE-inhibitors
Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE-inhibitors. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE-inhibitors.
Aspirin
When ibuprofen tablets are administered with aspirin, its protein binding is reduced, although the clearance of free ibuprofen tablets is not altered. The clinical significance of this interaction is not known; however, as with other NSAIDs, concomitant administration of ibuprofen and aspirin is not generally recommended because of the potential for increased adverse effects.
Diuretics
Clinical studies, as well as post marketing observations, have shown that ibuprofen tablets can reduce the natriuretic effect-of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis. During concomitant therapy with NSAIDs, the patient should be observed closely for signs of renal failure (see WARNINGS , Renal Effects), as well as to assure diuretic efficacy.
Lithium
Ibuprofen produced an elevation of plasma lithium levels and a reduction in renal lithium clearance in a study of eleven normal volunteers. The mean minimum lithium concentration increased 15%and the renal clearance of lithium was decreased by 19%during this period of concomitant drug administration. This effect has been attributed to inhibition of renal prostaglandin synthesis by ibuprofen. Thus, when ibuprofen and lithium are administered concurrently, subjects should be observed carefully for signs of lithium toxicity. (Read circulars for lithium preparation before use of such concurrent therapy.)
Methotrexate
NSAIDs have been reported to competitively inhibit methotrexate accumulation in rabbit kidney slices. This may indicate that they could enhance the toxicity of methotrexate. Caution should be used when NSAIDs are administered concomitantly with methotrexate.
Warfarin-type anticoagulants
Several short-term controlled studies failed to show that ibuprofen tablets significantly affected prothrombin times or a variety of other clotting factors when administered to individuals on coumarin-type anticoagulants. However, because bleeding has been reported when ibuprofen tablets and other NSAIDs have been administered to patients on coumarin-type anticoagulants, the physician should be cautious when administering ibuprofen tablets to patients on anticoagulants. The effects of warfarin and NSAIDs on GI bleeding are synergistic, such that the users of both drugs together have a risk of serious GI bleeding higher than users of either drug alone.
Pregnancy
Teratogenic Effects
Reproductive studies conducted in rats and rabbits have not demonstrated evidence of developmental abnormalities. However, animal reproduction studies are not always predictive of human response. There are no adequate and well-controlled studies in pregnant women. Ibuprofen tablets should be used in pregnancy only if the potential benefit justifies the potential risk to the fetus .
Labor And Delivery
In rat studies with NSAIDs, as with other drugs known to inhibit prostaglandin synthesis, an increased incidence of dystocia , delayed parturition , and decreased pup survival occurred. The effects of ibuprofen tablets on labor and delivery in pregnant women are unknown.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human-milk and because of the potential for serious adverse reactions in nursing infants from ibuprofen tablets, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness of ibuprofen tablets in pediatric patients have not been established.
Geriatric Use
As with any NSAIDs, caution should be exercised in treating the elderly (65 years and older).
Hepatic effects
Borderline elevations of one or more liver tests may occur in up to 15%of patients taking NSAIDs, including ibuprofen tablets. These laboratory abnormalities may progress, may remain unchanged, or may be transient with continuing therapy. Notable elevations of ALT or AST (approximately three or more times the upper limit of normal) have been reported in approximately 1%of patients in clinical trials with NSAIDs. In addition, rare cases of severe hepatic reactions, including jaundice , fulminant hepatitis , liver necrosis, and hepatic failure, some of them with fatal outcomes have been reported.
A patient with symptoms and/or signs suggesting liver dysfunction, or with abnormal liver test values, should be evaluated for evidence of the development of a more severe hepatic reaction while on therapy with ibuprofen tablets. If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia , rash, etc.), ibuprofen tablets should be discontinued.
Hematological effects
Anemia is sometimes seen in patients receiving NSAIDs, including ibuprofen tablets. This may be due to fluid retention, occult or gross GI blood loss, or an incompletely described effect upon erythropoiesis. Patients on long-term treatment with NSAIDs, including ibuprofen tablets, should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia.
In two postmarketing clinical studies the incidence of a decreased hemoglobin level was greater than previously reported. Decrease in hemoglobin of 1 gram or more was observed in 17.1%of 193 patients on 1600 mg ibuprofen daily ( osteoarthritis ), and in 22.8%of 189 patients taking 2400 mg of ibuprofen daily ( rheumatoid arthritis ). Positive stool occult blood tests and elevated serum creatinine levels were also observed in these studies.
NSAIDs inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Unlike aspirin, their effect on platelet function is quantitatively less, of shorter duration, and reversible.
Patients receiving ibuprofen tablets who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants should be carefully monitored.
Preexisting asthma
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm, which can be fatal. Since cross reactivity, including bronchospasm, between aspirin and NSAIDs has been reported in such aspirin-sensitive patients, ibuprofen tablets should not be administered to patients with this form of aspirin sensitivity and should be used with caution in patients with preexisting asthma.
Ophthalmological effects
Blurred and/or diminished vision, scotomata, and/or changes in color vision have been reported. If a patient develops such complaints while receiving ibuprofen tablets, the drug should be discontinued, and the patient should have an ophthalmologic examination which includes central visual fields and color vision testing.
Aseptic meningitis
Aseptic meningitis with fever and coma has been observed on rare occasions in patients on ibuprofen therapy. Although it is probably more likely to occur in patients with systemic lupus erythematosus and related connective tissue diseases, it has been reported in patients who do not have an underlying chronic disease . If signs or symptoms of meningitis develop in a patient on ibuprofen tablets, the possibility of its being related to ibuprofen tablets should be considered.
-
ADVERSE REACTIONS
The most frequent type of adverse reaction occurring with ibuprofen tablets is gastrointestinal . In controlled clinical trials the percentage of patients reporting one or more gastrointestinal complaints ranged from 4%to 16%.
In controlled studies when ibuprofen tablets were compared to aspirin and indomethacin in equally effective doses, the overall incidence of gastrointestinal complaints was about half that seen in either the aspirin- or indomethacin-treated patients.
Adverse reactions observed during controlled clinical trials at an incidence greater than 1%are listed in the table. Those reactions listed in Column one encompass observations in approximately 3,000 patients. More than 500 of these patients were treated for periods of at least 54 weeks.
Still other reactions occurring less frequently than 1 in 100 were reported in controlled clinical trials and from marketing experience. These reactions have been divided into two categories: Column two of the table lists reactions with therapy with ibuprofen tablets where the probability of a causal relationship exists: for the reactions in Column three, a causal relationship with ibuprofen tablets has not been established.
Reported side effects were higher at doses of 3200 mg/day than at doses of 2400 mg or less per day in clinical trials of patients with rheumatoid arthritis . The increases in incidence were slight and still within the ranges reported in the table.
* Reactions are classified under "Probable Causal Relationship (PCR)" if there has been one positive rechallenge or if three or more cases occur which might be causally related. Reactions are classified under "Causal Relationship Unknown" if seven or more events have been reported but the criteria for PCR have not been met.
† Reactions occurring in 3% to 9% of patients treated with ibuprofen tablets. (Those reactions occurring in less than 3% of the patients are unmarked).
Incidence Greater than 1% (but less than 3%) probable casual Relationship
Precise Incidence Unknown (but less than 1%) probable Casual Relationship* Precise Incidence Unknown (but less than 1%) Casual Relationship Unknown*
GASTROINTESTINAL
Nausea†, epigastric paint†, heartburn, diarrhea, abdominal distress, nausea and vomiting, indigestion, constipation, abdominal cramps or Pain, fullness of GI tract (bloating and flatulence)
Gastric or duodenal ulcer with bleeding and/or perforation, gastrointestinal hemorrhage, melena, gastritis, hepatitis, jaundice, abnormal liver function tests; pancreatis
CENTRAL NERVOUS SYSTEM
Dizziness†, headache, nervousness
Depression, insomnia, confusion, emotional liability, somnolence, aseptic meningitis with fever and coma (see PRECAUTIONS)
Paresthesias, hallucinations, dream abnormalities, pseudo-tumor cerebri
DERMATOLOGIC
Rash† (including maculopapular type), pruritis
Vesiculobullous eruptions, urticaria, erythema multiforme, Stevens-Johnson syndrome, alopecia
Toxic epidermal necrolysis, photoallergic skin reactions
SPECIAL SENSES
Tinnitus
Hearing loss, amblyopia (blurred and/or diminished vision, scotomata and/or changes in color vision) (see PRECAUTIONS)
Conjuctivitis, diplopia, optic neuritis, cataracts
HEMATOLOGIC
Neutropenia, agranulocytosis, aplastic anemia, hemolytic anemia (sometimes Coombs positive), thrombocytopenia with or without purpura, eosinophilia, decreases in hemoglobin and hematocrit (see PRECAUTIONS)
Bleeding episodes (eg epistaxis, menorrhagia)
METABOLIC/ENDOCRINE
Decreased appetite
Gynecomastia, hypoglycemic reaction, acidosis
CARDIOVASCULAR
Edema, fluid retention (generally responds promptly to drug discontinuation) (see PRECAUTIONS)
Congestive heart failure in patients with marginal cardiac function elevated blood pressure, palpitations
Arrhythmias (sinus tachycardia, sinus bradycardia)
ALLERGIC
Syndrome of abdominal pain, fever, chills, nausea and vomiting; anaphylaxis; bronchospasm (see CONTRADICATIONS)
Serum sickness, lupuserythematosus syndrome, Henoch-Schonlein vasculitis, angioedema
RENAL
Acute renal failure see PRECAUTIONS ), decreased creatinine clearance, polyuria, azotemia, cystitis, Hematuria
Renal papillary necrosis
MISCELLANEOUS
Dry eyes and mouth, gingival ulcer, rhinitis
-
OVERDOSAGE
Approximately 1½ hours after the reported ingestion of from 7 to 10 ibuprofen tablets (400 mg), a 19-month old child weighing 12 kg was seen in the hospital emergency room, apneic and cyanotic , responding only to painful stimuli. This type of stimulus, however, was sufficient to induce respiration . Oxygen and parenteral fluids were given; a greenish-yellow fluid was aspirated from the stomach with no evidence to indicate the presence of ibuprofen . Two hours after ingestion the child's condition seemed stable; she still responded only to painful stimuli and continued to have periods of apnea lasting from 5 to 10 seconds. She was admitted to intensive care and sodium bicarbonate was administered as well as infusions of dextrose and normal saline. By four hours post-ingestion she could be aroused easily, sit by herself and respond to spoken commands. Blood level of ibuprofen was 102.9 μg/mL approximately 8½ hours after accidental ingestion. At 12 hours she appeared to be completely recovered.
In two other reported cases where children (each weighing approximately 10 kg) accidentally, acutely ingested approximately 120 mg/kg, there were no signs of acute intoxication or late sequelae . Blood level in one child 90 minutes after ingestion was 700 μg/mL — about 10 times the peak levels seen in absorption-excretion studies.
A 19-year old male who had taken 8,000 mg of ibuprofen over a period of a few hours complained of dizziness , and nystagmus was noted. After hospitalization, parenteral hydration and three days bed rest, he recovered with no reported sequelae.
In cases of acute overdosage, the stomach should be emptied by vomiting or lavage, though little drug will likely be recovered if more than an hour has elapsed since ingestion. Because the drug is acidic and is excreted in the urine, it is theoretically beneficial to administer alkali and induce diuresis . In addition to supportive measures, the use of oral activated charcoal may help to reduce the absorption and reabsorption of ibuprofen tablets.
-
DOSAGE AND ADMINISTRATION
Carefully consider the potential benefits and risks of ibuprofen tablets and other treatment options before deciding to use ibuprofen tablets. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS).
After observing the response to initial therapy with ibuprofen tablets, the dose and frequency should be adjusted to suit an individual patient's needs.
Do not exceed 3200 mg total daily dose. If gastrointestinal complaints occur, administer ibuprofen tablets with meals or milk.
Rheumatoid arthritis and osteoarthritis, including flare-ups of chronic disease
Suggested Dosage: 1200 mg-3200 mg daily (300 mg qid; 400 mg, 600 mg or 800 mg tid or qid). Individual patients may show a better response to 3200 mg daily, as compared with 2400 mg, although in well-controlled clinical trials patients on 3200 mg did not show a better mean response in terms of efficacy. Therefore, when treating patients with 3200 mg/day, the physician should observe sufficient increased clinical benefits to offset potential increased risk.
The dose should be tailored to each patient, and may be lowered or raised depending on the severity of symptoms either at time of initiating drug therapy or as the patient responds or fails to respond.
In general, patients with rheumatoid arthritis seem to require higher doses of ibuprofen tablets than do patients with osteoarthritis.
The smallest dose of ibuprofen tablets that yields acceptable control should be employed. A linear blood level dose-response relationship exists with single doses up to 800 mg (See CLINICAL PHARMACOLOGY for effects of food on rate of absorption).
The availability of four tablet strengths facilitates dosage adjustment.
In chronic conditions , a therapeutic response to therapy with ibuprofen tablets is sometimes seen in a few days to a week but most often is observed by two weeks. After a satisfactory response has been achieved, the patient's dose should be reviewed and adjusted as required.
Mild to moderate pain: 400 mg every 4 to 6 hours as necessary for relief of pain.
In controlled analgesic clinical trials, doses of ibuprofen tablets greater than 400 mg were no more effective than the 400 mg dose.
-
HOW SUPPLIED
Ibuprofen tablets are available in the following strengths:
400 mg (white to off white, round, biconvex, film coated tablets debossed with ‘121' on one side and plain on other side)
Blister strips of 5 (NDC 55607-400-10)
Store at controlled room temperature 20° to 25°C (68° to 77°F) [see USP].
Rx only
Manufactured for:
Health Science Funding, LLC
Morristown, NJ 07960 USAManufactured by:
Marksans Pharma Ltd.
Verna, Goa-403 722, IndiaIss. 03/14
-
SPL Medguide
Medication Guide for Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
(See the end of this Medication Guide for a list of prescription NSAID medicines)
What is the most important information I should know about medicines called Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)?
NSAID medicines may increase the chance of a heart attack or stroke that can lead to death. This chance increases:
- with longer use of NSAID medicines
- in people who have heart disease
NSAID medicines should never be used right before or after a heart surgery called a "coronary artery bypass graft (CABG)."
NSAID medicines can cause ulcers and bleeding in the stomach and intestines at any time during treatment. Ulcers and bleeding:
- can happen without warning symptoms
- may cause death
The chance of a person getting an ulcer or bleeding increases with:
- taking medicines called "corticosteroids" and "anticoagulants"
- longer use
- smoking
- drinking alcohol
- older age
- having poor health
NSAID medicines should only be used:
- exactly as prescribed
- at the lowest dose possible for your treatment
- for the shortest time needed
What are Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)?
NSAID medicines are used to treat pain and redness, swelling, and heat (inflammation) from medical conditions such as:
- different types of arthritis
- menstrual cramps and other types of short-term pain
Who should not take a Non-Steroidal Anti-Inflammatory Drug (NSAID)?
Do not take an NSAID medicine:
- if you had an asthma attack, hives, or other allergic reaction with aspirin or any other NSAID medicine
- for pain right before or after heart bypass surgery
Tell your healthcare provider:
- about all of your medical conditions
- about all of the medicines you take. NSAIDs and some other medicines can interact with each other and cause serious side effects. Keep a list of your medicines to show to your healthcare provider and pharmacist.
- if you are pregnant. NSAID medicines should not be used by pregnant women late in their pregnancy.
- if you are breastfeeding. Talk to your doctor.
What are the possible side effects of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)?
Serious side effects include:
- heart attack
- stroke
- high blood pressure
- heart failure from body swelling (fluid retention)
- kidney problems including kidney failure
- bleeding and ulcers in the stomach and intestine
- low red blood cells (anemia)
- life-threatening skin reactions
- life-threatening allergic reactions
- liver problems including liver failure
- asthma attacks in people who have asthma
Other side effects include:
- stomach pain
- constipation
- diarrhea
- gas
- heartburn
- nausea
- vomiting
- dizziness
Get emergency help right away if you have any of the following symptoms:
- shortness of breath or trouble breathing
- chest pain
- weakness in one part or side of your body
- slurred speech
- swelling of the face or throat
Stop your NSAID medicine and call your healthcare provider right away if you have any of the following symptoms:
- nausea
- more tired or weaker than usual
- itching
- your skin or eyes look yellow
- stomach pain
- flu-like symptoms
- vomit blood
- there is blood in your bowel movement or it is black and sticky like tar
- unusual weight gain
- skin rash or blisters with fever
- swelling of the arms and legs, hands and feet
These are not all the side effects with NSAID medicines. Talk to your healthcare provider or pharmacist for more information about NSAID medicines.
Other information about Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
- Aspirin is an NSAID medicine but it does not increase the chance of a heart attack. Aspirin can cause bleeding in the brain, stomach, and intestines. Aspirin can also cause ulcers in the stomach and intestines.
- Some of these NSAID medicines are sold in lower doses without a prescription (over-the-counter). Talk to your healthcare provider before using over-the-counter NSAIDs for more than 10 days.
NSAID medicines that need a prescription
Generic Name
Tradename
Celecoxib
Celebrex
Diclofenac
Cataflam, Volataren, Arthrotec (combined with misoprostol)
Diflunisal
Dolobid
Etodolac
Lodine, Lodine XL
Fenoprofen
Nalfon, Nalfon 200
Flurbiprofen
Ansaid
Ibuprofen
Motrin, Tab-Profen, Vicoprofen* (combined with hydrocodone), combunox (combined with oxycodone)
Indomethacin
Indocin, Indocin SR, Indo-Lemmon, Indomethagan
Ketoprofen
Oruvail
Ketorolac
Toradol
Mefenamic Acid
Ponstel
Meloxicam
Mobic
Nabumetone
Relafen
Naproxen
Naprosyn, Anaprox, Anaprox DS, EC-Naproxyn, Naprelan, Naprapac (copackaged with lansoprazole)
Oxaprozin
Daypro
Piroxicam
Feldene
Sulindac
Clinoril
Tolmetin
Tolectin, Tolectin DS, Tolectin 600*Vicoprofen contains the same dose of ibuprofen as over-the-counter (OTC) NSAIDs, and is usually used for less than 10 days to treat pain. The OTC NSAID label warns that long term continuous use may increase the risk of heart attack or stroke.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Health Science Funding, LLC
Morristown, NJ 07960 USAManufactured by:
Marksans Pharma Ltd.
Verna, Goa-403 722, IndiaIss. 03/14
- Principal Display Panel - Capsule Label
- Principal Display Panel - Blister Label
- Principal Display Panel - Kit Label
-
INGREDIENTS AND APPEARANCE
PRASTERA
prasterone and ibuprofen kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55607-400 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55607-400-10 1 in 1 KIT; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 2 BLISTER PACK 30 Part 2 1 BLISTER PACK 5 Part 1 of 2 PRASTERONE
prasterone capsule, liquid filledProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Prasterone (UNII: 459AG36T1B) (Prasterone - UNII:459AG36T1B) Prasterone 200 mg Inactive Ingredients Ingredient Name Strength soybean oil (UNII: 241ATL177A) lecithin, soybean (UNII: 1DI56QDM62) gelatin (UNII: 2G86QN327L) yellow wax (UNII: 2ZA36H0S2V) titanium dioxide (UNII: 15FIX9V2JP) polysorbate 80 (UNII: 6OZP39ZG8H) silicon dioxide (UNII: ETJ7Z6XBU4) FD&C Blue No. 1 (UNII: H3R47K3TBD) Product Characteristics Color blue (blue) Score no score Shape OVAL (OVAL) Size 23mm Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 15 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date unapproved drug other 04/04/2014 Part 2 of 2 IBUPROFEN
ibuprofen tabletProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ibuprofen (UNII: WK2XYI10QM) (ibuprofen - UNII:WK2XYI10QM) ibuprofen 400 mg Inactive Ingredients Ingredient Name Strength COLLOIDAL SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOLS (UNII: 3WJQ0SDW1A) POLYVINYL ALCOHOL (UNII: 532B59J990) STARCH, CORN (UNII: O8232NY3SJ) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color white (white) Score no score Shape ROUND (ROUND) Size 13mm Flavor Imprint Code 121 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 5 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090796 04/04/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date unapproved drug other 04/04/2014 Labeler - Health Science Funding, LLC (078694644)