Label: ARGATROBAN injection

- NDC Code(s): 42023-182-01
- Packager: Par Pharmaceutical, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated August 10, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ARGATROBAN INJECTION safely and effectively. See full prescribing information for ARGATROBAN INJECTION.
ARGATROBAN INJECTION, for intravenous infusion only
Initial U.S. Approval: 2000RECENT MAJOR CHANGES
Dosing and Administration, Dosing in Pediatric Patients with Heparin Induced Thrombocytopenia-Heparin Induced Thrombocytopenia and Thrombosis Syndrome (2.5) Removed 8/2017
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
Argatroban Injection must be diluted 100-fold by mixing with 0.9% Sodium Chloride Injection, 5% Dextrose Injection or Lactated Ringer's Injection to a final concentration of 1 mg/mL. (2.1).
Heparin-Induced Thrombocytopenia (2.2)
The dose for heparin-induced thrombocytopenia without hepatic impairment is 2 mcg/kg/min administered as a continuous infusion (2.2)
Percutaneous Coronary Intervention (2.3)
The dose for patients with or at risk for heparin-induced thrombocytopenia undergoing percutaneous coronary intervention is started at 25 mcg/kg/min and a bolus of 350 mcg/kg administered via a large bore intravenous line over 3 to 5 minutes (2.3)
DOSAGE FORMS AND STRENGTHS
250 mg/2.5 mL single-dose vial (3)
WARNINGS AND PRECAUTIONS
- Hemorrhage can occur. Unexplained fall in hematocrit or blood pressure may indicate hemorrhage. (5.1)
- Hepatic impairment: Adjust starting dose and titrate carefully in patients with HIT who have moderate or severe hepatic impairment. Avoid use in PCI in patients with clinically significant hepatic impairment. (5.2)
ADVERSE REACTIONS
- HIT patients: The most common (greater than 5%) adverse reactions were dyspnea, hypotension, fever, diarrhea, sepsis, and cardiac arrest. (6.1)
- PCI patients: The most common (greater than 5%) adverse reactions were chest pain, hypotension, back pain, nausea, vomiting and headache. (6.1)
For medical advice about adverse reactions contact your medical professional. To report SUSPECTED ADVERSE REACTIONS, contact Par Pharmaceutical at 1-800-828-9393 or FDA at 1-800-FDA-1088 (1-800-332-1088) or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Heparin: Allow sufficient time for heparin's effect on activated partial thromboplastin time (aPTT) to decrease before initiating Argatroban Injection therapy. (7.1)
- Warfarin: Concomitant use results in increased prolongation of PT and INR. (7.2)
- Thrombolytic agents or glycoprotein IIb/IIIa antagonists: Safety and effectiveness of concomitant use with argatroban have not been established. (7.4, 7.5)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
RECENT MAJOR CHANGES
1 INDICATIONS AND USAGE
1.1 Heparin-Induced Thrombocytopenia
1.2 Percutaneous Coronary Intervention
2 DOSAGE AND ADMINISTRATION
2.1 Preparation for Intravenous Administration
2.2 Dosing in Patients with Heparin-Induced Thrombocytopenia
2.3 Dosing in Patients Undergoing Percutaneous Coronary Interventions
2.4 Dosing in Patients with Hepatic Impairment
2.5 Conversion to Oral Anticoagulant Therapy
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Hemorrhage
5.2 Use in Hepatic Impairment
5.3 Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Heparin
7.2 Oral Anticoagulant Agents
7.3 Aspirin/Acetaminophen
7.4 Thrombolytic Agents
7.5 Glycoprotein IIb/IIIa Antagonists
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Heparin-Induced Thrombocytopenia
14.2 Percutaneous Coronary Interventions (PCI) Patients with or at Risk for HIT
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Preparation for Intravenous Administration
Argatroban Injection must be diluted 100-fold prior to infusion. Argatroban Injection should not be mixed with other drugs prior to dilution.
Argatroban Injection should be diluted in 0.9% Sodium Chloride Injection, 5% Dextrose Injection, or Lactated Ringer's Injection to a final concentration of 1 mg/mL. The contents of each 2.5-mL vial should be diluted 100-fold by mixing with 250 mL of diluent. Use 250 mg (2.5 mL) per 250 mL of diluent or 500 mg (5 mL) per 500 mL of diluent.
The constituted solution must be mixed by repeated inversion of the diluent bag for 1 minute. Upon preparation, the solution may show slight but brief haziness due to the formation of microprecipitates that rapidly dissolve upon mixing. Use of diluent at room temperature is recommended. Colder temperatures can slow down the rate of dissolution of precipitates. The final solution must be clear before use. The pH of the intravenous solution prepared as recommended is 3.2 to 7.5. Solutions prepared as recommended are stable at controlled room temperature, 20°C to 25°C (68°F to 77°F) (see USP) in ambient indoor light for 24 hours; therefore, light-resistant measures such as foil protection for intravenous lines are unnecessary. Solutions are physically and chemically stable for up to 96 hours when protected from light and stored at controlled room temperature, 20°C to 25°C (68°F to 77°F) (see USP), or at refrigerated conditions, 5°C ± 3°C (41°F ± 5°F). Prepared solutions should not be exposed to direct sunlight. No significant potency losses have been noted following simulated delivery of the solution through intravenous tubing.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit.
2.2 Dosing in Patients with Heparin-Induced Thrombocytopenia
Initial Dosage:
Before administering argatroban, discontinue heparin therapy and obtain a baseline activated partial thromboplastin time (aPTT). The recommended initial dose of argatroban for adult patients without hepatic impairment is 2 mcg/kg/min, administered as a continuous infusion (see Table 1).
Table 1. Recommended Doses and Infusion Rates for 2 mcg/kg/min Dose of Argatroban for Patients With HIT*and Without Hepatic Impairment (1 mg/mL Final Concentration) Body Weight (kg) Dose (mcg/min) Infusion Rate (mL/hr) - *
- with or without thrombosis
50
100
6
60
120
7
70
140
8
80
160
10
90
180
11
100
200
12
110
220
13
120
240
14
130
260
16
140
280
17
Monitoring Therapy:
For use in HIT, therapy with Argatroban Injection is monitored using the aPTT with a target range of 1.5 to 3 times the initial baseline value (not to exceed 100 seconds). Tests of anticoagulant effects (including the aPTT) typically attain steady-state levels within 1 to 3 hours following initiation of Argatroban Injection. Check the aPTT 2 hours after initiation of therapy and after any dose change to confirm that the patient has attained the desired therapeutic range.
Dosage Adjustment:
After the initiation of Argatroban Injection, adjust the dose (not to exceed 10 mcg/kg/min) as necessary to obtain a steady-state aPTT in the target range [see Clinical Studies (14.1)].
2.3 Dosing in Patients Undergoing Percutaneous Coronary Interventions
Initial Dosage:
Initiate an infusion of Argatroban Injection at 25 mcg/kg/min and administer a bolus of 350 mcg/kg via a large bore intravenous line over 3 to 5 minutes (see Table 2). Check an activated clotting time (ACT) 5 to 10 minutes after the bolus dose is completed. The PCI procedure may proceed if the ACT is greater than 300 seconds.
Dosage Adjustment:
If the ACT is less than 300 seconds, an additional intravenous bolus dose of 150 mcg/kg should be administered, the infusion dose increased to 30 mcg/kg/min, and the ACT checked 5 to 10 minutes later (see Table 3).
If the ACT is greater than 450 seconds, decrease the infusion rate to 15 mcg/kg/min, and check the ACT 5 to 10 minutes later (Table 3).
Continue titrating the dose until a therapeutic ACT (between 300 and 450 seconds) has been achieved; continue the same infusion rate for the duration of the PCI procedure.
In case of dissection, impending abrupt closure, thrombus formation during the procedure, or inability to achieve or maintain an ACT over 300 seconds, additional bolus doses of 150 mcg/kg may be administered and the infusion dose increased to 40 mcg/kg/min. Check the ACT after each additional bolus or change in the rate of infusion.
Table 2. Recommended Starting and Maintenance Doses (Within the Target ACT Range) of Argatroban Injection in Patients Undergoing PCI Without Hepatic Impairment (1 mg/mL Final Concentration) Body Weight
(kg)Starting Bolus Dose
(350 mcg/kg)Starting and Maintenance Continuous Infusion Dosing For ACT
300-450 seconds
25 mcg/kg/minBolus Dose
(mcg)Bolus Volume
(mL)Continuous Infusion Dose
(mcg/min)Continuous Infusion Rate
(mL/hr)NOTE: 1 mg = 1000 mcg; 1 kg = 2.2 lbs 50
17500
18
1250
75
60
21000
21
1500
90
70
24500
25
1750
105
80
28000
28
2000
120
90
31500
32
2250
135
100
35000
35
2500
150
110
38500
39
2750
165
120
42000
42
3000
180
130
45500
46
3250
195
140
49000
49
3500
210
Table 3. Recommended Dose Adjustments of Argatroban Injection for Patients Outside of ACT Target Range Undergoing PCI Without Hepatic Impairment (1 mg/mL Final Concentration) Body Weight (kg) If ACT Less than 300 seconds
Dosage Adjustment*
30 mcg/kg/minIf ACT
Greater than 450 seconds
Dosage Adjustment†
15 mcg/kg/minAdditional
Bolus Dose
(mcg)
Bolus Volume
(mL)
Continuous Infusion
Dose
(mcg/min)
Continuous Infusion
Rate
(mL/hr)Continuous Infusion
Dose
(mcg/min)Continuous Infusion
Rate
(mL/hr)50 7500 8 1500 90 750 45 60 9000 9 1800 108 900 54 70 10500 11 2100 126 1050 63 80 12000 12 2400 144 1200 72 90 13500 14 2700 162 1350 81 100 15000 15 3000 180 1500 90 110 16500 17 3300 198 1650 99 120 18000 18 3600 216 1800 108 130 19500 20 3900 234 1950 117 140 21000 21 4200 252 2100 126 NOTE: 1 mg = 1000 mcg; 1 kg = 2.2 lbs
Monitoring Therapy:
For use in PCI, therapy with Argatroban Injection is monitored using ACT. Obtain ACTs before dosing, 5 to 10 minutes after bolus dosing, following adjustments in the infusion rate, and at the end of the PCI procedure.
Obtain additional ACTs every 20 to 30 minutes during a prolonged procedure.
Continued Anticoagulation after PCI:
If a patient requires anticoagulation after the procedure, Argatroban Injection may be continued, but at a rate of 2 mcg/kg/min and adjusted as needed to maintain the aPTT in the desired range [see Dosage and Administration (2.1)].
2.4 Dosing in Patients with Hepatic Impairment
Initial Dosage:
For adult patients with HIT and moderate or severe hepatic impairment (based on Child-Pugh classification), an initial dose of 0.5 mcg/kg/min is recommended, based on the approximately 4-fold decrease in argatroban clearance relative to those with normal hepatic function. Monitor the aPTT closely, and adjust the dosage as clinically indicated.
Monitoring Therapy:
Achievement of steady state aPTT levels may take longer and require more dose adjustments in patients with hepatic impairment compared to patients with normal hepatic function.
For patients with hepatic impairment undergoing PCI and who have HIT or are at risk for HIT, carefully titrate argatroban until the desired level of anticoagulation is achieved. Use of argatroban in PCI patients with clinically significant hepatic disease or AST/ALT levels greater than or equal to 3 times the upper limit of normal should be avoided [see Warnings and Precautions (5.2)].
2.5 Conversion to Oral Anticoagulant Therapy
Initiating Oral Anticoagulant Therapy:
When converting patients from argatroban to oral anticoagulant therapy, consider the potential for combined effects on INR with coadministration of argatroban and warfarin. A loading dose of warfarin should not be used. Initiate therapy using the expected daily dose of warfarin. To avoid prothrombotic effects and to ensure continuous anticoagulation when initiating warfarin, it is suggested that argatroban and warfarin therapy be overlapped. There are insufficient data available to recommend the duration of the overlap.
Coadministration of Warfarin and Argatroban Injection at Doses up to 2 mcg/kg/min:
Measure INR daily while Argatroban Injection and warfarin are coadministered. In general, with doses of Argatroban Injection up to 2 mcg/kg/min, Argatroban Injection can be discontinued when the INR is greater than 4 on combined therapy. After Argatroban Injection is discontinued, repeat the INR measurement in 4 to 6 hours. If the repeat INR is below the desired therapeutic range, resume the infusion of Argatroban Injection and repeat the procedure daily until the desired therapeutic range on warfarin alone is reached.
Coadministration of Warfarin and Argatroban Injection at Doses Greater than 2 mcg/kg/min:
For doses of argatroban greater than 2 mcg/kg/min, the relationship of INR between warfarin alone to the INR on warfarin plus argatroban is less predictable. In this case, in order to predict the INR on warfarin alone, temporarily reduce the dose of Argatroban Injection to a dose of 2 mcg/kg/min. Repeat the INR on Argatroban Injection and warfarin 4 to 6 hours after reduction of the Argatroban Injection dose and follow the process outlined above for administering Argatroban Injection at doses up to 2 mcg/kg/min.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Hemorrhage
Hemorrhage can occur at any site in the body in patients receiving argatroban. Unexplained fall in hematocrit or blood pressure may indicate hemorrhage. Intracranial and retroperitoneal hemorrhage [see Adverse Reactions (6.1)] has been reported. The risk of hemorrhage with argatroban may be increased in severe hypertension, immediately following lumbar puncture, spinal anesthesia, major surgery (especially involving the brain, spinal cord, or eye), hematologic conditions associated with increased bleeding tendencies such as congenital or acquired bleeding disorders, and gastrointestinal lesions such as ulcerations.
Concomitant use of argatroban with antiplatelet agents, thrombolytics, and other anticoagulants may increase the risk of bleeding.
5.2 Use in Hepatic Impairment
When administering argatroban to patients with hepatic impairment, start with a lower dose and carefully titrate until the desired level of anticoagulation is achieved. Achievement of steady state aPTT levels may take longer and require more argatroban dose adjustments in patients with hepatic impairment compared to patients with normal hepatic function [see Use in Specific Populations (8.6)]. Also, upon cessation of argatroban infusion in the hepatically impaired patient, full reversal of anticoagulant effects may require longer than 4 hours due to decreased clearance and increased elimination half-life of argatroban [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)]. Avoid the use of high doses of argatroban in patients undergoing PCI who have clinically significant hepatic disease or AST/ALT levels greater than or equal to 3 times the upper limit of normal.
5.3 Laboratory Tests
Anticoagulation effects associated with argatroban infusion at doses up to 40 mcg/kg/min correlate with increases of the aPTT. Although other global clot-based tests including prothrombin time (PT), the International Normalized Ratio (INR), and thrombin time (TT) are affected by argatroban, the therapeutic ranges for these tests have not been identified for argatroban therapy. In clinical trials in PCI, the ACT was used for monitoring argatroban anticoagulant activity during the procedure. The concomitant use of argatroban and warfarin results in prolongation of the PT and INR beyond that produced by warfarin alone [see Dosage and Administration (2.5) and Clinical Pharmacology (12.2)].
-
6 ADVERSE REACTIONS
The following adverse reaction is also discussed in other sections of the labeling:
- Risk of Hemorrhage [see Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Adverse Events in Patients with HIT (with or without Thrombosis)
Because clinical trials are conducted under widely varying conditions, adverse event rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The following safety information is based on all 568 patients treated with argatroban in Study 1 and Study 2. The safety profile of the patients from these studies is compared with that of 193 historical controls in which the adverse events were collected retrospectively. Adverse events are separated into hemorrhagic and non-hemorrhagic events.
Major bleeding was defined as bleeding that was overt and associated with a hemoglobin decrease greater than or equal to 2 g/dL, that led to a transfusion of greater than or equal to 2 units, or that was intracranial, retroperitoneal, or into a major prosthetic joint. Minor bleeding was overt bleeding that did not meet the criteria for major bleeding.
Table 4 gives an overview of the most frequently observed hemorrhagic events, presented separately by major and minor bleeding, sorted by decreasing occurrence among argatroban-treated patients with HIT (with or without thrombosis).
Table 4. Major and Minor Hemorrhagic Adverse Events in Patients With HIT* - *
- with or without thrombosis
- †
- Patients may have experienced more than 1 adverse event.
- ‡
-
The historical control group consisted of patients with a clinical diagnosis of HIT (with or without thrombosis) that were considered eligible by an independent medical panel.
DIC = disseminated intravascular coagulation.
BKA = below-the-knee amputation.
- §
- One patient experienced intracranial hemorrhage 4 days after discontinuation of argatroban and following therapy with urokinase and oral anticoagulation.
Major Hemorrhagic Events†
Argatroban-Treated Patients
(Study 1 and Study 2)
(n = 568)
%Historical Control‡
(n = 193)
%Overall bleeding
5.3
6.7
Gastrointestinal
2.3
1.6
Genitourinary and hematuria
0.9
0.5
Decrease in hemoglobin and hematocrit
0.7
0
Multisystem hemorrhage and DIC
0.5
1
Limb and BKA stump
0.5
0
Intracranial hemorrhage
0§
0.5
Minor Hemorrhagic Events†
Argatroban-Treated Patients
(Study 1 and Study 2)
(n = 568)
%Historical Control‡
(n = 193)
%Gastrointestinal 14.4
18.1
Genitourinary and hematuria 11.6
0.8
Decrease in hemoglobin and hematocrit 10.4
0 Groin 5.4
3.1
Hemoptysis 2.9
0.8
Brachial 2.4
0.8
Table 5 gives an overview of the most frequently observed non-hemorrhagic events sorted by decreasing frequency of occurrence (greater than or equal to 2%) among argatroban-treated HIT/HITTS patients.
Table 5. Non-hemorrhagic Adverse Events in Patients* With HIT† Argatroban-Treated Patients
(Study 1 and Study 2)
(n = 568)
%Historical Control‡
(n = 193)
%Dyspnea
8.1
8.8
Hypotension
7.2
2.6
Fever
6.9
2.1
Diarrhea
6.2
1.6
Sepsis
6.0
12.4
Cardiac arrest
5.8
3.1
Nausea
4.8
0.5
Ventricular tachycardia
4.8
3.1
Pain
4.6
3.1
Urinary tract infection
4.6
5.2
Vomiting
4.2
0
Infection
3.7
3.6
Pneumonia
3.3
9.3
Atrial fibrillation
3.0
11.4
Coughing
2.8
1.6
Abnormal renal function
2.8
4.7
Abdominal pain
2.6
1.6
Cerebrovascular disorder
2.3
4.1
Adverse Events in Patients with or at Risk for HIT Undergoing PCI
The following safety information is based on 91 patients initially treated with argatroban and 21 patients subsequently re-exposed to argatroban for a total of 112 PCIs with argatroban anticoagulation. Adverse events are separated into hemorrhagic (Table 6) and non-hemorrhagic (Table 7) events.
Major bleeding was defined as bleeding that was overt and associated with a hemoglobin decrease greater than or equal to 5 g/dL, that led to a transfusion of greater than or equal to 2 units, or that was intracranial, retroperitoneal, or into a major prosthetic joint.
The rate of major bleeding events in patients treated with argatroban in the PCI trials was 1.8%.
Table 6. Major and Minor Hemorrhagic Adverse Events in Patients With HIT Undergoing PCI Major Hemorrhagic Events*
Argatroban-Treated Patients
(n = 112)†
%
Retroperitoneal
0.9
Gastrointestinal
0.9
Intracranial
0
Minor Hemorrhagic Events*
Argatroban-Treated Patients
(n = 112)†
%
Groin (bleeding or hematoma)
3.6
Gastrointestinal (includes hematemesis)
2.6
Genitourinary (includes hematuria)
1.8
Decrease in hemoglobin and/or hematocrit
1.8
CABG (coronary arteries)
1.8
Access site
0.9
Hemoptysis
0.9
Other
0.9
CABG = coronary artery bypass graft.
Table 7 gives an overview of the most frequently observed non-hemorrhagic events (greater than 2%), sorted by decreasing frequency of occurrence among argatroban-treated PCI patients.
Table 7. Non-hemorrhagic Adverse Events* in Patients With HIT Undergoing PCI Argatroban Procedures*
(n = 112)†
%
Chest pain
15.2
Hypotension
10.7
Back pain
8.0
Nausea
7.1
Vomiting
6.3
Headache
5.4
Bradycardia
4.5
Abdominal pain
3.6
Fever
3.6
Myocardial infarction
3.6
There were 22 serious adverse events in 17 PCI patients (19.6% in 112 interventions). Table 8 lists the serious adverse events occurring in argatroban-treated patients with or at risk for HIT undergoing PCI.
Table 8. Serious Adverse Events in Patients With HIT Undergoing PCI* Coded Term
Argatroban Procedures†
(n = 112)
Myocardial infarction
4 (3.5%)
Angina pectoris
2 (1.8%)
Coronary thrombosis
2 (1.8%)
Myocardial ischemia
2 (1.8%)
Occlusion coronary
2 (1.8%)
Chest pain
1 (0.9%)
Fever
1 (0.9%)
Retroperitoneal hemorrhage
1 (0.9%)
Aortic stenosis
1 (0.9%)
Arterial thrombosis
1 (0.9%)
Gastrointestinal hemorrhage
1 (0.9%)
Gastrointestinal disorder (GERD)
1 (0.9%)
Cerebrovascular disorder
1 (0.9%)
Lung edema
1 (0.9%)
Vascular disorder
1 (0.9%)
Intracranial Bleeding in Other Populations
Increased risks for intracranial bleeding have been observed in investigational studies of argatroban for other uses. In a study of patients with acute myocardial infarction receiving both argatroban and thrombolytic therapy (streptokinase or tissue plasminogen activator), the overall frequency of intracranial bleeding was 1% (8 out of 810 patients). Intracranial bleeding was not observed in 317 subjects or patients who did not receive concomitant thrombolysis [see Drug Interactions (7.4)].
The safety and effectiveness of argatroban for cardiac indications other than PCI in patients with HIT have not been established.
Intracranial bleeding was also observed in a prospective, placebo-controlled study of argatroban in patients who had onset of acute stroke within 12 hours of study entry. Symptomatic intracranial hemorrhage was reported in 5 of 117 patients (4.3%) who received argatroban at 1 to 3 mcg/kg/min and in none of the 54 patients who received placebo. Asymptomatic intracranial hemorrhage occurred in 5 (4.3%) and 2 (3.7%) of the patients, respectively.
Allergic Reactions
One hundred fifty-six allergic reactions or suspected allergic reactions were observed in 1,127 individuals who were treated with argatroban in clinical pharmacology studies or for various clinical indications. About 95% (148/156) of these reactions occurred in patients who concomitantly received thrombolytic therapy (e.g., streptokinase) or contrast media.
Allergic reactions or suspected allergic reactions in populations other than patients with HIT (with or without thrombosis) include (in descending order of frequency):
- Airway reactions (coughing, dyspnea): 10% or more
- Skin reactions (rash, bullous eruption): 1 to <10%
- General reactions (vasodilation): 1 to 10%
Limited data are available on the potential formation of drug-related antibodies. Plasma from 12 healthy volunteers treated with argatroban over 6 days showed no evidence of neutralizing antibodies. No loss of anticoagulant activity was noted with repeated administration of argatroban to more than 40 patients.
-
7 DRUG INTERACTIONS
7.1 Heparin
If argatroban is to be initiated after cessation of heparin therapy, allow sufficient time for heparin's effect on the aPTT to decrease prior to initiation of argatroban therapy.
7.2 Oral Anticoagulant Agents
Pharmacokinetic drug-drug interactions between argatroban and warfarin (7.5 mg single oral dose) have not been demonstrated. However, the concomitant use of argatroban and warfarin (5 to 7.5 mg initial oral dose, followed by 2.5 to 6 mg/day orally for 6 to 10 days) results in prolongation of the prothrombin time (PT) and International Normalized Ratio (INR) [see Dosage and Administration (2.5) and Clinical Pharmacology (12.2)].
7.3 Aspirin/Acetaminophen
No drug-drug interactions have been demonstrated between argatroban and concomitantly administered aspirin or acetaminophen [see Clinical Pharmacology (12.3)].
7.4 Thrombolytic Agents
The safety and effectiveness of argatroban with thrombolytic agents have not been established [see Adverse Reactions (6.1)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited data from published literature and postmarketing reports do not suggest an association between argatroban and adverse fetal developmental outcomes. There are risks to the mother associated with untreated thrombosis in pregnancy and a risk of hemorrhage in the mother and fetus associated with use of anticoagulants (see Clinical Considerations). In animal reproduction studies, there was no evidence of adverse developmental outcomes with intravenous administration of argatroban during organogenesis in rats and rabbits at doses up to 0.3 and 0.2 times, respectively, the maximum recommended human dose (MHRD) (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnancy confers an increased risk for thromboembolism that is higher for women with underlying thromboembolic disease and certain high risk pregnancy conditions. Published data describe that women with a previous history of venous thrombosis are at high risk for recurrence during pregnancy.
Fetal/Neonatal Adverse Reactions
Use of anticoagulants, including argatroban, may increase the risk of bleeding in the fetus and neonate. Monitor neonates for bleeding [see Warnings and Precautions (5.1, 5.3)].
Labor or Delivery
All patients receiving anticoagulants, including pregnant women, are at risk for bleeding. Pregnant women receiving argatroban should be carefully monitored for evidence of excessive bleeding or unexpected changes in coagulation parameters [see Warnings and Precautions (5.1, 5.3)].
Data
Animal Data
Developmental studies performed in rats with argatroban at intravenous doses up to 27 mg/kg/day (0.3 times the maximum recommended human dose, based on body surface area) and in rabbits at intravenous doses up to 10.8 mg/kg/day (0.2 times the maximum recommended human dose, based on body surface area) have revealed no evidence of harm to the fetus.
8.2 Lactation
Risk Summary
There are no data on the presence of argatroban in human milk, or its effects on milk production. Argatroban is present in rat milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Argatroban and any potential adverse effects on the breastfed infant from Argatroban or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness have not been established in pediatric patients.
Argatroban was studied among 18 seriously ill pediatric patients who required an alternative to heparin anticoagulation. Most patients were diagnosed with HIT or suspected HIT. Age ranges of patients were < 6 months, n = 8; six months to < 8 years, n = 6; 8 to 16 years, n = 4. All patients had serious underlying conditions and were receiving multiple concomitant medications. Thirteen patients received argatroban solely as a continuous infusion (no bolus dose). Dosing was initiated in the majority of these 13 patients at 1 mcg/kg/min. Dosing was titrated as needed to achieve and maintain an aPTT of 1.5 to 3 times the baseline value. Most patients required multiple dose adjustments to maintain anticoagulation parameters within the desired range. During the 30-day study period, thrombotic events occurred during argatroban administration to two patients: and following argatroban discontinuation in three other patients. Major bleeding occurred among two patients; one patient experienced an intracranial hemorrhage after 4 days of argatroban therapy in the setting of sepsis and thrombocytopenia and another patient experienced an intracranial hemorrhage after receiving argatroban for greater than 14 days. The study findings did not establish the safe and effective use of argatroban in pediatric patients and the dosing of 1 mcg/kg/in was not supported by the pharmacokinetic data described below.
Pediatric Pharmacokinetics (PK) and Pharmacodynamics (PD)
PK parameters of argatroban were characterized in population PK/PD analysis model with sparse data from 15 seriously ill pediatric patients. Argatroban clearance in these seriously ill pediatric patients (0.16 L/hr/kg) was 50% lower compared to argatroban clearance in healthy adults (0.31 L/hr/kg). Four pediatric patients with elevated bilirubin (secondary to cardiac complications or hepatic impairment) had, on average, 80% lower clearance (0.03 L/hr/kg) when compared to pediatric patients with normal bilirubin levels.
These PK/PD analysis models based on a goal of aPTT prolongation of 1.5 to 3 times the baseline value and avoidance of an aPTT greater than 100 seconds for seriously ill pediatric patients with HIT/HITTS who require an alternative to heparin suggested the following:
-
For patients with normal hepatic function, a starting infusion rate of 0.75 mcg/kg/min may have comparable aPTT responses as a starting dose of 2 mcg/kg/min in healthy adults. Additionally, based on an evaluation of aPTT every two hours, increasing the dosage by 0.1 to 0.25 mcg/kg/min could achieve additional aPTT responses.
-
For patients with hepatic impairment, a starting infusion rate of 0.2 mcg/kg/min with increasing dosing by increments of 0.05 mcg/kg/min may have comparable argatroban exposure as expected with adult doses.
The safety and effectiveness of argatroban with the above dosing have not been adequately assessed in pediatric patients and the safety and effectiveness of argatroban is not established in pediatric patients. In addition, the described dosage did not take into account multiple factors that could affect the dosage such as current aPTT, target aPTT, and the clinical status of the patient.
8.5 Geriatric Use
Of the total number of subjects (1340) in clinical studies of argatroban, 35% were 65 and over. In the clinical studies of adult patients with HIT (with or without thrombosis), the effectiveness of argatroban was not affected by age. No trends were observed across age groups for both aPTT and the ACT. The safety analysis did suggest that older patients had increased underlying conditions, which may predispose them to events. The studies were not sized appropriately to detect differences in safety between age groups.
8.6 Hepatic Impairment
Dose reduction and careful titration are required when administering argatroban to patients with hepatic impairment. Reversal of anticoagulant effect may be prolonged in this population [see Dosage and Administration (2.3), Warning and Precautions (5.2), Clinical Pharmacology (12.3)].
-
-
10 OVERDOSAGE
Excessive anticoagulation, with or without bleeding, may be controlled by discontinuing argatroban or by decreasing the argatroban dose. In clinical studies, anticoagulation parameters generally returned from therapeutic levels to baseline within 2 to 4 hours after discontinuation of the drug. Reversal of anticoagulant effect may take longer in patients with hepatic impairment.
No specific antidote to argatroban is available; if life-threatening bleeding occurs and excessive plasma levels of argatroban are suspected, discontinue argatroban immediately and measure aPTT and other coagulation parameters. When argatroban was administered as a continuous infusion (2 mcg/kg/min) prior to and during a 4-hour hemodialysis session, approximately 20% of argatroban was cleared through dialysis.
Single intravenous doses of argatroban at 200, 124, 150, and 200 mg/kg were lethal to mice, rats, rabbits, and dogs, respectively. The symptoms of acute toxicity were loss of righting reflex, tremors, clonic convulsions, paralysis of hind limbs, and coma.
-
11 DESCRIPTION
Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5-[(aminoiminomethyl)amino]-1-oxo-2-[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4-methyl-2-piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S∙H2O. Its molecular weight is 526.66 g/mol. The structural formula is:
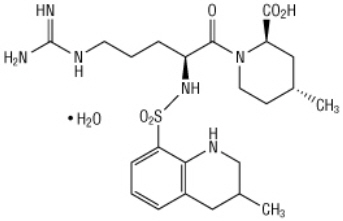
Argatroban is a white, odorless crystalline powder that is freely soluble in glacial acetic acid, slightly soluble in ethanol, and insoluble in acetone, ethyl acetate, and ether.
Argatroban Injection is a sterile clear, colorless to pale yellow, slightly viscous solution in a single-use amber vial containing 250 mg/2.5 mL of argatroban. Each mL of sterile, nonpyrogenic solution contains 100 mg argatroban, 300 mg D-sorbitol, and 400 mg dehydrated alcohol in water for injection.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Argatroban is a direct thrombin inhibitor that reversibly binds to the thrombin active site. Argatroban does not require the co-factor antithrombin III for antithrombotic activity. Argatroban exerts its anticoagulant effects by inhibiting thrombin-catalyzed or -induced reactions, including fibrin formation; activation of coagulation factors V, VIII, and XIII; activation of protein C; and platelet aggregation.
Argatroban inhibits thrombin with an inhibition constant (Ki) of 0.04 µM. At therapeutic concentrations, argatroban has little or no effect on related serine proteases (trypsin, factor Xa, plasmin, and kallikrein).
Argatroban is capable of inhibiting the action of both free and clot-associated thrombin.
12.2 Pharmacodynamics
When argatroban is administered by continuous infusion, anticoagulant effects and plasma concentrations of argatroban follow similar, predictable temporal response profiles, with low intersubject variability. Immediately upon initiation of argatroban infusion, anticoagulant effects are produced as plasma argatroban concentrations begin to rise. Steady-state levels of both drug and anticoagulant effect are typically attained within 1 to 3 hours and are maintained until the infusion is discontinued or the dosage adjusted. Steady-state plasma argatroban concentrations increase proportionally with dose (for infusion doses up to 40 mcg/kg/min in healthy subjects) and are well correlated with steady-state anticoagulant effects. For infusion doses up to 40 mcg/kg/min, argatroban increases in a dose-dependent fashion, the aPTT, the ACT, the prothrombin time (PT), the International Normalized Ratio (INR), and the thrombin time (TT) in healthy volunteers and cardiac patients.
Representative steady-state plasma argatroban concentrations and anticoagulant effects are shown below for argatroban infusion doses up to 10 mcg/kg/min (see Figure 1).
Figure 1. Relationship at Steady State Between Argatroban Dose, Plasma Argatroban Concentration and Anticoagulant Effect 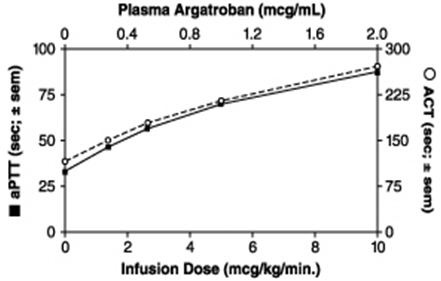
Effect on International Normalized Ratio (INR): Because argatroban is a direct thrombin inhibitor, coadministration of argatroban and warfarin produces a combined effect on the laboratory measurement of the INR. However, concurrent therapy, compared to warfarin monotherapy, exerts no additional effect on vitamin K–dependent factor Xa activity.
The relationship between INR on co-therapy and warfarin alone is dependent on both the dose of argatroban and the thromboplastin reagent used. This relationship is influenced by the International Sensitivity Index (ISI) of the thromboplastin. Data for 2 commonly utilized thromboplastins with ISI values of 0.88 (Innovin, Dade) and 1.78 (Thromboplastin C Plus, Dade) are presented in Figure 2 for an argatroban dose of 2 mcg/kg/min. Thromboplastins with higher ISI values than shown result in higher INRs on combined therapy of warfarin and argatroban. These data are based on results obtained in normal individuals [see Drug Interactions (7.2), Dosage and Administration (2.5)].
Figure 2. INR Relationship of Argatroban Plus Warfarin Versus Warfarin Alone 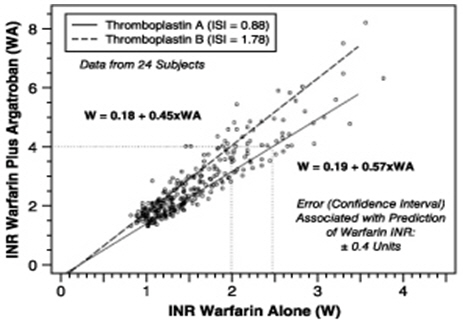
Figure 2 demonstrates the relationship between INR for warfarin alone and INR for warfarin coadministered with argatroban at a dose of 2 mcg/kg/min. To calculate INR for warfarin alone (INRW), based on INR for co-therapy of warfarin and argatroban (INRWA), when the argatroban dose is 2 mcg/kg/min, use the equation next to the appropriate curve. Example: At a dose of 2 mcg/kg/min and an INR performed with Thromboplastin A, the equation 0.19 + 0.57 (INRWA) = INRW would allow a prediction of the INR on warfarin alone (INRW). Thus, using an INRWA value of 4.0 obtained on combined therapy: INRW = 0.19 + 0.57 (4) = 2.47 as the value for INR on warfarin alone. The error (confidence interval) associated with a prediction is ± 0.4 units. Similar linear relationships and prediction errors exist for argatroban at a dose of 1 mcg/kg/min. Thus, for argatroban doses of 1 or 2 mcg/kg/min, INRW can be predicted from INRWA. For argatroban doses greater than 2 mcg/kg/min, the error associated with predicting INRW from INRWA is ± 1. Thus, INRW cannot be reliably predicted from INRWA at doses greater than 2 mcg/kg/min.
12.3 Pharmacokinetics
Distribution:
Argatroban distributes mainly in the extra cellular fluid as evidenced by an apparent steady-state volume of distribution of 174 mL/kg (12.18 L in a 70 kg adult). Argatroban is 54% bound to human serum proteins, with binding to albumin and α1-acid glycoprotein being 20% and 34%, respectively.
Metabolism:
The main route of argatroban metabolism is hydroxylation and aromatization of the 3-methyltetrahydroquinoline ring in the liver. The formation of each of the 4 known metabolites is catalyzed in vitro by the human liver microsomal cytochrome P450 enzymes CYP3A4/5. The primary metabolite (M1) exerts 3- to 5-fold weaker anticoagulant effects than argatroban. Unchanged argatroban is the major component in plasma. The plasma concentrations of M1 range between 0% and 20% of that of the parent drug. The other metabolites (M2 to M4) are found only in very low quantities in the urine and have not been detected in plasma or feces. These data, together with the lack of effect of erythromycin (a potent CYP3A4/5 inhibitor) on argatroban pharmacokinetics, suggest that CYP3A4/5-mediated metabolism is not an important elimination pathway in vivo.
Total body clearance is approximately 5.1 mL/kg/min (0.31 L/kg/hr) for infusion doses up to 40 mcg/kg/min. The terminal elimination half-life of argatroban ranges between 39 and 51 minutes.
There is no interconversion of the 21–(R):21–(S) diastereoisomers. The plasma ratio of these diastereoisomers is unchanged by metabolism or hepatic impairment, remaining constant at 65:35 (± 2%).
Excretion:
Argatroban is excreted primarily in the feces, presumably through biliary secretion. In a study in which 14C-argatroban (5 mcg/kg/min) was infused for 4 hours into healthy subjects, approximately 65% of the radioactivity was recovered in the feces within 6 days of the start of infusion with little or no radioactivity subsequently detected. Approximately 22% of the radioactivity appeared in the urine within 12 hours of the start of infusion. Little or no additional urinary radioactivity was subsequently detected. Average percent recovery of unchanged drug, relative to total dose, was 16% in urine and at least 14% in feces.
Special Populations:
Hepatic Impairment: The dosage of argatroban should be decreased in patients with hepatic impairment [see Dosage and Administration (2.3) and Warnings and Precautions (5.2)]. Patients with hepatic impairment were not studied in percutaneous coronary intervention (PCI) trials. At a dose of 2.5 mcg/kg/min, hepatic impairment is associated with decreased clearance and increased elimination half-life of argatroban (to 1.9 mL/kg/min and 181 minutes, respectively, for patients with a Child-Pugh score greater than 6).
Renal Impairment: No dosage adjustment is necessary in patients with renal dysfunction. The effect of renal disease on the pharmacokinetics of argatroban was studied in 6 subjects with normal renal function (mean Clcr = 95 ± 16 mL/min) and in 18 subjects with mild (mean Clcr = 64 ± 10 mL/min), moderate (mean Clcr = 41 ± 5.8 mL/min), and severe (mean Clcr = 5 ± 7 mL/min) renal impairment. The pharmacokinetics and pharmacodynamics of argatroban at dosages up to 5 mcg/kg/min were not significantly affected by renal dysfunction.
Use of argatroban was evaluated in a study of 12 patients with stable end-stage renal disease undergoing chronic intermittent hemodialysis. Argatroban was administered at a rate of 2 to 3 mcg/kg/min (begun at least 4 hours prior to dialysis) or as a bolus dose of 250 mcg/kg at the start of dialysis followed by a continuous infusion of 2 mcg/kg/min. Although these regimens did not achieve the goal of maintaining ACT values at 1.8 times the baseline value throughout most of the hemodialysis period, the hemodialysis sessions were successfully completed with both of these regimens. The mean ACTs produced in this study ranged from 1.39 to 1.82 times baseline, and the mean aPTTs ranged from 1.96 to 3.4 times baseline. When argatroban was administered as a continuous infusion of 2 mcg/kg/min prior to and during a 4-hour hemodialysis session, approximately 20% was cleared through dialysis.
Drug-Drug Interactions:
Digoxin: In 12 healthy volunteers, intravenous infusion of argatroban (2 mcg/kg/min) over 5 days (study days 11 to 15) did not affect the steady-state pharmacokinetics of oral digoxin (0.375 mg daily for 15 days).
Erythromycin: In 10 healthy subjects, orally administered erythromycin (a potent inhibitor of CYP3A4/5) at 500 mg four times daily for 7 days had no effect on the pharmacokinetics of argatroban at a dose of 1 mcg/kg/min for 5 hours. These data suggest oxidative metabolism by CYP3A4/5 is not an important elimination pathway in vivo for argatroban.
Aspirin and Acetaminophen: Drug-drug interactions have not been demonstrated between argatroban and concomitantly administered aspirin (162.5 mg orally given 26 and 2 hours prior to initiation of argatroban 1 mcg/kg/min over 4 hours) or acetaminophen (1,000 mg orally given 12, 6, and 0 hours prior to, and 6 and 12 hours subsequent to, initiation of argatroban 1.5 mcg/kg/min over 18 hours).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with argatroban have not been performed.
Argatroban was not genotoxic in the Ames test, the Chinese hamster ovary cell (CHO/HGPRT) forward mutation test, the Chinese hamster lung fibroblast chromosome aberration test, the rat hepatocyte, and WI-38 human fetal lung cell unscheduled DNA synthesis (UDS) tests, or the mouse micronucleus test.
Argatroban at intravenous doses up to 27 mg/kg/day (0.3 times the recommended maximum human dose based on body surface area) had no effect on fertility and reproductive function of male and female rats.
-
14 CLINICAL STUDIES
14.1 Heparin-Induced Thrombocytopenia
The safety and efficacy of argatroban were evaluated in a historically controlled efficacy and safety study (Study 1) and a follow-on efficacy and safety study (Study 2). These studies were comparable with regard to study design, study objectives, dosing regimens as well as study outline, conduct, and monitoring.
In these studies, 568 adult patients were treated with argatroban and 193 adult patients made up the historical control group. Patients had a clinical diagnosis of heparin-induced thrombocytopenia, either without thrombosis (HIT) or with thrombosis (HITTS [heparin-induced thrombocytopenia and thrombosis syndrome]) and were males or non-pregnant females between the age of 18 and 80 years old. HIT/HITTS was defined by a fall in platelet count to less than 100,000/µL or a 50% decrease in platelets after the initiation of heparin therapy with no apparent explanation other than HIT. Patients with HITTS also had an arterial or venous thrombosis documented by appropriate imaging techniques or supported by clinical evidence such as acute myocardial infarction, stroke, pulmonary embolism, or other clinical indications of vascular occlusion. Patients who had documented histories of positive heparin-dependent antibody tests without current thrombocytopenia or heparin challenge (e.g., patients with latent disease) were also included if they required anticoagulation.
These studies did not include patients with documented unexplained aPTT greater than 200% of control at baseline, documented coagulation disorder or bleeding diathesis unrelated to HIT, a lumbar puncture within the past 7 days or a history of previous aneurysm, hemorrhagic stroke, or a thrombotic stroke within the past 6 months unrelated to HIT.
The initial dose of argatroban was 2 mcg/kg/min. Two hours after the start of the argatroban infusion, an aPTT level was obtained and dose adjustments were made (up to a maximum of 10 mcg/kg/min) to achieve a steady-state aPTT value that was 1.5 to 3.0 times the baseline value, not to exceed 100 seconds. Overall the mean aPTT level for HIT and HITTS patients during the argatroban infusion increased from baseline values of 34 and 38 seconds, respectively, to 62.5 and 64.5 seconds, respectively.
The primary efficacy analysis was based on a comparison of event rates for a composite endpoint that included death (all causes), amputation (all causes) or new thrombosis during the treatment and follow-up period (study days 0 to 37). Secondary analyses included evaluation of the event rates for the components of the composite endpoint as well as time-to-event analyses.
In Study 1, a total of 304 patients were enrolled as follows: active HIT (n=129), active HITTS (n=144), or latent disease (n=31). Among the 193 historical controls, 139 (72%) had active HIT, 46 (24%) had active HITTS, and 8 (4%) had latent disease. Within each group, those with active HIT and those with latent disease were analyzed together. Positive laboratory confirmation of HIT/HITTS by the heparin-induced platelet aggregation test or serotonin release assay was demonstrated in 174 of 304 (57%) argatroban-treated patients (i.e., in 80 with HIT or latent disease and 94 with HITTS) and in 149 of 193 (77%) historical controls (i.e., in 119 with HIT or latent disease and 30 with HITTS). The test results for the remainder of the patients and controls were either negative or not determined.
There was a significant improvement in the composite outcome in patients with HIT and HITTS treated with argatroban versus those in the historical control group (see Table 9). The components of the composite endpoint are shown in Table 9.
Table 9. Efficacy Results of Study 1: Composite Endpointa and Individual Components, Ranked by Severityb HIT HITTS HIT/HITTS Parameter,
N (%)Control
n = 147Argatroban
n = 160Control
n = 46Argatroban
n = 144Control
n = 193Argatroban
n = 304Composite
Endpoint57 (38.8) 41 (25.6) 26 (56.5) 63 (43.8) 83 (43.0) 104 (34.2) Individual Componentsb: Death 32 (21.8) 27 (16.9) 13 (28.3) 26 (18.1) 45 (23.3) 53 (17.4) Amputation 3 (2.0) 3 (1.9) 4 (8.7) 16 (11.1) 7 (3.6) 19 (6.2) New Thrombosis 22 (15.0) 11 (6.9) 9 (19.6) 21 (14.6) 31 (16.1) 32 (10.5) a) Death (all cause), amputation (all cause), or new thrombosis within 37-day study period
b) Reported as the most severe outcome among the components of composite endpoint (severity ranking: death greater than amputation greater than new thrombosis); patients may have had multiple outcomes.Time-to-event analyses showed significant improvements in the time-to-first event in patients with HIT or HITTS treated with argatroban versus those in the historical control group. The between-group differences in the proportion of patients who remained free of death, amputation, or new thrombosis were statistically significant in favor of argatroban by these analyses.
A time-to-event analysis for the composite endpoint is shown in Figure 3 for patients with HIT and Figure 4 for patients with HITTS.
STUDY 1
Figure 3. Time to First Event for the Composite Efficacy Endpoint: HIT Patients *Censored indicates no clinical endpoint (defined as death, amputation, or new thrombosis) was observed during the follow-up period (maximum period of follow-up was 37 days). 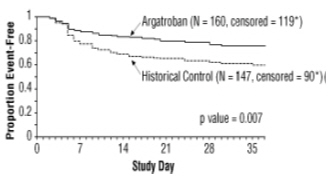
STUDY 1
Figure 4. Time to First Event for the Composite Efficacy Endpoint: HITTS Patients *Censored indicates no clinical endpoint (defined as death, amputation, or new thrombosis) was observed during the follow-up period (maximum period of follow-up was 37 days). 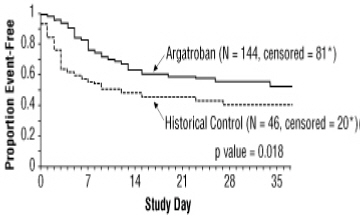
In Study 2, a total of 264 patients were enrolled as follows: HIT (n=125) or HITTS (n=139). There was a significant improvement in the composite efficacy outcome for argatroban-treated patients, versus the same historical control group from Study 1, among patients having HIT (25.6% vs. 38.8%), patients having HITTS (41.0% vs. 56.5%), and patients having either HIT or HITTS (33.7% vs. 43.0%). Time-to-event analyses showed significant improvements in the time-to-first event in patients with HIT or HITTS treated with argatroban versus those in the historical control group. The between-group differences in the proportion of patients who remained free of death, amputation, or new thrombosis were statistically significant in favor of argatroban.
Anticoagulant Effect:
In Study 1, the mean (± SE) dose of argatroban administered was 2.0 ± 0.1 mcg/kg/min in the HIT arm and 1.9 ± 0.1 mcg/kg/min in the HITTS arm. Seventy-six percent of patients with HIT and 81% of patients with HITTS achieved a target aPTT at least 1.5-fold greater than the baseline aPTT at the first assessment occurring on average at 4.6 hours (HIT) and 3.9 hours (HITTS) following initiation of argatroban therapy.
No enhancement of aPTT response was observed in subjects receiving repeated administration of argatroban.
Platelet Count Recovery:
In Study 1, 53% of patients with HIT and 58% of patients with HITTS, had a recovery of platelet count by Day 3. Platelet Count Recovery was defined as an increase in platelet count to greater than 100,000/µL or to at least 1.5-fold greater than the baseline count (platelet count at study initiation) by Day 3 of the study.
14.2 Percutaneous Coronary Interventions (PCI) Patients with or at Risk for HIT
In 3 similarly designed trials, argatroban was administered to 91 patients with current or previous clinical diagnosis of HIT or heparin-dependent antibodies, who underwent a total of 112 percutaneous coronary interventions (PCIs) including percutaneous transluminal coronary angioplasty (PTCA), coronary stent placement, or atherectomy. Among the 91 patients undergoing their first PCI with argatroban, notable ongoing or recent medical history included myocardial infarction (n = 35), unstable angina (n = 23), and chronic angina (n = 34). There were 33 females and 58 males. The average age was 67.6 years (median 70.7, range 44 to 86), and the average weight was 82.5 kg (median 81.0 kg, range 49 to 141).
Twenty-one of the 91 patients had a repeat PCI using argatroban an average of 150 days after their initial PCI. Seven of 91 patients received glycoprotein IIb/IIIa inhibitors. Safety and efficacy were assessed against historical control populations who had been anticoagulated with heparin.
All patients received oral aspirin (325 mg) 2 to 24 hours prior to the interventional procedure. After venous or arterial sheaths were in place, anticoagulation was initiated with a bolus of argatroban of 350 mcg/kg via a large-bore intravenous line or through the venous sheath over 3 to 5 minutes. Simultaneously, a maintenance infusion of 25 mcg/kg/min was initiated to achieve a therapeutic ACT of 300 to 450 seconds. If necessary to achieve this therapeutic range, the maintenance infusion dose was titrated (15 to 40 mcg/kg/min) and/or an additional bolus dose of 150 mcg/kg could be given. Each patient's ACT was checked 5 to 10 minutes following the bolus dose. The ACT was checked as clinically indicated. Arterial and venous sheaths were removed no sooner than 2 hours after discontinuation of argatroban and when the ACT was less than 160 seconds.
If a patient required anticoagulation after the procedure, argatroban could be continued, but at a lower infusion dose between 2.5 and 5 mcg/kg/min. An aPTT was drawn 2 hours after this dose reduction and the dose of argatroban then was adjusted as clinically indicated (not to exceed 10 mcg/kg/min), to reach an aPTT between 1.5 and 3 times baseline value (not to exceed 100 seconds).
In 92 of the 112 interventions (82%), the patient received the initial bolus of 350 mcg/kg and an initial infusion dose of 25 mcg/kg/min. The majority of patients did not require additional bolus dosing during the PCI procedure. The mean value for the initial ACT measurement after the start of dosing for all interventions was 379 sec (median 338 sec; 5th percentile-95th percentile 238 to 675 sec). The mean ACT value per intervention over all measurements taken during the procedure was 416 sec (median 390 sec; 5th percentile-95th percentile 261 to 698 sec). About 65% of patients had ACTs within the recommended range of 300 to 450 seconds throughout the procedure. The investigators did not achieve anticoagulation within the recommended range in about 23% of patients. However, in this small sample, patients with ACTs below 300 seconds did not have more coronary thrombotic events, and patients with ACTs over 450 seconds did not have higher bleeding rates.
Acute procedural success was defined as lack of death, emergent coronary artery bypass graft (CABG), or Q-wave myocardial infarction. Acute procedural success was reported in 98.2% of patients who underwent PCIs with argatroban anticoagulation compared with 94.3% of historical control patients anticoagulated with heparin (p = NS). Among the 112 interventions, 2 patients had emergency CABGs, 3 had repeat PTCAs, 4 had non-Q-wave myocardial infarctions, 3 had myocardial ischemia, 1 had an abrupt closure, and 1 had an impending closure (some patients may have experienced more than 1 event). No patients died.
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Inform patients of the risks associated with Argatroban Injection as well as the plan for regular monitoring during administration of the drug. Specifically, inform patients to report:
- the use of any other products known to affect bleeding.
- any medical history that may increase the risk for bleeding, including a history of severe hypertension; recent lumbar puncture or spinal anesthesia; major surgery, especially involving the brain, spinal cord, or eye; hematologic conditions associated with increased bleeding tendencies such as congenital or acquired bleeding disorders and gastrointestinal lesions such as ulcerations.
- any bleeding signs or symptoms.
- the occurrence of any signs or symptoms of allergic reactions (e.g., airway reactions, skin reactions and vasodilation reactions).
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ARGATROBAN
argatroban injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:42023-182 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ARGATROBAN (UNII: IY90U61Z3S) (ARGATROBAN ANHYDROUS - UNII:OCY3U280Y3) ARGATROBAN ANHYDROUS 250 mg in 2.5 mL Inactive Ingredients Ingredient Name Strength SORBITOL (UNII: 506T60A25R) ALCOHOL (UNII: 3K9958V90M) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:42023-182-01 1 in 1 CARTON 06/01/2015 1 2.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091665 06/01/2015 Labeler - Par Pharmaceutical, Inc. (092733690)