Label: FOSAMAX PLUS D- alendronate sodium and cholecalciferol tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 54868-5480-0 - Packager: Physicians Total Care, Inc.
- This is a repackaged label.
- Source NDC Code(s): 0006-0710
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated December 20, 2011
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FOSAMAX PLUS D safely and effectively. See full prescribing information for FOSAMAX PLUS D.
FOSAMAX® PLUS D
(alendronate sodium/cholecalciferol) tablets
Initial U.S. Approval: 2005RECENT MAJOR CHANGES
INDICATIONS AND USAGE
FOSAMAX PLUS D is a combination of a bisphosphonate and vitamin D indicated for:
- Treatment of osteoporosis in postmenopausal women (1.1)
- Treatment to increase bone mass in men with osteoporosis (1.2)
FOSAMAX PLUS D alone should not be used to treat vitamin D deficiency. (1.3)
The optimal duration of use has not been determined. Patients should have the need for continued therapy re-evaluated on a periodic basis.
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 70 mg/2800 IU and 70 mg/5600 IU (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Severe irritation of upper gastrointestinal mucosa can occur. Follow dosing instructions. Use caution in patients with active upper GI disease. Discontinue if new or worsening symptoms occur. (5.1)
- Hypocalcemia can worsen and must be corrected prior to use. (5.2)
- Severe bone, joint, muscle pain may occur. Discontinue use if severe symptoms develop. (5.3)
- Osteonecrosis of the jaw has been reported rarely. (5.4)
- Atypical femur fractures have been reported. Patients with new thigh or groin pain should be evaluated to rule out an incomplete femoral fracture. (5.5)
ADVERSE REACTIONS
The most common adverse reactions for alendronate (incidence ≥3%) are: abdominal pain, acid regurgitation, constipation, diarrhea, dyspepsia, musculoskeletal pain, nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- Calcium supplements/antacids and some medications will likely interfere with absorption of alendronate and should be taken at least 30 minutes after FOSAMAX PLUS D. (2.3, 7.1)
- Aspirin and nonsteroidal anti-inflammatory drug use may worsen gastrointestinal irritation; caution should be used. (7.2, 7.3)
- Some drugs may impair the absorption or increase the catabolism of cholecalciferol (vitamin D3). Additional vitamin D supplementation should be considered. (7.4, 7.5, 12.3)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2011
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Treatment of Osteoporosis in Postmenopausal Women
1.3 Important Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Treatment of Osteoporosis in Postmenopausal Women
2.2 Treatment to Increase Bone Mass in Men with Osteoporosis
2.3 Dosing Instructions
2.4 Recommendations for Calcium and Vitamin D Supplementation
2.5 Dosing in Elderly and Renal Insufficiency
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Upper Gastrointestinal Adverse Reactions
5.2 Mineral Metabolism
5.3 Musculoskeletal Pain
5.4 Osteonecrosis of the Jaw
5.5 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
5.6 Renal Insufficiency
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Calcium Supplements/Antacids
7.2 Aspirin
7.3 Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
7.4 Drugs that May Impair the Absorption of Cholecalciferol
7.5 Drugs that May Increase the Catabolism of Cholecalciferol
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Treatment of Postmenopausal Osteoporosis
14.2 Treatment to Increase Bone Mass in Men with Osteoporosis
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Osteoporosis Recommendations, Including Calcium and Vitamin D Supplementation
17.2 Dosing Instructions
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
FOSAMAX® PLUS D is indicated for:
1.1 Treatment of Osteoporosis in Postmenopausal Women
For the treatment of osteoporosis, FOSAMAX PLUS D increases bone mass and reduces the incidence of fractures, including those of the hip and spine (vertebral compression fractures).
1.3 Important Limitations of Use
FOSAMAX PLUS D alone should not be used to treat vitamin D deficiency.
The safety and effectiveness of FOSAMAX PLUS D for the treatment of osteoporosis are based on clinical data of four years duration. The optimal duration of use has not been determined. All patients on bisphosphonate therapy should have the need for continued therapy re-evaluated on a periodic basis.
-
2 DOSAGE AND ADMINISTRATION
2.1 Treatment of Osteoporosis in Postmenopausal Women
The recommended dosage is one 70 mg alendronate/2800 IU vitamin D3 or one 70 mg alendronate/5600 IU vitamin D3 tablet once weekly. For most osteoporotic women, the appropriate dose is FOSAMAX PLUS D (70 mg alendronate/5600 IU vitamin D3) once weekly.
2.2 Treatment to Increase Bone Mass in Men with Osteoporosis
The recommended dosage is one 70 mg alendronate/2800 IU vitamin D3 or one 70 mg alendronate/5600 IU vitamin D3 tablet once weekly. For most osteoporotic men, the appropriate dose is FOSAMAX PLUS D (70 mg alendronate/5600 IU vitamin D3) once weekly.
2.3 Dosing Instructions
FOSAMAX PLUS D must be taken at least one-half hour before the first food, beverage, or medication of the day with plain water only [see Medication Guide]. Other beverages (including mineral water), food, and some medications are likely to reduce the absorption of alendronate [see Drug Interactions (7.1)]. Waiting less than 30 minutes, or taking FOSAMAX PLUS D with food, beverages (other than plain water) or other medications will lessen the effect of alendronate by decreasing its absorption into the body.
To facilitate delivery to the stomach and thus reduce the potential for esophageal irritation, FOSAMAX PLUS D should only be swallowed upon arising for the day with a full glass of water (6-8 oz) and patients should not lie down for at least 30 minutes and until after their first food of the day. FOSAMAX PLUS D should not be taken at bedtime or before arising for the day. Failure to follow these instructions may increase the risk of esophageal adverse experiences [see Warnings and Precautions (5.1); Medication Guide].
2.4 Recommendations for Calcium and Vitamin D Supplementation
Patients should receive supplemental calcium if dietary intake is inadequate [see Warnings and Precautions (5.2)]. Patients at increased risk for vitamin D insufficiency (e.g., over the age of 70 years, nursing home bound, or chronically ill) may need additional vitamin D supplementation. Patients with gastrointestinal malabsorption syndromes may require higher doses of vitamin D supplementation and measurement of 25-hydroxyvitamin D should be considered.
The recommended intake of vitamin D is 400 IU-800 IU daily. FOSAMAX PLUS D 70 mg/2800 IU and 70 mg/5600 IU are intended to provide seven days’ worth of 400 and 800 IU daily vitamin D in a single, once-weekly dose, respectively.
Causes of osteoporosis other than estrogen deficiency, aging, and glucocorticoid use should be considered.
2.5 Dosing in Elderly and Renal Insufficiency
No dosage adjustment is necessary for the elderly or for patients with mild-to-moderate renal insufficiency (creatinine clearance 35 to 60 mL/min). FOSAMAX PLUS D is not recommended for patients with more severe renal insufficiency (creatinine clearance <35 mL/min) due to lack of experience.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
- Abnormalities of the esophagus which delay esophageal emptying such as stricture or achalasia [see Warnings and Precautions (5.1)]
- Inability to stand or sit upright for at least 30 minutes [see Dosage and Administration (2.3), Warnings and Precautions (5.1)]
- Hypocalcemia [see Warnings and Precautions (5.2)]
- Hypersensitivity to any component of this product. Hypersensitivity reactions including urticaria and angioedema have been reported [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Upper Gastrointestinal Adverse Reactions
FOSAMAX PLUS D, like other bisphosphonates administered orally, may cause local irritation of the upper gastrointestinal mucosa. Because of these possible irritant effects and a potential for worsening of the underlying disease, caution should be used when FOSAMAX PLUS D is given to patients with active upper gastrointestinal problems (such as known Barrett's esophagus, dysphagia, other esophageal diseases, gastritis, duodenitis, or ulcers).
Esophageal adverse experiences, such as esophagitis, esophageal ulcers and esophageal erosions, occasionally with bleeding and rarely followed by esophageal stricture or perforation, have been reported in patients receiving treatment with oral bisphosphonates including FOSAMAX PLUS D. In some cases these have been severe and required hospitalization. Physicians should therefore be alert to any signs or symptoms signaling a possible esophageal reaction and patients should be instructed to discontinue FOSAMAX PLUS D and seek medical attention if they develop dysphagia, odynophagia, retrosternal pain or new or worsening heartburn.
The risk of severe esophageal adverse experiences appears to be greater in patients who lie down after taking oral bisphosphonates including FOSAMAX PLUS D and/or who fail to swallow oral bisphosphonates including FOSAMAX PLUS D with the recommended full glass (6-8 oz) of water, and/or who continue to take oral bisphosphonates including FOSAMAX PLUS D after developing symptoms suggestive of esophageal irritation. Therefore, it is very important that the full dosing instructions are provided to, and understood by, the patient [see Dosage and Administration (2.3)]. In patients who cannot comply with dosing instructions due to mental disability, therapy with FOSAMAX PLUS D should be used under appropriate supervision.
There have been post-marketing reports of gastric and duodenal ulcers with oral bisphosphonate use, some severe and with complications, although no increased risk was observed in controlled clinical trials [see Adverse Reactions (6.2)].
5.2 Mineral Metabolism
Alendronate Sodium
Hypocalcemia must be corrected before initiating therapy with FOSAMAX PLUS D [see Contraindications (4)]. Other disorders affecting mineral metabolism (such as vitamin D deficiency) should also be effectively treated. In patients with these conditions, serum calcium and symptoms of hypocalcemia should be monitored during therapy with FOSAMAX PLUS D.
Presumably due to the effects of alendronate on increasing bone mineral, small, asymptomatic decreases in serum calcium and phosphate may occur.
Cholecalciferol
FOSAMAX PLUS D alone should not be used to treat vitamin D deficiency (commonly defined as 25-hydroxyvitamin D level below 9 ng/mL). Patients at increased risk for vitamin D insufficiency may require higher doses of vitamin D supplementation [see Dosage and Administration (2.4)]. Patients with gastrointestinal malabsorption syndromes may require higher doses of vitamin D supplementation and measurement of 25-hydroxyvitamin D should be considered.
Vitamin D3 supplementation may worsen hypercalcemia and/or hypercalciuria when administered to patients with diseases associated with unregulated overproduction of 1,25 dihydroxyvitamin D (e.g., leukemia, lymphoma, sarcoidosis). Urine and serum calcium should be monitored in these patients.
5.3 Musculoskeletal Pain
In post-marketing experience, severe and occasionally incapacitating bone, joint, and/or muscle pain has been reported in patients taking bisphosphonates that are approved for the prevention and treatment of osteoporosis [see Adverse Reactions (6.2)]. This category of drugs includes alendronate. Most of the patients were postmenopausal women. The time to onset of symptoms varied from one day to several months after starting the drug. Discontinue use if severe symptoms develop. Most patients had relief of symptoms after stopping. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate.
In placebo-controlled clinical studies of FOSAMAX, the percentages of patients with these symptoms were similar in the FOSAMAX and placebo groups.
5.4 Osteonecrosis of the Jaw
Osteonecrosis of the jaw (ONJ), which can occur spontaneously, is generally associated with tooth extraction and/or local infection with delayed healing, and has been reported in patients taking bisphosphonates, including FOSAMAX PLUS D. Known risk factors for osteonecrosis of the jaw include invasive dental procedures (e.g., tooth extraction, dental implants, boney surgery), diagnosis of cancer, concomitant therapies (e.g., chemotherapy, corticosteroids), poor oral hygiene, and co-morbid disorders (e.g., periodontal and/or other pre-existing dental disease, anemia, coagulopathy, infection, ill-fitting dentures).
For patients requiring invasive dental procedures, discontinuation of bisphosphonate treatment may reduce the risk for ONJ. Clinical judgment of the treating physician and/or oral surgeon should guide the management plan of each patient based on individual benefit/risk assessment.
Patients who develop osteonecrosis of the jaw while on bisphosphonate therapy should receive care by an oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition. Discontinuation of bisphosphonate therapy should be considered based on individual benefit/risk assessment.
5.5 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
Atypical, low-energy, or low trauma fractures of the femoral shaft have been reported in bisphosphonate-treated patients. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Causality has not been established as these fractures also occur in osteoporotic patients who have not been treated with bisphosphonates.
Atypical femur fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs. A number of reports note that patients were also receiving treatment with glucocorticoids (e.g. prednisone) at the time of fracture.
Any patient with a history of bisphosphonate exposure who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patients presenting with an atypical fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of bisphosphonate therapy should be considered, pending a risk/benefit assessment, on an individual basis.
5.6 Renal Insufficiency
FOSAMAX PLUS D is not recommended for patients with renal insufficiency (creatinine clearance <35 mL/min). [See Dosage and Administration (2.5).]
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
FOSAMAX
FOSAMAX has been evaluated for safety in approximately 8000 postmenopausal women in clinical studies.
Postmenopausal Women
FOSAMAX daily
In two identically designed, three-year, placebo-controlled, double-blind, multicenter studies (United States and Multinational; n=994), discontinuation of therapy due to any clinical adverse experience occurred in 4.1% of 196 patients treated with FOSAMAX 10 mg/day and 6.0% of 397 patients treated with placebo. In the Fracture Intervention Trial (n=6459), discontinuation of therapy due to any clinical adverse experience occurred in 9.1% of 3236 patients treated with FOSAMAX 5 mg/day for 2 years and 10 mg/day for either one or two additional years and 10.1% of 3223 patients treated with placebo. Discontinuations due to upper gastrointestinal adverse experiences were: FOSAMAX, 3.2%; placebo, 2.7%. In these study populations, 49-54% had a history of gastrointestinal disorders at baseline and 54-89% used nonsteroidal anti-inflammatory drugs or aspirin at some time during the studies. Adverse experiences from these studies considered by the investigators as possibly, probably, or definitely drug related in ≥1% of patients treated with either FOSAMAX or placebo are presented in Table 1.
Table 1: Osteoporosis Treatment Studies in Postmenopausal Women: Adverse Experiences Considered Possibly, Probably, or Definitely Drug Related by the Investigators and Reported in ≥1% of Patients United States/Multinational Studies Fracture Intervention Trial FOSAMAX
%
(n=196)Placebo
%
(n=397)FOSAMAX
%
(n=3236)Placebo
%
(n=3223)Gastrointestinal
abdominal pain
nausea
dyspepsia
constipation
diarrhea
flatulence
acid regurgitation
esophageal ulcer
vomiting
dysphagia
abdominal distention
gastritis
6.6
3.6
3.6
3.1
3.1
2.6
2.0
1.5
1.0
1.0
1.0
0.5
4.8
4.0
3.5
1.8
1.8
0.5
4.3
0.0
1.5
0.0
0.8
1.3
1.5
1.1
1.1
0.0
0.6
0.2
1.1
0.1
0.2
0.1
0.0
0.6
1.5
1.5
1.2
0.2
0.3
0.3
0.9
0.1
0.3
0.1
0.0
0.7Musculoskeletal
musculoskeletal (bone, muscle or joint) pain
muscle cramp
4.1
0.0
2.5
1.0
0.4
0.2
0.3
0.1Nervous System/Psychiatric
headache
dizziness
2.6
0.0
1.5
1.0
0.2
0.0
0.2
0.1Special Senses
taste perversion
0.5
1.0
0.1
0.0Rarely, rash and erythema have occurred.
The adverse experience profile was similar for the 401 patients treated with either 5- or 20-mg doses of FOSAMAX in the United States and Multinational studies. The adverse experience profile for the 296 patients who received continued treatment with either 5- or 10-mg doses of FOSAMAX in the two-year extension of these studies (treatment years 4 and 5) was similar to that observed during the three-year placebo-controlled period. During the extension period, of the 151 patients treated with FOSAMAX 10 mg/day, the proportion of patients who discontinued therapy due to any clinical adverse experience was similar to that during the first three years of the study.
FOSAMAX Once-Weekly
In a one-year, double-blind, multicenter study, the overall safety and tolerability profiles of once weekly FOSAMAX 70 mg and FOSAMAX 10 mg daily were similar. The adverse experiences considered by the investigators as possibly, probably, or definitely drug related in ≥1% of patients in either treatment group are presented in Table 2.
Table 2: Osteoporosis Treatment Studies in Postmenopausal Women: Adverse Experiences Considered Possibly, Probably, or Definitely Drug Related by the Investigators and Reported in ≥1% of Patients Once Weekly FOSAMAX
70 mg
%
(n=519)FOSAMAX
10 mg/day
%
(n=370)Gastrointestinal
abdominal pain
dyspepsia
acid regurgitation
nausea
abdominal distention
constipation
flatulence
gastritis
gastric ulcer
3.7
2.7
1.9
1.9
1.0
0.8
0.4
0.2
0.0
3.0
2.2
2.4
2.4
1.4
1.6
1.6
1.1
1.1Musculoskeletal
musculoskeletal (bone, muscle, joint) pain
muscle cramp
2.9
0.2
3.2
1.1Concomitant Use With Estrogen or Estrogen/Progestin Products
In two studies (of one and two years’ duration) of postmenopausal osteoporotic women (total: n=853), the safety and tolerability profile of combined treatment with FOSAMAX 10 mg once daily and estrogen ± progestin (n=354) was consistent with those of the individual treatments.
Men
In two placebo-controlled, double-blind, multicenter studies in men (a two-year study of FOSAMAX 10 mg/day and a one-year study of once weekly FOSAMAX 70 mg) the rates of discontinuation of therapy due to any clinical adverse experience were 2.7% for FOSAMAX 10 mg/day vs. 10.5% for placebo, and 6.4% for once weekly FOSAMAX 70 mg vs. 8.6% for placebo. The adverse experiences considered by the investigators as possibly, probably, or definitely drug related in ≥2% of patients treated with either FOSAMAX or placebo are presented in Table 3.
Table 3: Osteoporosis Studies in Men: Adverse Experiences Considered Possibly, Probably, or Definitely Drug Related by the Investigators and Reported in ≥2% of Patients Two-year Study One-year Study
FOSAMAX 10 mg/day
%
(n=146)
Placebo
%
(n=95)Once Weekly FOSAMAX 70 mg
%
(n=109)
Placebo
%
(n=58)Gastrointestinal
acid regurgitation
flatulence
gastroesophageal reflux disease
dyspepsia
diarrhea
abdominal pain
nausea
4.1
4.1
0.7
3.4
1.4
2.1
2.1
3.2
1.1
3.2
0.0
1.1
1.1
0.0
0.0
0.0
2.8
2.8
2.8
0.9
0.0
0.0
0.0
0.0
1.7
0.0
3.4
0.0Laboratory Test Findings
In double-blind, multicenter, controlled studies, asymptomatic, mild, and transient decreases in serum calcium and phosphate were observed in approximately 18% and 10%, respectively, of patients taking FOSAMAX versus approximately 12% and 3% of those taking placebo. However, the incidences of decreases in serum calcium to <8.0 mg/dL (2.0 mM) and serum phosphate to ≤2.0 mg/dL (0.65 mM) were similar in both treatment groups.
FOSAMAX PLUS D
In a fifteen-week double-blind, multinational study in osteoporotic postmenopausal women (n=682) and men (n=35), the safety profile of FOSAMAX PLUS D (70 mg/2800 IU) was similar to that of FOSAMAX once weekly 70 mg. In the 24-week double-blind extension study in women (n=619) and men (n=33), the safety profile of FOSAMAX PLUS D (70 mg/2800 IU) administered with an additional 2800 IU vitamin D3 was similar to that of FOSAMAX PLUS D (70 mg/2800 IU).
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of FOSAMAX and FOSAMAX PLUS D. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a Whole: hypersensitivity reactions including urticaria and rarely angioedema. Transient symptoms of myalgia, malaise, asthenia and rarely, fever have been reported with alendronate, typically in association with initiation of treatment. Rarely, symptomatic hypocalcemia has occurred, generally in association with predisposing conditions. Rarely, peripheral edema.
Gastrointestinal: esophagitis, esophageal erosions, esophageal ulcers, rarely esophageal stricture or perforation, and oropharyngeal ulceration. Gastric or duodenal ulcers, some severe and with complications have also been reported [see Dosage and Administration (2.3); Warnings and Precautions (5.1); Medication Guide].
Localized osteonecrosis of the jaw, generally associated with tooth extraction and/or local infection with delayed healing, has been reported rarely [see Warnings and Precautions (5.4)].
Musculoskeletal: bone, joint, and/or muscle pain, occasionally severe, and rarely incapacitating [see Warnings and Precautions (5.3)]; joint swelling; low-energy femoral shaft and subtrochanteric fractures [see Warnings and Precautions (5.5)].
Nervous System: dizziness and vertigo.
Skin: rash (occasionally with photosensitivity), pruritus, alopecia, rarely severe skin reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis.
Special Senses: rarely uveitis, scleritis or episcleritis.
-
7 DRUG INTERACTIONS
7.1 Calcium Supplements/Antacids
It is likely that calcium supplements, antacids, and some oral medications will interfere with absorption of alendronate. Therefore, patients must wait at least one-half hour after taking FOSAMAX PLUS D before taking any other oral medications.
7.2 Aspirin
In clinical studies, the incidence of upper gastrointestinal adverse events was increased in patients receiving concomitant therapy with daily doses of FOSAMAX greater than 10 mg and aspirin-containing products.
7.3 Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
FOSAMAX PLUS D may be administered to patients taking NSAIDs. In a 3-year, controlled, clinical study (n=2027) during which a majority of patients received concomitant NSAIDs, the incidence of upper gastrointestinal adverse events was similar in patients taking FOSAMAX 5 or 10 mg/day compared to those taking placebo. However, since NSAID use is associated with gastrointestinal irritation, caution should be used during concomitant use with FOSAMAX PLUS D.
7.4 Drugs that May Impair the Absorption of Cholecalciferol
Olestra, mineral oils, orlistat, and bile acid sequestrants (e.g., cholestyramine, colestipol) may impair the absorption of vitamin D. Additional vitamin D supplementation should be considered [see Clinical Pharmacology (12.3)].
7.5 Drugs that May Increase the Catabolism of Cholecalciferol
Anticonvulsants, cimetidine, and thiazides may increase the catabolism of vitamin D. Additional vitamin D supplementation should be considered [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C:
Alendronate Sodium
Reproduction studies in rats showed decreased postimplantation survival at 2 mg/kg/day and decreased body weight gain in normal pups at 1 mg/kg/day. Sites of incomplete fetal ossification were statistically significantly increased in rats beginning at 10 mg/kg/day in vertebral (cervical, thoracic, and lumbar), skull, and sternebral bones. The above doses ranged from one time (1 mg/kg) to 10 times (10 mg/kg) a maximum recommended daily dose of 10 mg/day based on surface area, mg/m2. No similar fetal effects were seen when pregnant rabbits were treated at doses up to 35 mg/kg/day (40 times a 10 mg human daily dose based on surface area, mg/m2).
Both total and ionized calcium decreased in pregnant rats at 15 mg/kg/day (13 times a 10-mg human daily dose based on surface area, mg/m2) resulting in delays and failures of delivery. Protracted parturition due to maternal hypocalcemia occurred in rats at doses as low as 0.5 mg/kg/day (0.5 times a 10 mg human daily dose based on surface area, mg/m2) when rats were treated from before mating through gestation. Maternotoxicity (late pregnancy deaths) occurred in the female rats treated with 15 mg/kg/day for varying periods of time ranging from treatment only during pre-mating to treatment only during early, middle, or late gestation; these deaths were lessened but not eliminated by cessation of treatment. Calcium supplementation either in the drinking water or by minipump could not ameliorate the hypocalcemia or prevent maternal and neonatal deaths due to delays in delivery; calcium supplementation IV prevented maternal, but not fetal deaths.
Bisphosphonates are incorporated into the bone matrix, from which they are gradually released over a period of years. The amount of bisphosphonate incorporated into adult bone, and hence, the amount available for release back into the systemic circulation, is directly related to the dose and duration of bisphosphonate use. There are no data on fetal risk in humans. However, there is a theoretical risk of fetal harm, predominantly skeletal, if a woman becomes pregnant after completing a course of bisphosphonate therapy. The impact of variables such as time between cessation of bisphosphonate therapy to conception, the particular bisphosphonate used, and the route of administration (intravenous versus oral) on the risk has not been studied.
Cholecalciferol
No data are available for cholecalciferol (vitamin D3). Administration of high doses (≥10,000 IU/every other day) of ergocalciferol (vitamin D2) to pregnant rabbits resulted in abortions and an increased incidence of fetal aortic stenosis. Administration of vitamin D2 (40,000 IU/day) to pregnant rats resulted in neonatal death, decreased fetal weight, and impaired osteogenesis of long bones postnatally.
There are no studies in pregnant women. FOSAMAX PLUS D should be used during pregnancy only if the potential benefit justifies the potential risk to the mother and fetus.
8.3 Nursing Mothers
Cholecalciferol and some of its active metabolites pass into breast milk. It is not known whether alendronate is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when FOSAMAX PLUS D is administered to nursing women.
8.4 Pediatric Use
FOSAMAX PLUS D is not indicated for use in children.
The efficacy and safety of alendronate were examined in a randomized, double-blind, placebo-controlled two-year study of 139 pediatric patients, aged 4-18 years, with severe osteogenesis imperfecta. One-hundred-and-nine patients were randomized to 5 mg alendronate daily (weight <40 kg) or 10 mg alendronate daily (weight ≥40 kg) and 30 patients to placebo. The mean baseline lumbar spine BMD Z-score of the patients was -4.5. The mean change in lumbar spine BMD Z-score from baseline to Month 24 was 1.3 in the alendronate-treated patients and 0.1 in the placebo-treated patients. Treatment with alendronate did not reduce the risk of fracture. Sixteen percent of the alendronate patients who sustained a radiologically-confirmed fracture by Month 12 of the study had delayed fracture healing (callus remodeling) or fracture non-union when assessed radiographically at Month 24 compared with 9% of the placebo-treated patients. In alendronate-treated patients, bone histomorphometry data obtained at Month 24 demonstrated decreased bone turnover and delayed mineralization time; however, there were no mineralization defects. There were no statistically significant differences between the alendronate and placebo groups in reduction of bone pain.
8.5 Geriatric Use
Of the patients receiving FOSAMAX in the Fracture Intervention Trial (FIT), 71% (n=2302) were ≥65 years of age and 17% (n=550) were ≥75 years of age. Of the patients receiving FOSAMAX in the United States and Multinational osteoporosis treatment studies in women, and osteoporosis studies in men [see Clinical Studies (14.1, 14.2)], 45% and 54%, respectively, were 65 years of age or over. No overall differences in efficacy or safety were observed between these patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Dietary requirements of vitamin D3 are increased in the elderly.
-
10 OVERDOSAGE
Alendronate Sodium
Significant lethality after single oral doses with alendronate was seen in female rats and mice at 552 mg/kg (3256 mg/m2) and 966 mg/kg (2898 mg/m2), respectively. In males, these values were slightly higher, 626 and 1280 mg/kg, respectively. There was no lethality in dogs at oral doses up to 200 mg/kg (4000 mg/m2).
No specific information is available on the treatment of overdosage with alendronate. Hypocalcemia, hypophosphatemia, and upper gastrointestinal adverse events, such as upset stomach, heartburn, esophagitis, gastritis, or ulcer, may result from oral overdosage. Milk or antacids should be given to bind alendronate. Due to the risk of esophageal irritation, vomiting should not be induced and the patient should remain fully upright.
Dialysis would not be beneficial.
Cholecalciferol
Significant lethality occurred in mice treated with a single high oral dose of calcitriol (4 mg/kg), the hormonal metabolite of cholecalciferol.
There is limited information regarding doses of cholecalciferol associated with acute toxicity, although intermittent (yearly or twice yearly) single doses of ergocalciferol (vitamin D2) as high as 600,000 IU have been given without reports of toxicity. Signs and symptoms of vitamin D toxicity include hypercalcemia, hypercalciuria, anorexia, nausea, vomiting, polyuria, polydipsia, weakness, and lethargy. Serum and urine calcium levels should be monitored in patients with suspected vitamin D toxicity. Standard therapy includes restriction of dietary calcium, hydration, and systemic glucocorticoids in patients with severe hypercalcemia.
Dialysis to remove vitamin D would not be beneficial.
-
11 DESCRIPTION
FOSAMAX PLUS D contains alendronate sodium, a bisphosphonate, and cholecalciferol (vitamin D3).
Alendronate sodium is a bisphosphonate that acts as a specific inhibitor of osteoclast-mediated bone resorption. Bisphosphonates are synthetic analogs of pyrophosphate that bind to the hydroxyapatite found in bone.
Alendronate sodium is chemically described as (4-amino-1-hydroxybutylidene) bisphosphonic acid monosodium salt trihydrate.
The empirical formula of alendronate sodium is C4H12NNaO7P2•3H2O and its formula weight is 325.12. The structural formula is:
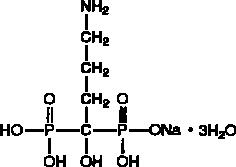
Alendronate sodium is a white, crystalline, nonhygroscopic powder. It is soluble in water, very slightly soluble in alcohol, and practically insoluble in chloroform.
Cholecalciferol (vitamin D3) is a secosterol that is the natural precursor of the calcium-regulating hormone calcitriol (1,25 dihydroxyvitamin D3).
The chemical name of cholecalciferol is (3β,5Z,7E)-9,10-secocholesta-5,7,10(19)-trien-3-ol. The empirical formula of cholecalciferol is C27H44O and its molecular weight is 384.6. The structural formula is:
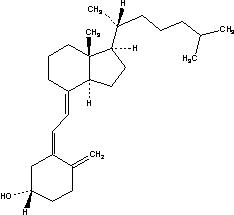
Cholecalciferol is a white, crystalline, odorless powder. Cholecalciferol is practically insoluble in water, freely soluble in usual organic solvents, and slightly soluble in vegetable oils.
FOSAMAX PLUS D for oral administration contains 91.37 mg of alendronate monosodium salt trihydrate, the molar equivalent of 70 mg of free acid, and 70 or 140 mcg of cholecalciferol, equivalent to 2800 or 5600 International Units (IU) vitamin D, respectively. Each tablet contains the following inactive ingredients: microcrystalline cellulose, lactose anhydrous, medium chain triglycerides, gelatin, croscarmellose sodium, sucrose, colloidal silicon dioxide, magnesium stearate, butylated hydroxytoluene, modified food starch, and sodium aluminum silicate.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Alendronate Sodium
Animal studies have indicated the following mode of action. At the cellular level, alendronate shows preferential localization to sites of bone resorption, specifically under osteoclasts. The osteoclasts adhere normally to the bone surface but lack the ruffled border that is indicative of active resorption. Alendronate does not interfere with osteoclast recruitment or attachment, but it does inhibit osteoclast activity. Studies in mice on the localization of radioactive [3H]alendronate in bone showed about 10‑fold higher uptake on osteoclast surfaces than on osteoblast surfaces. Bones examined 6 and 49 days after [3H]alendronate administration in rats and mice, respectively, showed that normal bone was formed on top of the alendronate, which was incorporated inside the matrix. While incorporated in bone matrix, alendronate is not pharmacologically active. Thus, alendronate must be continuously administered to suppress osteoclasts on newly formed resorption surfaces. Histomorphometry in baboons and rats showed that alendronate treatment reduces bone turnover (i.e., the number of sites at which bone is remodeled). In addition, bone formation exceeds bone resorption at these remodeling sites, leading to progressive gains in bone mass.
Cholecalciferol
Vitamin D3 is produced in the skin by photochemical conversion of 7-dehydrocholesterol to previtamin D3 by ultraviolet light. This is followed by non-enzymatic isomerization to vitamin D3. In the absence of adequate sunlight exposure, vitamin D3 is an essential dietary nutrient. Vitamin D3 in skin and dietary vitamin D3 (absorbed into chylomicrons) is converted to 25‑hydroxyvitamin D3 in the liver. Conversion to the active calcium-mobilizing hormone 1,25-dihydroxyvitamin D3 (calcitriol) in the kidney is stimulated by both parathyroid hormone and hypophosphatemia. The principal action of 1,25‑dihydroxyvitamin D3 is to increase intestinal absorption of both calcium and phosphate as well as regulate serum calcium, renal calcium and phosphate excretion, bone formation and bone resorption.
Vitamin D is required for normal bone formation. Vitamin D insufficiency develops when both sunlight exposure and dietary intake are inadequate. Insufficiency is associated with negative calcium balance, increased parathyroid hormone levels, bone loss, and increased risk of skeletal fracture. In severe cases, deficiency results in more severe hyperparathyroidism, hypophosphatemia, proximal muscle weakness, bone pain and osteomalacia.
12.2 Pharmacodynamics
Alendronate Sodium
Alendronate is a bisphosphonate that binds to bone hydroxyapatite and specifically inhibits the activity of osteoclasts, the bone-resorbing cells. Alendronate reduces bone resorption with no direct effect on bone formation, although the latter process is ultimately reduced because bone resorption and formation are coupled during bone turnover.
Daily oral doses of alendronate (5, 20, and 40 mg for six weeks) in postmenopausal women produced biochemical changes indicative of dose-dependent inhibition of bone resorption, including decreases in urinary calcium and urinary markers of bone collagen degradation (such as deoxypyridinoline and cross-linked N‑telopeptides of type I collagen). These biochemical changes tended to return toward baseline values as early as 3 weeks following the discontinuation of therapy with alendronate and did not differ from placebo after 7 months.
Long-term treatment of osteoporosis with FOSAMAX 10 mg/day (for up to five years) reduced urinary excretion of markers of bone resorption, deoxypyridinoline and cross-linked N‑telopeptides of type l collagen, by approximately 50% and 70%, respectively, to reach levels similar to those seen in healthy premenopausal women. The decrease in the rate of bone resorption indicated by these markers was evident as early as one month and at three to six months reached a plateau that was maintained for the entire duration of treatment with FOSAMAX. In osteoporosis treatment studies FOSAMAX 10 mg/day decreased the markers of bone formation, osteocalcin and bone specific alkaline phosphatase by approximately 50%, and total serum alkaline phosphatase by approximately 25 to 30% to reach a plateau after 6 to 12 months. Similar reductions in the rate of bone turnover were observed in postmenopausal women during one-year studies with once weekly FOSAMAX 70 mg for the treatment of osteoporosis. These data indicate that the rate of bone turnover reached a new steady-state, despite the progressive increase in the total amount of alendronate deposited within bone.
As a result of inhibition of bone resorption, asymptomatic reductions in serum calcium and phosphate concentrations were also observed following treatment with FOSAMAX. In the long-term studies, reductions from baseline in serum calcium (approximately 2%) and phosphate (approximately 4 to 6%) were evident the first month after the initiation of FOSAMAX 10 mg. No further decreases in serum calcium were observed for the five-year duration of treatment; however, serum phosphate returned toward prestudy levels during years three through five. In one-year studies with once weekly FOSAMAX 70 mg, similar reductions were observed at 6 and 12 months. The reduction in serum phosphate may reflect not only the positive bone mineral balance due to FOSAMAX but also a decrease in renal phosphate reabsorption.
Osteoporosis in Men
Treatment of men with osteoporosis with FOSAMAX 10 mg/day for two years reduced urinary excretion of cross-linked N-telopeptides of type I collagen by approximately 60% and bone-specific alkaline phosphatase by approximately 40%. Similar reductions were observed in a one-year study in men with osteoporosis receiving once weekly FOSAMAX 70 mg.
Cholecalciferol
Vitamin D is required for normal bone formation. Vitamin D insufficiency is associated with negative calcium balance, leading to increased parathyroid hormone levels and worsening of bone loss associated with osteoporosis. When taken without vitamin D, alendronate is also associated with a reduction in serum calcium concentrations and increased parathyroid hormone levels. In a 15‑week trial, 717 postmenopausal women and men, mean age 67 years, with osteoporosis (lumbar spine bone mineral density [BMD] of at least 2.5 standard deviations below the premenopausal mean) were randomized to receive either weekly FOSAMAX PLUS D 70 mg/2800 IU vitamin D or weekly FOSAMAX 70 mg alone with no vitamin D supplementation. Patients who were vitamin D deficient (25‑hydroxyvitamin D <9 ng/mL) at baseline were excluded. Treatment with FOSAMAX PLUS D 70 mg/2800 IU resulted in a smaller reduction in serum calcium levels (-0.9%) when compared to FOSAMAX 70 mg alone (-1.4%). As well, treatment with FOSAMAX PLUS D 70 mg/2800 IU resulted in a significantly smaller increase in parathyroid hormone levels when compared to FOSAMAX 70 mg alone (14% and 24%, respectively).
The sufficiency of patients’ vitamin D status is best assessed by measuring 25-hydroxyvitamin D levels. In the 15‑week trial mentioned above, baseline 25-hydroxyvitamin D levels were 22.2 ng/mL in the FOSAMAX PLUS D group and 22.1 ng/mL in the FOSAMAX only group. After 15 weeks of treatment, the mean levels were 23.1 ng/mL and 18.4 ng/mL in the FOSAMAX PLUS D and FOSAMAX only groups, respectively. The final levels of 25-hydroxyvitamin D at Week 15 are summarized in Table 4.
Table 4: 25-hydroxyvitamin D Levels after Treatment with FOSAMAX PLUS D (70 mg/2800 IU) or FOSAMAX 70 mg at Week 15 Number (%) of Patients 25-hydroxyvitamin D Ranges (ng/mL) <9 9-14 15-19 20-24 25-29 30-62 FOSAMAX PLUS D
(70 mg/2800 IU)
(N=357)4 (1.1) 37 (10.4) 87 (24.4) 84 (23.5) 82 (23.0) 63 (17.7) FOSAMAX 70 mg
(N=351)46 (13.1) 66 (18.8) 108 (30.8) 58 (16.5) 37 (10.5) 36 (10.3) Patients (n=652) who completed the above 15-week trial continued in a 24-week extension in which all received FOSAMAX PLUS D (70 mg/2800 IU) and were randomly assigned to receive either additional once weekly vitamin D3 2800 IU (Vitamin D3 5600 IU group) or matching placebo (Vitamin D3 2800 IU group). After 24 weeks of extended treatment (Week 39 from original baseline), the mean levels of 25-hydroxyvitamin D were 27.9 ng/mL and 25.6 ng/mL in the vitamin D3 5600 IU group and vitamin D3 2800 IU group, respectively. The percentage of patients with hypercalciuria at Week 39 was not statistically different between treatment groups.
The distribution of the final levels of 25-hydroxyvitamin D at Week 39 is summarized in Table 5.
Table 5: 25-hydroxyvitamin D Levels after Treatment with FOSAMAX PLUS D at Week 39 Number (%) of Patients 25-hydroxyvitamin D Ranges (ng/mL) <9 9-14 15-19 20-24 25-29 30-59 FOSAMAX PLUS D (Vitamin D3 5600 IU group) (N=321) 0 10 (3.1) 29 (9.0) 79 (24.6) 87 (27.1) 116 (36.1) FOSAMAX PLUS D (Vitamin D3 2800 IU group) (N=320) 1 (0.3) 17 (5.3) 56 (17.5) 80 (25.0) 74 (23.1) 92 (28.8) 12.3 Pharmacokinetics
Absorption
Alendronate Sodium
Relative to an intravenous (IV) reference dose, the mean oral bioavailability of alendronate in women was 0.64% for doses ranging from 5 to 70 mg when administered after an overnight fast and two hours before a standardized breakfast. Oral bioavailability of the 10-mg tablet in men (0.59%) was similar to that in women when administered after an overnight fast and 2 hours before breakfast.
In a study, the alendronate in the FOSAMAX PLUS D (70 mg/2800 IU) tablet and the FOSAMAX (alendronate sodium) 70-mg tablet were found to be equally bioavailable. In a separate study, the alendronate in the FOSAMAX PLUS D (70 mg/5600 IU) tablet was found to be equally bioavailable to the alendronate in the FOSAMAX (alendronate sodium) 70-mg tablet.
A study examining the effect of timing of a meal on the bioavailability of alendronate was performed in 49 postmenopausal women. Bioavailability was decreased (by approximately 40%) when 10 mg alendronate was administered either 0.5 or 1 hour before a standardized breakfast, when compared to dosing 2 hours before eating. In studies of treatment and prevention of osteoporosis, alendronate was effective when administered at least 30 minutes before breakfast.
Bioavailability was negligible whether alendronate was administered with or up to two hours after a standardized breakfast. Concomitant administration of alendronate with coffee or orange juice reduced bioavailability by approximately 60%.
Cholecalciferol
Following administration of FOSAMAX PLUS D (70 mg/2800 IU) after an overnight fast and two hours before a standard meal, the baseline adjusted mean area under the serum-concentration-time curve (AUC0-120 hrs) for vitamin D3 was 120.7 ng-hr/mL. The baseline adjusted mean maximal serum concentration (Cmax) of vitamin D3 was 4.0 ng/mL, and the baseline adjusted mean time to maximal serum concentration (Tmax) was 10.6 hrs. The bioavailability of the 2800 IU vitamin D3 in FOSAMAX PLUS D is similar to 2800 IU vitamin D3 administered alone.
In a separate study, the baseline adjusted mean AUC0-80 hrs and baseline adjusted mean Cmax for vitamin D3 were 355.6 ng-hr/mL and 10.8 ng/mL, respectively. The baseline adjusted mean Tmax was 9.2 hrs. The bioavailability of the 5600 IU vitamin D3 in the FOSAMAX PLUS D is similar to 5600 IU vitamin D3 administered as two 2800 IU vitamin D3 tablets.
Distribution
Alendronate Sodium
Preclinical studies (in male rats) show that alendronate transiently distributes to soft tissues following 1 mg/kg IV administration but is then rapidly redistributed to bone or excreted in the urine. The mean steady-state volume of distribution, exclusive of bone, is at least 28 L in humans. Concentrations of drug in plasma following therapeutic oral doses are too low (less than 5 ng/mL) for analytical detection. Protein binding in human plasma is approximately 78%.
Cholecalciferol
Following absorption, vitamin D3 enters the blood as part of chylomicrons. Vitamin D3 is rapidly distributed mostly to the liver where it undergoes metabolism to 25-hydroxyvitamin D3, the major storage form. Lesser amounts are distributed to adipose tissue and stored as vitamin D3 at these sites for later release into the circulation. Circulating vitamin D3 is bound to vitamin D-binding protein.
Metabolism
Alendronate Sodium
There is no evidence that alendronate is metabolized in animals or humans.
Cholecalciferol
Vitamin D3 is rapidly metabolized by hydroxylation in the liver to 25-hydroxyvitamin D3, and subsequently metabolized in the kidney to 1,25-dihydroxyvitamin D3, which represents the biologically active form. Further hydroxylation occurs prior to elimination. A small percentage of vitamin D3 undergoes glucuronidation prior to elimination.
Excretion
Alendronate Sodium
Following a single IV dose of [14C]alendronate, approximately 50% of the radioactivity was excreted in the urine within 72 hours and little or no radioactivity was recovered in the feces. Following a single 10-mg IV dose, the renal clearance of alendronate was 71 mL/min (64, 78; 90% confidence interval [CI]), and systemic clearance did not exceed 200 mL/min. Plasma concentrations fell by more than 95% within 6 hours following IV administration. The terminal half-life in humans is estimated to exceed 10 years, probably reflecting release of alendronate from the skeleton. Based on the above, it is estimated that after 10 years of oral treatment with FOSAMAX (10 mg daily) the amount of alendronate released daily from the skeleton is approximately 25% of that absorbed from the gastrointestinal tract.
Cholecalciferol
When radioactive vitamin D3 was intravenously administered to healthy subjects, the mean urinary excretion of radioactivity after 48 hours was 2.4% of the administered dose, and the mean fecal excretion of radioactivity after 48 hours was 4.9% of the administered dose. In both cases, the excreted radioactivity was almost exclusively as metabolites of the parent. The mean half-life of baseline adjusted vitamin D3 in the serum following an oral dose of FOSAMAX PLUS D is approximately 14 hours.
Special Populations
Pediatric: The oral bioavailability of alendronate in children was similar to that observed in adults; however, FOSAMAX PLUS D is not indicated for use in children [see Use in Specific Populations (8.4)].
Gender: Bioavailability and the fraction of an IV dose of alendronate excreted in urine were similar in men and women.
Geriatric:
Alendronate Sodium
Bioavailability and disposition of alendronate (urinary excretion) were similar in elderly and younger patients. No dosage adjustment of alendronate is necessary [see Dosage and Administration (2.5)].
Cholecalciferol
Dietary requirements of vitamin D3 are increased in the elderly.
Race: Pharmacokinetic differences due to race have not been studied.
Renal Insufficiency:
Alendronate Sodium
Preclinical studies show that, in rats with kidney failure, increasing amounts of drug are present in plasma, kidney, spleen, and tibia. In healthy controls, drug that is not deposited in bone is rapidly excreted in the urine. No evidence of saturation of bone uptake was found after 3 weeks dosing with cumulative IV doses of 35 mg/kg in young male rats. Although no clinical information is available, it is likely that, as in animals, elimination of alendronate via the kidney will be reduced in patients with impaired renal function. Therefore, somewhat greater accumulation of alendronate in bone might be expected in patients with impaired renal function.
No dosage adjustment is necessary for patients with mild-to-moderate renal insufficiency (creatinine clearance 35 to 60 mL/min). FOSAMAX PLUS D is not recommended for patients with more severe renal insufficiency (creatinine clearance <35 mL/min) due to lack of experience with alendronate in renal failure.
Cholecalciferol
Patients with renal insufficiency will have decreased ability to form the active 1,25-dihydroxyvitamin D3 metabolite.
Hepatic Insufficiency:
Alendronate Sodium
As there is evidence that alendronate is not metabolized or excreted in the bile, no studies were conducted in patients with hepatic insufficiency. No dosage adjustment is necessary.
Cholecalciferol
Vitamin D3 may not be adequately absorbed in patients who have malabsorption due to inadequate bile production.
Drug Interactions
Alendronate Sodium
Intravenous ranitidine was shown to double the bioavailability of oral alendronate. The clinical significance of this increased bioavailability and whether similar increases will occur in patients given oral H2-antagonists is unknown.
In healthy subjects, oral prednisone (20 mg three times daily for five days) did not produce a clinically meaningful change in the oral bioavailability of alendronate (a mean increase ranging from 20 to 44%).
Products containing calcium and other multivalent cations are likely to interfere with absorption of alendronate.
Cholecalciferol
Olestra, mineral oils, orlistat, and bile acid sequestrants (e.g., cholestyramine, colestipol) may impair the absorption of vitamin D. Anticonvulsants, cimetidine, and thiazides may increase the catabolism of vitamin D.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The following data are based on findings for the individual components of FOSAMAX PLUS D.
Alendronate Sodium
Harderian gland (a retro-orbital gland not present in humans) adenomas were increased in high-dose female mice (p=0.003) in a 92‑week oral carcinogenicity study at doses of alendronate of 1, 3, and 10 mg/kg/day (males) or 1, 2, and 5 mg/kg/day (females). These doses are equivalent to 0.5 to 4 times a maximum recommended daily dose of 10 mg based on surface area, mg/m2. The relevance of this finding to humans is unknown.
Parafollicular cell (thyroid) adenomas were increased in high-dose male rats (p=0.003) in a 2‑year oral carcinogenicity study at doses of 1 and 3.75 mg/kg body weight. These doses are equivalent to 1 and 4 times a 10-mg human daily dose based on surface area, mg/m2. The relevance of this finding to humans is unknown.
Alendronate was not genotoxic in the in vitro microbial mutagenesis assay with and without metabolic activation, in an in vitro mammalian cell mutagenesis assay, in an in vitro alkaline elution assay in rat hepatocytes, and in an in vivo chromosomal aberration assay in mice. In an in vitro chromosomal aberration assay in Chinese hamster ovary cells, however, alendronate gave equivocal results.
Alendronate had no effect on fertility (male or female) in rats at oral doses up to 5 mg/kg/day (4 times a 10-mg human daily dose based on surface area, mg/m2).
Cholecalciferol
The carcinogenic potential of cholecalciferol (vitamin D3) has not been studied in rodents. Calcitriol, the hormonal metabolite of cholecalciferol, was not genotoxic in the Ames microbial mutagenesis assay with or without metabolic activation, and in an in vivo micronucleus assay in mice.
Ergocalciferol (vitamin D2) at high doses (150,000 to 200,000 IU/kg/day) administered prior to mating resulted in altered estrous cycle and inhibition of pregnancy in rats. The potential effect of cholecalciferol on male fertility is unknown in rats.
13.2 Animal Toxicology and/or Pharmacology
The relative inhibitory activities on bone resorption and mineralization of alendronate and etidronate were compared in the Schenk assay, which is based on histological examination of the epiphyses of growing rats. In this assay, the lowest dose of alendronate that interfered with bone mineralization (leading to osteomalacia) was 6000‑fold the antiresorptive dose. The corresponding ratio for etidronate was one to one. These data suggest that alendronate administered in therapeutic doses is highly unlikely to induce osteomalacia.
-
14 CLINICAL STUDIES
14.1 Treatment of Postmenopausal Osteoporosis
Effect on Fracture Incidence
Data on the effects of FOSAMAX on fracture incidence are derived from three clinical studies of postmenopausal women, 44 to 84 years of age, with osteoporosis: 1) U.S. and Multinational combined: a study of patients with a lumbar spine BMD T-score at or below minus 2.5 with or without a prior vertebral fracture, 2) Three-Year Study of the Fracture Intervention Trial (FIT): a study of patients with at least one baseline vertebral fracture, and 3) Four-Year Study of FIT: a study of patients with low bone mass but without a baseline vertebral fracture.
To assess the effects of FOSAMAX on the incidence of vertebral fractures (detected by digitized radiography; approximately one third of these were clinically symptomatic), the U.S. (478 patients) and Multinational (516 patients in 15 countries) studies (of virtually identical design) were combined in an analysis that compared placebo to the pooled dosage groups of FOSAMAX (5 or 10 mg for three years or 20 mg for two years followed by 5 mg for one year). There was a statistically significant reduction in the proportion of patients treated with FOSAMAX experiencing one or more new vertebral fractures relative to those treated with placebo (3.2% vs. 6.2%; a 48% relative risk reduction). A reduction in the total number of new vertebral fractures (4.2 vs. 11.3 per 100 patients) was also observed. In the pooled analysis, patients who received FOSAMAX had a loss in stature that was statistically significantly less than was observed in those who received placebo (-3.0 mm vs. -4.6 mm).
The Fracture Intervention Trial (FIT) consisted of two studies in postmenopausal women: the Three-Year Study of patients who had at least one baseline radiographic vertebral fracture and the Four-Year Study of patients with low bone mass but without a baseline vertebral fracture. In both studies of FIT, 96% of randomized patients completed the studies (i.e., had a closeout visit at the scheduled end of the study); approximately 80% of patients were still taking study medication upon completion.
Fracture Intervention Trial: Three-Year Study (patients with at least one baseline radiographic vertebral fracture)
This randomized, double-blind, placebo-controlled, 2027-patient study (FOSAMAX, n=1022; placebo, n=1005) demonstrated that treatment with FOSAMAX resulted in statistically significant reductions in fracture incidence at three years as shown in Table 6.
Table 6: Effect of FOSAMAX on Fracture Incidence in the Three-Year Study of FIT (patients with vertebral fracture at baseline) Percent of Patients
FOSAMAX
(n=1022)
Placebo
(n=1005)Absolute
Reduction
in Fracture
IncidenceRelative
Reduction in Fracture
Risk %Patients with: Vertebral fractures (diagnosed by X-ray) ≥1 new vertebral fracture 7.9 15.0 7.1 47* ≥2 new vertebral fractures 0.5 4.9 4.4 90* Clinical (symptomatic) fractures Any clinical (symptomatic) fracture 13.8 18.1 4.3 26 ≥1 clinical (symptomatic) vertebral fracture 2.3 5.0 2.7 54 Hip fracture 1.1 2.2 1.1 51† Wrist (forearm) fracture 2.2 4.1 1.9 48† Furthermore, in this population of patients with baseline vertebral fracture, treatment with FOSAMAX significantly reduced the incidence of hospitalizations (25.0% vs. 30.7%).
In the Three-Year Study of FIT, fractures of the hip occurred in 22 (2.2%) of 1005 patients on placebo and 11 (1.1%) of 1022 patients on FOSAMAX, p=0.047. Figure 1 displays the cumulative incidence of hip fractures in this study.
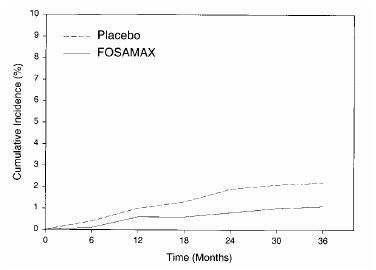
Figure 1: Cumulative Incidence of Hip Fractures in the Three-Year Study of FIT (patients with radiographic vertebral fracture at baseline)
Fracture Intervention Trial: Four-Year Study (patients with low bone mass but without a baseline radiographic vertebral fracture)
This randomized, double-blind, placebo-controlled, 4432-patient study (FOSAMAX, n=2214; placebo, n=2218) further investigated the reduction in fracture incidence due to FOSAMAX. The intent of the study was to recruit women with osteoporosis, defined as a baseline femoral neck BMD at least two standard deviations below the mean for young adult women. However, due to subsequent revisions to the normative values for femoral neck BMD, 31% of patients were found not to meet this entry criterion and thus this study included both osteoporotic and non-osteoporotic women. The results are shown in Table 7 below for the patients with osteoporosis.
Table 7: Effect of FOSAMAX on Fracture Incidence in Osteoporotic Patients in the Four-Year Study of FIT (patients without vertebral fracture at baseline) Percent of Patients
FOSAMAX
(n=1545)
Placebo
(n=1521)Absolute
Reduction
in Fracture
IncidenceRelative
Reduction in Fracture Risk (%)- *
- Not significant. This study was not powered to detect differences at these sites.
Patients with: Vertebral fractures (diagnosed by X-ray) ≥1 new vertebral fracture 2.5 4.8 2.3 48 ≥2 new vertebral fractures 0.1 0.6 0.5 78 Clinical (symptomatic) fractures Any clinical (symptomatic) fracture 12.9 16.2 3.3 22 ≥1 clinical (symptomatic) vertebral fracture 1.0 1.6 0.6 41 (NS)* Hip fracture 1.0 1.4 0.4 29 (NS)* Wrist (forearm) fracture 3.9 3.8 -0.1 NS* Fracture Results Across Studies
In the Three-Year Study of FIT, FOSAMAX reduced the percentage of women experiencing at least one new radiographic vertebral fracture from 15.0% to 7.9% (47% relative risk reduction, p<0.001); in the Four-Year Study of FIT, the percentage was reduced from 3.8% to 2.1% (44% relative risk reduction, p=0.001); and in the combined U.S./Multinational studies, from 6.2% to 3.2% (48% relative risk reduction, p=0.034).
FOSAMAX reduced the percentage of women experiencing multiple (two or more) new vertebral fractures from 4.2% to 0.6% (87% relative risk reduction, p<0.001) in the combined U.S./Multinational studies and from 4.9% to 0.5% (90% relative risk reduction, p<0.001) in the Three-Year Study of FIT. In the Four-Year Study of FIT, FOSAMAX reduced the percentage of osteoporotic women experiencing multiple vertebral fractures from 0.6% to 0.1% (78% relative risk reduction, p=0.035).
Thus, FOSAMAX reduced the incidence of radiographic vertebral fractures in osteoporotic women whether or not they had a previous radiographic vertebral fracture.
FOSAMAX, over a three- or four-year period, was associated with statistically significant reductions in loss of height vs. placebo in patients with and without baseline radiographic vertebral fractures. At the end of the FIT studies the between-treatment group differences were 3.2 mm in the Three-Year Study and 1.3 mm in the Four-Year Study.
Effect on Bone Mineral Density
The efficacy of FOSAMAX 10 mg once daily in postmenopausal women with osteoporosis (lumbar spine bone mineral density [BMD] of at least 2 standard deviations below the premenopausal mean) was demonstrated in four double-blind, placebo-controlled clinical studies of two or three years’ duration. These included two three-year, multicenter studies of virtually identical design, one performed in the United States (U.S.) and the other in 15 different countries (Multinational), which enrolled 478 and 516 patients, respectively. Figure 2 shows the mean increases in BMD of the lumbar spine, femoral neck, and trochanter in patients receiving FOSAMAX 10 mg/day relative to placebo-treated patients at three years for each of these studies.
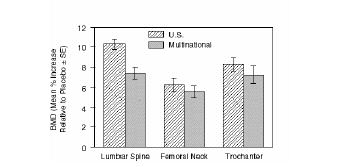
Figure 2: Osteoporosis Treatment Studies in Postmenopausal Women: Increase in BMD: FOSAMAX 10 mg/day at Three Years
At three years significant increases in BMD, relative both to baseline and placebo, were seen at each measurement site in each study in patients who received FOSAMAX 10 mg/day. Total body BMD also increased significantly in each study, suggesting that the increases in bone mass of the spine and hip did not occur at the expense of other skeletal sites. Increases in BMD were evident as early as three months and continued throughout the three years of treatment. (See figure 3 for lumbar spine results.) In the two-year extension of these studies, treatment of 147 patients with FOSAMAX 10 mg/day resulted in continued increases in BMD at the lumbar spine and trochanter (absolute additional increases between years 3 and 5: lumbar spine, 0.94%; trochanter, 0.88%). BMD at the femoral neck, forearm and total body were maintained. FOSAMAX was similarly effective regardless of age, race, baseline rate of bone turnover, and baseline BMD in the range studied (at least 2 standard deviations below the premenopausal mean).
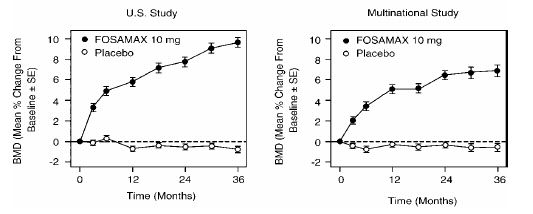
Figure 3: Osteoporosis Treatment Studies in Postmenopausal Women: Time Course of Effect of FOSAMAX 10 mg/day Versus Placebo: Lumbar Spine BMD Percent Change From Baseline
In patients with postmenopausal osteoporosis treated with FOSAMAX 10 mg/day for one or two years, the effects of treatment withdrawal were assessed. Following discontinuation, there were no further increases in bone mass and the rates of bone loss were similar to those of the placebo groups.
The therapeutic equivalence of once weekly FOSAMAX 70 mg (n=519) and FOSAMAX 10 mg daily (n=370) was demonstrated in a one-year, double-blind, multicenter study of postmenopausal women with osteoporosis. In the primary analysis of completers, the mean increases from baseline in lumbar spine BMD at one year were 5.1% (4.8, 5.4%; 95% CI) in the 70-mg once-weekly group (n=440) and 5.4% (5.0, 5.8%; 95% CI) in the 10-mg daily group (n=330). The two treatment groups were also similar with regard to BMD increases at other skeletal sites. The results of the intention-to-treat analysis were consistent with the primary analysis of completers.
Bone Histology
Bone histology in 270 postmenopausal patients with osteoporosis treated with FOSAMAX at doses ranging from 1 to 20 mg/day for one, two, or three years revealed normal mineralization and structure, as well as the expected decrease in bone turnover relative to placebo. These data, together with the normal bone histology and increased bone strength observed in rats and baboons exposed to long-term alendronate treatment, support the conclusion that bone formed during therapy with FOSAMAX is of normal quality.
Concomitant Use with Estrogen Hormone Replacement Therapy
The effects on BMD of treatment with FOSAMAX 10 mg once daily and conjugated estrogen (0.625 mg/day) either alone or in combination were assessed in a two-year, double-blind, placebo-controlled study of hysterectomized postmenopausal osteoporotic women (n=425). At two years, the increases in lumbar spine BMD from baseline were significantly greater with the combination (8.3%) than with either estrogen or FOSAMAX alone (both 6.0%).
The effects on BMD when FOSAMAX was added to stable doses (for at least one year) of HRT (estrogen ± progestin) were assessed in a one-year, double-blind, placebo-controlled study in postmenopausal osteoporotic women (n=428). The addition of FOSAMAX 10 mg once daily to HRT produced, at one year, significantly greater increases in lumbar spine BMD (3.7%) vs. HRT alone (1.1%).
In these studies, significant increases or favorable trends in BMD for combined therapy compared with HRT alone were seen at the total hip, femoral neck, and trochanter. No significant effect was seen for total body BMD.
Histomorphometric studies of transiliac biopsies in 92 subjects showed normal bone architecture. Compared to placebo there was a 98% suppression of bone turnover (as assessed by mineralizing surface) after 18 months of combined treatment with FOSAMAX and HRT, 94% on FOSAMAX alone, and 78% on HRT alone. The long-term effects of combined FOSAMAX and HRT on fracture occurrence and fracture healing have not been studied.
14.2 Treatment to Increase Bone Mass in Men with Osteoporosis
The efficacy of FOSAMAX in men with hypogonadal or idiopathic osteoporosis was demonstrated in two clinical studies.
A two-year, double-blind, placebo-controlled, multicenter study of FOSAMAX 10 mg once daily enrolled a total of 241 men between the ages of 31 and 87 (mean, 63). All patients in the trial had either: 1) a BMD T-score ≤‑2 at the femoral neck and ≤‑1 at the lumbar spine, or 2) a baseline osteoporotic fracture and a BMD T-score ≤‑1 at the femoral neck. At two years, the mean increases relative to placebo in BMD in men receiving FOSAMAX 10 mg/day were significant at the following sites: lumbar spine, 5.3%; femoral neck, 2.6%; trochanter, 3.1%; and total body, 1.6%. Treatment with FOSAMAX also reduced height loss (FOSAMAX, ‑0.6 mm vs. placebo, ‑2.4 mm).
A one-year, double-blind, placebo-controlled, multicenter study of once weekly FOSAMAX 70 mg enrolled a total of 167 men between the ages of 38 and 91 (mean, 66). Patients in the study had either: 1) a BMD T-score ≤-2 at the femoral neck and ≤-1 at the lumbar spine, 2) a BMD T-score ≤-2 at the lumbar spine and ≤-1 at the femoral neck, or 3) a baseline osteoporotic fracture and a BMD T-score ≤-1 at the femoral neck. At one year, the mean increases relative to placebo in BMD in men receiving FOSAMAX 70 mg once weekly were significant at the following sites: lumbar spine, 2.8%; femoral neck, 1.9%; trochanter, 2.0%; and total body, 1.2%. These increases in BMD were similar to those seen at one year in the 10 mg once-daily study.
In both studies, BMD responses were similar regardless of age (≥65 years vs. <65 years), gonadal function (baseline testosterone <9 ng/dL vs. ≥9 ng/dL), or baseline BMD (femoral neck and lumbar spine T-score ≤-2.5 vs. >-2.5).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
No. 3870 — Tablets FOSAMAX PLUS D 70 mg/2800 IU are white to off-white, modified capsule-shaped tablets with code 710 on one side and an outline of a bone image on the other. They are supplied as follows:
NDC 54868-5480-0 unit of use blister packages of 4.
Storage
Store at 20-25°C (68-77°F), excursions between 15-30°C (59-86°F) are allowed. [See USP Controlled Room Temperature.] Protect from moisture and light. Store tablets in the original blister package until use.
-
17 PATIENT COUNSELING INFORMATION
[See FDA-Approved Medication Guide.]
Physicians should instruct their patients to read the Medication Guide before starting therapy with FOSAMAX PLUS D and to reread it each time the prescription is renewed.
17.1 Osteoporosis Recommendations, Including Calcium and Vitamin D Supplementation
Patients should be instructed to take supplemental calcium if intake is inadequate. Patients at increased risk for vitamin D insufficiency (e.g., over the age of 70 years, nursing home bound, or chronically ill) should be instructed to take additional vitamin D if needed [see Dosage and Administration (2.3)]. Patients with gastrointestinal malabsorption syndromes should be informed that they may require additional vitamin D supplementation. Weight-bearing exercise should be considered along with the modification of certain behavioral factors, such as cigarette smoking and/or excessive alcohol consumption, if these factors exist.
17.2 Dosing Instructions
Patients should be instructed that the expected benefits of FOSAMAX PLUS D may only be obtained when it is taken with plain water the first thing upon arising for the day at least 30 minutes before the first food, beverage, or medication of the day. Even dosing with orange juice or coffee has been shown to markedly reduce the absorption of alendronate [see Clinical Pharmacology (12.3)].
To facilitate delivery to the stomach and thus reduce the potential for esophageal irritation, patients should be instructed to swallow each tablet of FOSAMAX PLUS D with a full glass of water (6‑8 oz) and not to lie down for at least 30 minutes and until after their first food of the day. Patients should not chew or suck on the tablet because of a potential for oropharyngeal ulceration. Patients should be specifically instructed not to take FOSAMAX PLUS D at bedtime or before arising for the day. Patients should be informed that failure to follow these instructions may increase their risk of esophageal problems. Patients should be instructed that if they develop symptoms of esophageal disease (such as difficulty or pain upon swallowing, retrosternal pain or new or worsening heartburn) they should stop taking FOSAMAX PLUS D and consult their physician.
Patients should be instructed that if they miss a dose of FOSAMAX PLUS D, they should take one tablet on the morning after they remember. They should not take two tablets on the same day but should return to taking one tablet once a week, as originally scheduled on their chosen day.
Manuf. for: Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC., Whitehouse Station, NJ 08889, USABy:
FROSST IBERICA, S.A.
28805 Alcalá de Henares
Madrid, SpainIssued July 2011
6013312
Copyright © 2005, 2007, 2010 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved -
MEDICATION GUIDE
MEDICATION GUIDE
FOSAMAX® PLUS D (FOSS-ah-max PLUS D)
(alendronate sodium/cholecalciferol)
TabletsRead the Medication Guide that comes with FOSAMAX® PLUS D before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment. Talk to your doctor if you have any questions about FOSAMAX PLUS D.
What is the most important information I should know about FOSAMAX PLUS D?
FOSAMAX PLUS D can cause serious side effects including:
- Esophagus problems
- Low calcium levels in your blood (hypocalcemia)
- Bone, joint, or muscle pain
- Severe jaw bone problems (osteonecrosis)
- Unusual thigh bone fractures
-
Esophagus problems.
Some people who take FOSAMAX PLUS D may develop problems in the esophagus (the tube that connects the mouth and the stomach). These problems include irritation, inflammation, or ulcers of the esophagus which may sometimes bleed.- It is important that you take FOSAMAX PLUS D exactly as prescribed to help lower your chance of getting esophagus problems. (See the section “How should I take FOSAMAX PLUS D?”)
-
Stop taking FOSAMAX PLUS D and call your doctor right away if you get chest pain, new or worsening heartburn, or have trouble or pain when you swallow.
-
Low calcium levels in your blood (hypocalcemia).
FOSAMAX PLUS D may lower the calcium levels in your blood. If you have low blood calcium before you start taking FOSAMAX PLUS D, it may get worse during treatment. Your low blood calcium must be treated before you take FOSAMAX PLUS D. Most people with low blood calcium levels do not have symptoms, but some people may have symptoms. Call your doctor right away if you have symptoms of low blood calcium such as:- Spasms, twitches, or cramps in your muscles
- Numbness or tingling in your fingers, toes, or around your mouth
Your doctor may prescribe calcium and vitamin D to help prevent low calcium levels in your blood, while you take FOSAMAX PLUS D. Take calcium and vitamin D as your doctor tells you to.
-
Bone, joint, or muscle pain.
Some people who take FOSAMAX PLUS D develop severe bone, joint, or muscle pain.
-
Severe jaw bone problems (osteonecrosis).
Severe jaw bone problems may happen when you take FOSAMAX PLUS D. Your doctor should examine your mouth before you start FOSAMAX PLUS D. Your doctor may tell you to see your dentist before you start FOSAMAX PLUS D. It is important for you to practice good mouth care during treatment with FOSAMAX PLUS D.
-
Unusual thigh bone fractures.
Some people have developed unusual fractures in their thigh bone. Symptoms of a fracture may include new or unusual pain in your hip, groin, or thigh.
Call your doctor right away if you have any of these side effects.
What is FOSAMAX PLUS D?
FOSAMAX PLUS D is a prescription medicine used to:
- Treat osteoporosis in women after menopause. FOSAMAX PLUS D helps increase bone mass and reduces the chance of having a hip or spinal fracture (break).
- Increase bone mass in men with osteoporosis.
FOSAMAX PLUS D should not be used to treat vitamin D deficiency.
It is not known how long FOSAMAX PLUS D works for the treatment and prevention of osteoporosis. You should see your doctor regularly to determine if FOSAMAX PLUS D is still right for you.
FOSAMAX PLUS D is not for use in children.
Who should not take FOSAMAX PLUS D?
Do not take FOSAMAX PLUS D if you:
- Have certain problems with your esophagus, the tube that connects your mouth with your stomach
- Cannot stand or sit upright for at least 30 minutes
- Have low levels of calcium in your blood
- Are allergic to FOSAMAX PLUS D or any of its ingredients. A list of ingredients is at the end of this leaflet.
What should I tell my doctor before taking FOSAMAX PLUS D?
Before you start FOSAMAX PLUS D, be sure to talk to your doctor if you:
- Have problems with swallowing
- Have stomach or digestive problems
- Have low blood calcium
- Plan to have dental surgery or teeth removed
- Have kidney problems
- Have sarcoidosis, leukemia, lymphoma. These conditions may cause changes in vitamin D.
- Have been told you have trouble absorbing minerals in your stomach or intestines (malabsorption syndrome)
- Are pregnant or plan to become pregnant. It is not known if FOSAMAX PLUS D can harm your unborn baby.
- Are breast-feeding or plan to breast-feed. It is not known if FOSAMAX PLUS D passes into your milk and may harm your baby.
Especially tell your doctor if you take:
- antacids
- aspirin
- Nonsteroidal Anti-Inflammatory (NSAID) medicines
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Certain medicines may affect how FOSAMAX PLUS D works.
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist each time you get a new medicine.
How should I take FOSAMAX PLUS D tablet?
- Take FOSAMAX PLUS D exactly as your doctor tells you.
- FOSAMAX PLUS D works only if taken on an empty stomach.
- Take 1 dose of FOSAMAX PLUS D 1 time a week, after you get up for the day and before taking your first food, drink, or other medicine.
- Take FOSAMAX PLUS D while you are sitting or standing.
- Take your FOSAMAX PLUS D tablet with a full glass (6-8 oz) of plain water.
- Do not chew or suck on a tablet of FOSAMAX PLUS D.
- Do not take FOSAMAX PLUS D with mineral water, coffee, tea, soda, or juice.
- Do not take FOSAMAX PLUS D at bedtime.
After swallowing FOSAMAX PLUS D, wait at least 30 minutes:
- Before you lie down. You may sit, stand or walk, and do normal activities like reading.
- Before you take your first food or drink except for plain water.
- Before you take other medicines, including antacids, calcium, and other supplements and vitamins.
Do not lie down for at least 30 minutes after you take FOSAMAX PLUS D and after you eat your first food of the day.
If you miss a dose of FOSAMAX PLUS D, do not take it later in the day. Take your missed dose on the next morning after you remember and then return to your normal schedule. Do not take 2 doses on the same day.
If you take too much FOSAMAX PLUS D, call your doctor. Do not try to vomit. Do not lie down.
What are the possible side effects of FOSAMAX PLUS D?
FOSAMAX PLUS D may cause serious side effects.
- See “What is the most important information I should know about FOSAMAX PLUS D?”
The most common side effects of FOSAMAX PLUS D are:
- Stomach area (abdominal) pain
- Heartburn
- Constipation
- Diarrhea
- Upset stomach
- Pain in your bones, joints, or muscles
- Nausea
You may get allergic reactions, such as hives or, in rare cases, swelling of your face, lips, tongue, or throat.
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of FOSAMAX PLUS D. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How do I store FOSAMAX PLUS D?
- Store FOSAMAX PLUS D at room temperature, 68°F to 77°F (20°C to 25°C).
- Keep FOSAMAX PLUS D away from light.
- Keep FOSAMAX PLUS D package and tablets dry.
- Store FOSAMAX PLUS D in the original package.
Keep FOSAMAX PLUS D and all medicines out of the reach of children.
General information about the safe and effective use of FOSAMAX PLUS D.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use FOSAMAX PLUS D for a condition for which it was not prescribed. Do not give FOSAMAX PLUS D to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about FOSAMAX PLUS D. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about FOSAMAX PLUS D that is written for health professionals. For more information, go to: www.fosamaxplusd.com or call 1-800-622-4477 (toll-free).
What are the ingredients in FOSAMAX PLUS D?
Active ingredients: alendronate sodium and cholecalciferol (vitamin D3).
Inactive ingredients: cellulose, lactose, medium chain triglycerides, gelatin, croscarmellose sodium, sucrose, colloidal silicon dioxide, magnesium stearate, butylated hydroxytoluene, modified food starch, and sodium aluminum silicate.
Manuf. for: Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC., Whitehouse Station, NJ 08889, USABy:
FROSST IBERICA, S.A.
28805 Alcalá de Henares
Madrid, SpainCopyright © 2010 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reservedRevised July 2011
6013312
This Medication Guide has been approved by the U.S. Food and Drug Administration.
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - BI FOLD CARD, OUTER 70 mg/2800 IU
FOSAMAX® PLUS D
(alendronate sodium/cholecalciferol) tablets
70 mg/2800 IU
NDC 54868-5480-0

Dispense the enclosed Medication Guide to each patient.
Each tablet contains 91.37 mg alendronate sodium (70 mg free acid equivalent) and 70 mcg cholecalciferol equivalent to 2800 IU vitamin D.
4 Tablets
No. 3870
Rx only
USUAL ADULT DOSAGE: ONE 70 mg/2800 IU TABLET ONCE WEEKLY
See accompanying circular for dosage information.
-
INGREDIENTS AND APPEARANCE
FOSAMAX PLUS D
alendronate sodium and cholecalciferol tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-5480(NDC:0006-0710) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength alendronate sodium (UNII: 2UY4M2U3RA) (alendronic acid - UNII:X1J18R4W8P) alendronic acid 70 mg cholecalciferol (UNII: 1C6V77QF41) (cholecalciferol - UNII:1C6V77QF41) cholecalciferol 2800 [iU] Inactive Ingredients Ingredient Name Strength butylated hydroxytoluene (UNII: 1P9D0Z171K) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) gelatin (UNII: 2G86QN327L) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) magnesium stearate (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) sucrose (UNII: C151H8M554) STARCH, CORN (UNII: O8232NY3SJ) MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) SODIUM ALUMINIUM SILICATE (UNII: 058TS43PSM) Product Characteristics Color WHITE (white to off-white) Score no score Shape OVAL (capsule-shaped) Size 12mm Flavor Imprint Code 710 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-5480-0 1 in 1 CARTON 1 4 in 1 BLISTER PACK Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021762 11/30/2005 Labeler - Physicians Total Care, Inc. (194123980) Establishment Name Address ID/FEI Business Operations Physicians Total Care, Inc. 194123980 relabel