Label: oncaspar- pegaspargase injection, solution for intramuscular and intravenous use
-
Contains inactivated NDC Code(s)
NDC Code(s): 57665-002-02
- Category: HUMAN PRESCRIPTION DRUG LABEL WITH HIGHLIGHTS
- DEA Schedule: None
Drug Label Information
Updated July 24, 2006
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- 2,500 IU/m2 intramuscularly (IM) or intravenously (IV) no more frequently than every 14 days. (2.1)
- For IM administration, limit the volume at a single injection site to 2 mL; if greater than 2 mL, use multiple injection sites. (2.2)
- For IV administration, give over a period of 1 to 2 hours in 100 mL of sodium chloride or dextrose injection 5%, through an infusion that is already running. (2.2)
- Do not administer Oncaspar® if drug has been frozen, stored at room temperature for more than 48 hours, or shaken or vigorously agitated. (2.3)
DOSAGE FORMS AND STRENGTHS
- 3,750 IU/5 mL single-use vial. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Most common adverse reactions (≥ 2%) are allergic reactions (including anaphylaxis), central nervous system (CNS) thrombosis, coagulopathy, elevated transaminases, hyperbilirubinemia, hyperglycemia, and pancreatitis. (6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2006
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 First Line Acute Lymphoblastic Leukemia (ALL)
1.2 Acute Lymphoblastic Leukemia and Hypersensitivity to Asparaginase
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Instructions for Administration
2.3 Preparation and Handling Precautions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis and Serious Allergic Reactions
5.2 Thrombosis
5.3 Pancreatitis
5.4 Glucose Intolerance
5.5 Coagulopathy
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Clinical Allergic Reactions
6.3 Immunogenicity
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 First-Line ALL
14.2 ALL Patients Hypersensitive to Asparaginase
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Serious Allergic Reactions
17.2 Thrombosis
17.3 Pancreatitis
17.4 Glucose Intolerance
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- N/A - Section Title Not Found In Database
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
The recommended dose of Oncaspar® is 2,500 IU/m2 intramuscularly (IM) or intravenously (IV). Oncaspar® should be administered no more frequently than every 14 days.
2.2 Instructions for Administration
When Oncaspar® is administered IM, the volume at a single injection site should be limited to 2 mL. If the volume to be administered is greater than 2 mL, multiple injection sites should be used.
When administered IV, Oncaspar® should be given over a period of 1 to 2 hours in 100 mL of sodium chloride or dextrose injection 5%, through an infusion that is already running.
2.3 Preparation and Handling Precautions
Do not administer Oncaspar® if drug has been:
- frozen
- stored at room temperature (+15°C to +25°C; 59°F to 77°F) for more than 48 hours
- shaken or vigorously agitated [see How Supplied/Storage and Handling (16)]
Parenteral drug products should be inspected visually for particulate matter, cloudiness, or discoloration prior to administration, whenever solution and container permit. If any of these are present, discard the vial.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis and Serious Allergic Reactions
Serious allergic reactions can occur in patients receiving Oncaspar®. The risk of serious allergic reactions is higher in patients with known hypersensitivity to other forms of L-asparaginase. Observe patients for 1 hour after administration of Oncaspar® in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis (for example, epinephrine, oxygen, intravenous steroids, antihistamines). Discontinue Oncaspar® in patients with serious allergic reactions.
5.2 Thrombosis
Serious thrombotic events, including sagittal sinus thrombosis can occur in patients receiving Oncaspar®. Discontinue Oncaspar® in patients with serious thrombotic events.
5.3 Pancreatitis
Pancreatitis can occur in patients receiving Oncaspar®. Evaluate patients with abdominal pain for evidence of pancreatitis. Discontinue Oncaspar® in patients with pancreatitis.
5.4 Glucose Intolerance
Glucose intolerance can occur in patients receiving Oncaspar®. In some cases, glucose intolerance is irreversible.
5.5 Coagulopathy
Increased prothrombin time, increased partial thromboplastin time, and hypofibrinogenemia can occur in patients receiving Oncaspar®. Monitor coagulation parameters at baseline and periodically during and after treatment. Initiate treatment with fresh-frozen plasma to replace coagulation factors in patients with severe or symptomatic coagulopathy.
-
6 ADVERSE REACTIONS
The following serious adverse reactions occur with Oncaspar® treatment [see Warnings and Precautions (5)]:
- Anaphylaxis and serious allergic reactions
- Serious thrombosis
- Pancreatitis
- Glucose intolerance
- Coagulopathy
The most common adverse reactions with Oncaspar® are allergic reactions (including anaphylaxis), hyperglycemia, pancreatitis, central nervous system (CNS) thrombosis, coagulopathy, hyperbilirubinemia, and elevated transaminases.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
First-Line ALL
The data presented below are derived from 2 studies in patients with standard-risk ALL who received Oncaspar® as a component of first-line multi-agent chemotherapy. Study 1 was a randomized (1:1), active-controlled study that enrolled 118 patients, with a median age of 4.7 years (1.1-9.9 years), of whom 54% were males and 65% White, 14% Hispanic, 8% Black, 8% Asian, and 6% other. Of the 59 patients in Study 1 who were randomized to Oncaspar®, 48 patients (81%) received all 3 planned doses of Oncaspar®, 6 (10%) received 2 doses, 4 (7%) received 1 dose, and 1 patient (2%) did not receive the assigned treatment. Study 2 is an ongoing, multi-factorial design study in which all patients received Oncaspar®as a component of various multi-agent chemotherapy regimens; interim safety data are available for 2,770 patients. Study participants had a median age of 4 years (1-10 years), and were 55% male, 68% White, 18% Hispanic, 4% Black, 3% Asian, and 7% other. Per protocol, the schedule of Oncaspar®varied by treatment arm, with intermittent doses of Oncaspar® for up to 10 months.
In Study 1, detailed safety information was collected for pre-specified adverse reactions identified as asparaginase-induced adverse reactions and for grade 3 and 4 non-hematologic adverse reactions according to the Children’s Cancer Group (CCG) Toxicity and Complication Criteria. The per-patient incidence, by treatment arm, for these selected adverse reactions occurring at a severity of grade 3 or 4 are presented in Table 1 below:
TABLE 1 STUDY 1: PER-PATIENT INCIDENCE OF SELECTED GRADE 3 AND 4 ADVERSE REACTIONS 1Aspartate aminotransferase, alanine aminotransferase.
2Prolonged prothrombin time or partial thromboplastin time; or hypofibrinogenemia.
Oncaspar®
(n=58)Native
E. coli
L‑Asparaginase
(n=59)Abnormal Liver Tests 3 (5%) 5 (8%) Elevated Transaminases1 2 (3%) 4 (7%) Hyperbilirubinemia 1 (2%) 1 (2%) Hyperglycemia 3 (5%) 2 (3%) Central Nervous System
Thrombosis2 (3%) 2 (3%) Coagulopathy2 1 (2%) 3 (5%) Pancreatitis 1 (2%) 1 (2%) Clinical Allergic Reactions to
Asparaginase1 (2%) 0 Safety data were collected in Study 2 only for National Cancer Institute Common Toxicity Criteria (NCI CTC) version 2.0, grade 3 and 4 non-hematologic toxicities. In this study, the per-patient incidence for the following adverse reactions occurring during treatment courses in which patients received Oncaspar® were: elevated transaminases, 11%; coagulopathy, 7%; hyperglycemia, 5%; CNS thrombosis/hemorrhage, 2%; pancreatitis, 2%; clinical allergic reaction, 1%; and hyperbilirubinemia, 1%. There were 3 deaths due to pancreatitis.
Previously Treated ALL
Adverse reaction information was obtained from 5 clinical trials that enrolled a total of 174 patients with relapsed ALL who received Oncaspar® as a single agent or in combination with multi-agent chemotherapy. The toxicity profile of Oncaspar® in patients with previously treated relapsed ALL is similar to that reported above with the exception of clinical allergic reactions (see Table 2). The most common adverse reactions of Oncaspar® were clinical allergic reactions, elevated transaminases, hyperbilirubinemia, and coagulopathies. The most common serious adverse events due to Oncaspar® treatment were thrombosis (4%), hyperglycemia requiring insulin therapy (3%), and pancreatitis (1%).
6.2 Clinical Allergic Reactions
Clinical allergic reactions include the following: bronchospasm, hypotension, laryngeal edema, local erythema or swelling, systemic rash, and urticaria.
First-Line ALL
Among 58 Oncaspar®-treated patients enrolled in Study 1, clinical allergic reactions were reported in 2 patients (3%). One patient experienced a grade 1 allergic reaction and the other grade 3 hives; both occurred during the first delayed intensification phase of the study (see Table 2).
Previously Treated ALL
Among 62 patients with relapsed ALL and prior hypersensitivity reactions to asparaginase, 35 patients (56%) had a history of clinical allergic reactions to native Escherichia (E.) coli L-asparaginase, and 27 patients (44%) had history of clinical allergic reactions to both native E. coli and native Erwinia L‑asparaginase. Twenty (32%) of these 62 patients experienced clinical allergic reactions to Oncaspar® (see Table 2).
Among 112 patients with relapsed ALL with no prior hypersensitivity reactions to asparaginase, 11 patients (10%) experienced clinical allergic reactions to Oncaspar® (see Table 2).
TABLE 2 INCIDENCE OF CLINICAL ALLERGIC REACTIONS, OVERALL AND BY SEVERITY GRADE Toxicity Grade, n (%) Patient Status 1 2 3 4 Total Previously
Hypersensitive
Patients (n=62)7 (11) 8 (13) 4 (6) 1 (2) 20 (32) Non-
Hypersensitive
Patients (n=112)5 (4) 4 (4) 1 (1) 1 (1) 11 (10) First Line (n=58) 1 (2) 0 1 (2) 0 2 (3) 6.3 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity, defined as development of binding and/or neutralizing antibodies to the product.
In Study 1, Oncaspar®-treated patients were assessed for evidence of binding antibodies using an enzyme-linked immunosorbent assay (ELISA) method. The incidence of protocol-specified “high-titer” antibody formation was 2% in Induction (n=48), 10% in Delayed Intensification 1 (n=50), and 11% in Delayed Intensification 2 (n=44). There is insufficient information to determine whether the development of antibodies is associated with an increased risk of clinical allergic reactions, altered pharmacokinetics, or loss of anti‑leukemic efficacy.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay, and the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, concomitant medications, and underlying disease. Therefore, comparison of the incidence of antibodies to Oncaspar® with the incidence of antibodies to other products may be misleading.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. Animal reproduction studies have not been conducted with Oncaspar®. It is also not known whether Oncaspar® can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Oncaspar® should be given to a pregnant woman only if clearly needed.
8.3 Nursing Mothers
It is not known whether Oncaspar® is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Oncaspar®, a decision should be made to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
-
10 OVERDOSAGE
Three patients received 10,000 IU/m2 of Oncaspar® as an intravenous infusion. One patient experienced a slight increase in liver enzymes. A second patient developed a rash 10 minutes after the start of the infusion, which was controlled with the administration of an antihistamine and by slowing down the infusion rate. A third patient did not experience any adverse reactions.
-
11 DESCRIPTION
Oncaspar® (pegaspargase) is a modified version of the enzyme L-asparaginase. To produce Oncaspar®, L-asparaginase is modified by covalently conjugating units of monomethoxypolyethylene glycol (PEG), molecular weight of 5,000, to the enzyme, forming the active ingredient PEG-L-asparaginase. The L‑asparaginase (L-asparagine amidohydrolase, type EC-2, EC 3.5.1.1) used in the manufacture of Oncaspar® is derived from E. coli and supplied by Ovation Pharmaceuticals (U.S. License No. 1688) under a shared manufacturing arrangement. Oncaspar® activity is expressed in International Units (IU) according to the recommendation of the International Union of Biochemistry. One IU of L-asparaginase is defined as that amount of enzyme required to generate 1 µmol of ammonia per minute at pH 7.3 and 37°C.
Oncaspar® is supplied as a clear, colorless, preservative-free, isotonic sterile solution in phosphate-buffered saline, pH 7.3. Each milliliter contains Oncaspar® 750 IU ± 20% (based on specific activity of at least 85 IU per milligram protein), 1.20 mg monobasic sodium phosphate, USP, 5.58 mg dibasic sodium phosphate, USP, and 8.50 mg sodium chloride, USP, in water for injection, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of Oncaspar® is thought to be based on selective killing of leukemic cells due to depletion of plasma asparagine. Some leukemic cells are unable to synthesize asparagine due to a lack of asparagine synthetase and are dependent on an exogenous source of asparagine for survival. Depletion of asparagine, which results from treatment with the enzyme L-asparaginase, kills the leukemic cells. Normal cells, however, are less affected by the depletion due to their ability to synthesize asparagine.
12.2 Pharmacodynamics
In Study 1, pharmacodynamics were assessed in 57 newly diagnosed pediatric patients with standard-risk ALL who received three IM doses of Oncaspar® (2,500 IU/m2), one each during induction and two delayed intensification treatment phases. Pharmacodynamic activity was assessed through serial measurements of asparagine in sera (n=57) and cerebrospinal fluid (CSF) (n=50). The data for asparagine depletion are presented in CLINICAL STUDIES [seeClinical Studies (14)].
12.3 Pharmacokinetics
Pharmacokinetic assessments were based on an enzymatic assay measuring asparaginase activity. Serum pharmacokinetics were assessed in 34 newly diagnosed pediatric patients with standard-risk ALL in Study 1 following IM administration of 2,500 IU/m2. The elimination half-life of Oncaspar® was approximately 5.8 days during the induction phase. Similar elimination half-lives were observed during Delayed Intensification 1 and Delayed Intensification 2. Concentrations greater than 0.1 IU/mL were observed in over 90% of the samples from patients treated with Oncaspar® during induction, Delayed Intensification 1, and Delayed Intensification 2 for approximately 20 days.
In 3 pharmacokinetic studies, 37 patients with relapsed ALL received Oncaspar®at 2,500 IU/m2 IM every 2 weeks. The plasma half-life of Oncaspar® was 3.2 ± 1.8 days in 9 patients who were previously hypersensitive to native E. coli L-asparaginase and 5.7 ± 3.2 days in 28 non-hypersensitive patients. The area under the plasma concentration-time curve (AUC) was 9.5 ± 4.0 IU/mL/day in the previously hypersensitive patients and 9.8 ± 6.0 IU/mL/day in the non-hypersensitive patients.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
- No long-term carcinogenicity studies in animals have been performed with Oncaspar®.
- No relevant studies addressing mutagenic potential have been conducted. Oncaspar® did not exhibit a mutagenic effect when tested against Salmonella typhimurium strains in the Ames assay.
- No studies have been performed on impairment of fertility.
-
14 CLINICAL STUDIES
14.1 First-Line ALL
The safety and effectiveness of Oncaspar® was evaluated in an open-label, multicenter, randomized, active-controlled study (Study 1). In this study, 118 pediatric patients aged 1 to 9 years with previously untreated standard-risk ALL were randomized 1:1 to Oncaspar® or native E. coli L-asparaginase as part of combination therapy. Oncaspar® was administered IM at a dose of 2,500 IU/m2 on Day 3 of the 4‑week induction phase and on Day 3 of each of two 8-week delayed intensification phases. Native E. coli L‑asparaginase was administered IM at a dose of 6,000 IU/m2 three times weekly for 9 doses during induction and for 6 doses during each delayed intensification phase.
The primary determination of effectiveness was based on demonstration of similar asparagine depletion (magnitude and duration) in the Oncaspar® and native E. coli L-asparaginase arms. The protocol-specified goal was achievement of asparagine depletion to a serum concentration of ≤1 μM. The proportion of patients with this level of depletion was similar between the 2 study arms during all 3 phases of treatment at the protocol-specified time points.
In all phases of treatment, serum asparagine concentrations decreased within 4 days of the first dose of asparaginase in the treatment phase and remained low for approximately 3 weeks for both Oncaspar® and native E. coli L-asparaginase arms. Serum asparagine concentrations during the induction phase are shown in Figure 1. The patterns of serum asparagine depletion in the 2 delayed intensification phases are similar to the pattern of serum asparagine depletion in the induction phase.
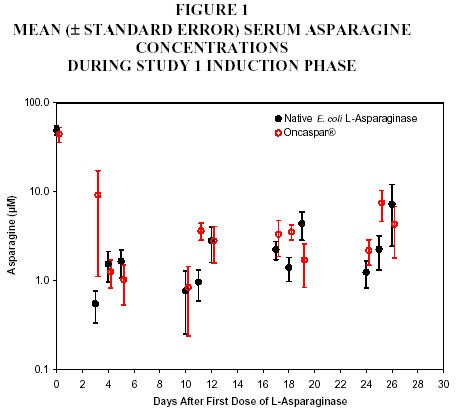
Note: Oncaspar® (2,500 IU/m2 IM) was administered on Day 3
of the 4-week induction phase. Native
E. coli L‑asparaginase (6,000 IU/m2 IM) was
administered 3 times weekly for 9 doses during induction.CSF asparagine concentrations were determined in 50 patients during the induction phase. CSF asparagine decreased from a mean pretreatment concentration of 3.1 µM to 1.7 µM on Day 4 ± 1 and 1.5 µM 25 ± 1 days after administration of Oncaspar®. These findings were similar to those observed in the native E. coli L-asparaginase treatment arm.
While the 3-year Event-Free Survival (EFS) for the Oncaspar®and native E. coli L‑asparaginase study arms were similar and in the range of 80%, Study 1 was not designed to evaluate for differences in EFS rates.
14.2 ALL Patients Hypersensitive to Asparaginase
The safety and effectiveness of Oncaspar® was evaluated in 4 open-label studies enrolling a total of 42 patients with multiply-relapsed, acute leukemia [39 (93%) with ALL] with a history of prior clinical allergic reaction to asparaginase. Hypersensitivity to asparaginase was defined by a history of systemic rash, urticaria, bronchospasm, laryngeal edema, hypotension, or local erythema, urticaria, or swelling, greater than 2 centimeters, for at least 10 minutes following administration of any form of native E. coli L-asparaginase. All patients received Oncaspar® at a dose of 2,000 or 2,500 IU/m2 administered IM or IV every 14 days. Patients received Oncaspar® as a single agent or in combination with multi-agent chemotherapy. The re‑induction response rate was 50% (95% confidence interval: 35%, 65%), based upon 36% complete remissions and 14% partial remissions. These results were similar to the overall response rates reported for patients with ALL receiving second-line, native E. coli L-asparaginase-containing re-induction chemotherapy. Anti-tumor activity was also observed with single-agent Oncaspar®. Three responses (1 complete remission and 2 partial remissions) were observed in 9 adult and pediatric patients with relapsed ALL and hypersensitivity to native E. coli L‑asparaginase.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Dosage Form
NDC 57665-002-02
3,750 IU/5 mL single-use vial individually packaged in a carton
Storage and Handling
Keep refrigerated at +2°C to +8ºC (36°F to 46°F).
Use only one dose per vial; do not re-enter the vial. Discard unused portions. Do not save unused drug for later administration.
Do not administer Oncaspar® if the drug:
- has been frozen
- has been stored at room temperature (+15°C to +25°C; 59°F to 77°F) for more than 48 hours
- has been shaken or vigorously agitated
- is cloudy, discolored, and precipitate is present.
-
17 PATIENT COUNSELING INFORMATION
17.1 Serious Allergic Reactions
Patients should be informed of the possibility of serious allergic reactions, including anaphylaxis, and to immediately report any swellings or difficulty breathing.
-
INGREDIENTS AND APPEARANCE
ONCASPAR
pegaspargase injection, solutionProduct Information Product Type Item Code (Source) NDC:57665-002 Route of Administration INTRAMUSCULAR, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength pegaspargase (UNII: 7D96IR0PPM) (pegaspargase - UNII:7D96IR0PPM) 750 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength sodium phosphate, monobasic (UNII: 3980JIH2SW) 1.2 mg in 1 mL sodium phosphate, dibasic (UNII: GR686LBA74) 5.58 mg in 1 mL sodium chloride (UNII: 451W47IQ8X) 8.5 mg in 1 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57665-002-02 1 in 1 CARTON 1 5 mL in 1 VIAL, SINGLE-USE