Label: TASIMELTEON capsule, gelatin coated

- NDC Code(s): 0480-4490-56
- Packager: Teva Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated May 31, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TASIMELTEON CAPSULES safely and effectively. See full prescribing information for TASIMELTEON CAPSULES.
TASIMELTEON capsules, for oral use
Initial U.S. Approval: 2014INDICATIONS AND USAGE
Tasimelteon is a melatonin receptor agonist.
Tasimelteon capsules are indicated for the treatment of:
- Non-24-Hour Sleep-Wake Disorder (Non-24) in adults (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Capsules: 20 mg (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
May cause somnolence: After taking tasimelteon capsules, patients should limit their activity to preparing for going to bed, because tasimelteon can impair the performance of activities requiring complete mental alertness (5.1)
ADVERSE REACTIONS
The most common adverse reactions (incidence >5% and at least twice as high on tasimelteon than on placebo) were headache, increased alanine aminotransferase, nightmares or unusual dreams, and upper respiratory or urinary tract infection (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Teva at 1-888-838-2872 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong CYP1A2 inhibitors (e.g., fluvoxamine): Avoid use of tasimelteon in combination with strong CYP1A2 inhibitors because of increased exposure (7.1, 12.3)
- Strong CYP3A4 inducers (e.g., rifampin): Avoid use of tasimelteon in combination with rifampin or other CYP3A4 inducers, because of decreased exposure (7.2, 12.3)
USE IN SPECIFIC POPULATIONS
Hepatic impairment: Tasimelteon has not been studied in patients with severe hepatic impairment and is not recommended in these patients (8.6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2022
- Non-24-Hour Sleep-Wake Disorder (Non-24) in adults (1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Non-24-Hour Sleep-Wake Disorder (Non-24)
2 DOSAGE AND ADMINISTRATION
2.2 Recommended Dosage for Tasimelteon Capsules for Non-24
2.4 Important Administration Information
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Somnolence
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Strong CYP1A2 Inhibitors (e.g., fluvoxamine)
7.2 Strong CYP3A4 Inducers (e.g., rifampin)
7.3 Beta-Adrenergic Receptor Antagonists (e.g., acebutolol, metoprolol)
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Smokers
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.2 Abuse
9.3 Dependence
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Non-24-Hour Sleep-Wake Disorder (Non-24)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
- 2 DOSAGE AND ADMINISTRATION
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
- 5 WARNINGS AND PRECAUTIONS
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
More than 2080 subjects have been treated with at least one dose of tasimelteon, of which more than 380 have been treated for > 26 weeks and more than 170 have been treated for > 1 year.
Non-24-Hour Sleep-Wake Disorder (Non-24)
A 26-week, parallel-arm placebo-controlled study (Study 1) evaluated tasimelteon (n=42) compared to placebo (n=42) in patients with Non-24. A randomized-withdrawal, placebo-controlled study of 8 weeks duration (Study 2) also evaluated tasimelteon (n=10), compared to placebo (n=10), in patients with Non-24.
In placebo-controlled studies, 6% of patients exposed to tasimelteon discontinued treatment due to an adverse event, compared with 4% of patients who received placebo.
Table 2 shows the incidence of adverse reactions from Study 1.
Table 2: Adverse Reactions in Study 1 Tasimelteon
N=42
Placebo
N=42
Headache
17%
7%
Alanine aminotransferase increased
10%
5%
Nightmare/abnormal dreams
10%
0%
Upper respiratory tract infection
7%
0%
Urinary tract infection
7%
2%
*Adverse reactions with an incidence >5% and at least twice as high on tasimelteon than on placebo are displayed.
-
7 DRUG INTERACTIONS
7.1 Strong CYP1A2 Inhibitors (e.g., fluvoxamine)
Avoid use of tasimelteon in combination with fluvoxamine or other strong CYP1A2 inhibitors because of a potentially large increase in tasimelteon exposure and greater risk of adverse reactions [see Clinical Pharmacology (12.3)].
7.2 Strong CYP3A4 Inducers (e.g., rifampin)
Avoid use of tasimelteon in combination with rifampin or other CYP3A4 inducers because of a potentially large decrease in tasimelteon exposure with reduced efficacy [see Clinical Pharmacology (12.3)].
7.3 Beta-Adrenergic Receptor Antagonists (e.g., acebutolol, metoprolol)
Beta-adrenergic receptor antagonists have been shown to reduce the production of melatonin via specific inhibition of beta-1 adrenergic receptors. Nighttime administration of beta- adrenergic receptor antagonists may reduce the efficacy of tasimelteon.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available postmarketing case reports with tasimelteon use in pregnant women are not sufficient to evaluate drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In pregnant rats, no embryofetal developmental toxicity was observed at exposures of 50 mg/kg/day, or up to 24 times higher than the human exposure at the maximum recommended human dose (MRHD) (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated populations are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In pregnant rats administered tasimelteon at oral doses of 5 mg/kg/day, 50 mg/kg/day, or 500 mg/kg/day during the period of organogenesis, there were no effects on embryofetal development. The highest dose tested is approximately 240 times the MRHD of 20 mg/day, based on mg/m2 body surface area.
In pregnant rabbits administered tasimelteon at oral doses of 5 mg/kg/day, 30 mg/kg/day, or 200 mg/kg/day during the period of organogenesis, embryolethality and embryofetal toxicity (reduced fetal body weight and delayed ossification) were observed at the highest dose tested. The highest dose is approximately 200 times the MRHD.
Oral administration of tasimelteon at 50 mg/kg/day, 150 mg/kg/day, or 450 mg/kg/day to rats throughout organogenesis resulted in persistent reductions in body weight, delayed sexual maturation, and physical development, and neurobehavioral impairment in offspring at the highest dose tested which is approximately 220 times the MRHD based on mg/m2 body surface area. Reduced body weight in offspring was also observed at the mid-dose. The no effect dose (NOEL), (50 mg/kg/day) is approximately 25 times the MRHD based on mg/m2 body surface area.
8.2 Lactation
Risk Summary
There are no data on the presence of tasimelteon or its metabolites in human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for tasimelteon and any potential adverse effects on the breastfed infant from tasimelteon or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of tasimelteon for the treatment of Non-24 in pediatric patients have not been established.
Juvenile Animal Toxicity Data
Juvenile rats received oral doses of tasimelteon at 50, 150, or 450 mg/kg from weaning (day 21) through adulthood (day 90). These doses are approximately 12 to 108 times the maximum recommended human dose (MRHD) of 20 mg based on a mg/m2 body surface area. Toxicity was observed mainly at the highest dose and included mortality (females only), tremors, unsteady gait, decrease in growth and development compared to controls. The former reflected as decreases in bone growth, bone mineral content, bone ossification, and a delay in attainment of sexual maturation. Tasimelteon had no effect on fertility, reproduction, or learning and memory. The No Observed Adverse Effect Level (NOAEL) is 150 mg/kg/day, which is approximately 178 times the MRHD based on AUC.
8.5 Geriatric Use
The risk of adverse reactions may be greater in elderly (>65 years) patients than younger patients because exposure to tasimelteon is increased by approximately 2-fold compared with younger patients.
8.6 Hepatic Impairment
Dose adjustment is not necessary in patients with mild or moderate hepatic impairment. Tasimelteon has not been studied in patients with severe hepatic impairment (Child-Pugh Class C). Therefore, tasimelteon is not recommended for use in patients with severe hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Smokers
Smoking causes induction of CYP1A2 levels. The exposure of tasimelteon in smokers was lower than in non-smokers and therefore the efficacy of tasimelteon may be reduced in smokers [see Clinical pharmacology (12.3)].
-
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
Tasimelteon is not a controlled substance under the Controlled Substances Act.
9.2 Abuse
Tasimelteon did not produce any abuse-related signals in animal behavioral studies. Rats did not self-administer tasimelteon, suggesting that the drug does not have rewarding properties. There were also no signs or symptoms indicative of abuse potential in clinical studies with tasimelteon.
-
10 OVERDOSAGE
There is limited premarketing clinical experience with the effects of an overdosage of tasimelteon.
As with the management of any overdose, general symptomatic and supportive measures should be used, along with immediate gastric lavage where appropriate. Intravenous fluids should be administered as needed. Respiration, pulse, blood pressure, and other appropriate vital signs should be monitored, and general supportive measures employed.
While hemodialysis was effective at clearing tasimelteon and the majority of its major metabolites in patients with renal impairment, it is not known if hemodialysis will effectively reduce exposure in the case of overdose.
As with the management of any overdose, the possibility of multiple drug ingestion should be considered. Contact a poison control center for current information on the management of overdose.
-
11 DESCRIPTION
Tasimelteon is a melatonin receptor agonist, chemically designated as (1R, 2R)-N-[2- (2,3-dihydrobenzofuran-4-yl)cyclopropylmethyl]propanamide, containing two chiral centers. The molecular formula is C15H19NO2, and the molecular weight is 245.32. The structural formula is:
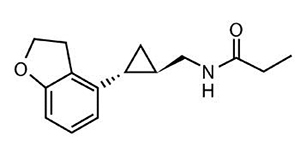
Tasimelteon is a white to off-white crystalline powder. It is very slightly soluble in cyclohexane, slightly soluble in water and 0.1 N hydrochloric acid, and freely soluble or very soluble in methanol, 95% ethanol, acetonitrile, isopropanol, polyethylene glycol 300, propylene glycol and ethyl acetate.
Tasimelteon is available in 20 mg strength capsules for oral administration. Inactive ingredients are colloidal silicon dioxide, croscarmellose sodium, lactose anhydrous, magnesium stearate, and microcrystalline cellulose. Each capsule shell consists of FD&C Blue No. 1, FD&C Red No. 40, gelatin, sodium lauryl sulfate, and titanium dioxide. The imprinting ink contains black iron oxide, potassium hydroxide, propylene glycol, and shellac.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism by which tasimelteon exerts its therapeutic effect in patients with Non-24 is unclear. However, tasimelteon is an agonist at melatonin MT1 and MT2 receptors which are thought to be involved in the control of circadian rhythms.
12.2 Pharmacodynamics
Tasimelteon is an agonist at MT1 and MT2 receptors with greater affinity for the MT2 as compared to the MT1 receptor (Ki = 0.304 nM and 0.07 nM, respectively). The major metabolites of tasimelteon have less than one-tenth of the binding affinity of the parent molecule for both the MT1 and MT2 receptors.
12.3 Pharmacokinetics
The pharmacokinetics of tasimelteon is linear over doses ranging from 3 to 300 mg (0.15 to 15 times the recommended daily dosage). The pharmacokinetics of tasimelteon and its metabolites did not change with repeated daily dosing.
Absorption
The absolute oral bioavailability is 38.3%. The peak concentration (Tmax) of tasimelteon occurred approximately 0.5 to 3 hours after fasted oral administration.
Effect of food
When administered with a high-fat meal, the Cmax of tasimelteon was 44% lower than when given in a fasted state, and the median Tmax was delayed by approximately 1.75 hours. Therefore, tasimelteon should be taken without food.
Distribution
The apparent oral volume of distribution of tasimelteon at steady state in young healthy subjects is approximately 59 L to 126 L. At therapeutic concentrations, tasimelteon is about 90% bound to proteins.
Metabolism
Tasimelteon is extensively metabolized. Metabolism of tasimelteon consists primarily of oxidation at multiple sites and oxidative dealkylation resulting in opening of the dihydrofuran ring followed by further oxidation to give a carboxylic acid. CYP1A2 and CYP3A4 are the major isozymes involved in the metabolism of tasimelteon.
Phenolic glucuronidation is the major phase II metabolic route.
Major metabolites had 13-fold or less activity at melatonin receptors compared to tasimelteon.
Elimination
Following oral administration of radiolabeled tasimelteon, 80% of total radioactivity was excreted in urine and approximately 4% in feces, resulting in a mean recovery of 84%. Less than 1% of the dose was excreted in urine as the parent compound.
The observed mean elimination half-life for tasimelteon is 1.3 ± 0.4 hours. The mean terminal elimination half-life ± standard deviation of the main metabolites ranges from 1.3 ± 0.5 to 3.7 ± 2.2.
Repeated once daily dosing with tasimelteon does not result in changes in pharmacokinetic parameters or significant accumulation of tasimelteon.
Studies in Specific Populations
Elderly
In elderly subjects, tasimelteon exposure increased by approximately two-fold compared with non-elderly adults.
Gender
The mean overall exposure of tasimelteon was approximately 20% to 30% greater in female than in male subjects.
Race
The effect of race on exposure of tasimelteon was not evaluated.
Hepatic Impairment
The pharmacokinetic profile of a 20 mg dose of tasimelteon was compared among eight subjects with mild hepatic impairment (Child-Pugh Score ≥5 and ≤6 points), eight subjects with moderate hepatic impairment (Child-Pugh Score ≥7 and ≤9 points), and 13 healthy matched controls. Tasimelteon exposure was increased less than two-fold in subjects with moderate hepatic impairment. Therefore, no dose adjustment is needed in patients with mild or moderate hepatic impairment. Tasimelteon has not been studied in patients with severe hepatic impairment (Child-Pugh Class C) and is not recommended in these patients.
Renal Impairment
The pharmacokinetic profile of a 20 mg dose of tasimelteon was compared among eight subjects with severe renal impairment (estimated glomerular filtration rate [eGFR] ≤29 mL/min/1.73 m2), eight subjects with end-stage renal disease (ESRD) (GFR <15 mL/min/1.73 m2) requiring hemodialysis, and sixteen healthy matched controls. There was no apparent relationship between tasimelteon CL/F and renal function, as measured by either estimated creatinine clearance or eGFR. Subjects with severe renal impairment had a 30% lower clearance, and clearance in subjects with ESRD was comparable to that of healthy subjects. No dose adjustment is necessary for patients with renal impairment.
Smokers (smoking is a moderate CYP1A2 inducer)
Tasimelteon exposure decreased by approximately 40% in smokers, compared to non-smokers [see Use in Specific Populations (8.7)].
Drug Interaction Studies
No potential drug interactions were identified in in vitro studies with CYP inducers or inhibitors of CYP1A1, CYP1A2, CYP2B6, CYP2C9/2C19, CYP2E1, CYP2D6, and transporters including P-glycoprotein, OATP1B1, OATP1B3, OCT2, OAT1, and OAT3.
Effect of Other Drugs on Tasimelteon
Drugs that inhibit CYP1A2 and CYP3A4 are expected to alter the metabolism of tasimelteon.
Fluvoxamine (strong CYP1A2 inhibitor): the AUC0-inf and Cmax of tasimelteon increased by 7-fold and 2-fold, respectively, when coadministered with fluvoxamine 50 mg (after 6 days of fluvoxamine 50 mg per day) [see Drug Interactions (7.1)].
Ketoconazole (strong CYP3A4 inhibitor): tasimelteon exposure increased by approximately 50% when coadministered with ketoconazole 400 mg (after 5 days of ketoconazole 400 mg per day) [see Drug Interactions (7.2)].
Rifampin (strong CYP3A4 and moderate CYP2C19 inducer): the exposure of tasimelteon decreased by approximately 90% when coadministered with rifampin 600 mg (after 11 days of rifampin 600 mg per day). Efficacy may be reduced when tasimelteon is used in combination with strong CYP3A4 inducers, such as rifampin [see Drug Interactions (7.2)].
Effect of Tasimelteon on Other Drugs
Midazolam (CYP3A4 substrate): Administration of tasimelteon 20 mg once a day for 14 days did not produce any significant changes in the Tmax, Cmax, or AUC of midazolam or 1-OH midazolam. This indicates there is no induction of CYP3A4 by tasimelteon at this dose.
Rosiglitazone (CYP2C8 substrate): Administration of tasimelteon 20 mg once a day for 16 days did not produce any clinically significant changes in the Tmax, Cmax, or AUC of rosiglitazone after oral administration of 4 mg. This indicates that there is no induction of CYP2C8 by tasimelteon at this dose.
Effect of Alcohol on Tasimelteon
In a study of 28 healthy volunteers, a single dose of ethanol (0.6 g/kg for women and 0.7 g/kg for men) was coadministered with a 20 mg dose of tasimelteon. There was a trend for an additive effect of tasimelteon and ethanol on some psychomotor tests.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Tasimelteon was administered orally for up to two years to mice (30 mg/kg/day, 100 mg/kg/day, and 300 mg/kg/day) and rats (20 mg/kg/day, 100 mg/kg/day, and 250 mg/kg/day). No evidence of carcinogenic potential was observed in mice; the highest dose tested is approximately 75 times the maximum recommended human dose (MRHD) of 20 mg/day, based on a mg/m2 body surface area. In rats, the incidence of liver tumors was increased in males (adenoma and carcinoma) and females (adenoma) at 100 mg/kg/day and 250 mg/kg/day; the incidence of tumors of the uterus (endometrial adenocarcinoma) and uterus and cervix (squamous cell carcinoma) were increased at 250 mg/kg/day. There was no increase in tumors at the lowest dose tested in rats, which is approximately 10 times the MRHD based on a mg/m2 body surface area.
Mutagenesis
Tasimelteon was negative in an in vitro bacterial reverse mutation (Ames) assay, an in vitro cytogenetics assay in primary human lymphocytes, and an in vivo micronucleus assay in rats.
Impairment of Fertility
When male and female rats were given tasimelteon at oral doses of 5 mg/kg/day, 50 mg/kg/day, or 500 mg/kg/day prior to and throughout mating and continuing in females to gestation day 7, estrus cycle disruption and decreased fertility were observed at all but the lowest dose tested. The no-effect dose for effects on female reproduction (5 mg/kg/day) is approximately 2 times the MRHD based on a mg/m2 body surface area.
-
14 CLINICAL STUDIES
14.1 Non-24-Hour Sleep-Wake Disorder (Non-24)
The effectiveness of tasimelteon in the treatment of Non-24-Hour Sleep-Wake Disorder (Non-24) was established in two randomized double-masked, placebo-controlled, multicenter, parallel-group studies (Studies 1 and 2) in totally blind patients with Non-24.
In study 1, 84 patients with Non-24 (median age 54 years) were randomized to receive tasimelteon 20 mg or placebo, one hour prior to bedtime, at the same time every night for up to 6 months.
Study 2 was a randomized withdrawal trial in 20 patients with Non-24 (median age 55 years) that was designed to evaluate the maintenance of efficacy of tasimelteon after 12-weeks. Patients were treated for approximately 12 weeks with tasimelteon 20 mg one hour prior to bedtime, at the same time every night. Patients in whom the calculated time of peak melatonin level (melatonin acrophase) occurred at approximately the same time of day (in contrast to the expected daily delay) during the run-in phase were randomized to receive placebo or continue treatment with tasimelteon 20 mg for 8 weeks.
Study 1 and Study 2 evaluated the duration and timing of nighttime sleep and daytime naps via patient-recorded diaries. During Study 1, patient diaries were recorded for an average of 88 days during screening, and 133 days during randomization. During Study 2, patient diaries were recorded for an average of 57 days during the run-in phase, and 59 days during the randomized-withdrawal phase.
Because symptoms of nighttime sleep disruption and daytime sleepiness are cyclical in patients with Non-24, with severity varying according to the state of alignment of the individual patient’s circadian rhythm with the 24-hour day (least severe when fully aligned, most severe when 12 hours out of alignment), efficacy endpoints for nighttime total sleep time and daytime nap duration were based on the 25% of nights with the least nighttime sleep, and the 25% of days with the most daytime nap time. In Study 1, patients in the tasimelteon group had, at baseline, an average 195 minutes of nighttime sleep and 137 minutes of daytime nap time on the 25% of most symptomatic nights and days, respectively. Treatment with tasimelteon resulted in a significant improvement, compared with placebo, for both of these endpoints in Study 1 and Study 2 (see Table 3).
Table 3: Effects of Tasimelteon 20 mg on Nighttime Sleep Time and Daytime Nap Time in Study 1 and Study 2 Study 1 Study 2 Change from Baseline Tasimelteon
20 mg
N=42Placebo
N=42Tasimelteon
20 mg
N=10Placebo
N=10Nighttime sleep time on 25% most symptomatic nights (minutes) 50 22 -7 -74 Daytime nap time on 25% most symptomatic days (minutes) -49 -22 -9 50 A responder analysis of patients with both ≥45 minutes increase in nighttime sleep and ≥45 minutes decrease in daytime nap time was conducted in Study 1: 29% (n=12) of patients treated with tasimelteon, compared with 12% (n=5) of patients treated with placebo met the responder criteria.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Tasimelteon capsules are available as follows:
20 mg – Each size 1 hard gelatin capsule with light blue opaque cap and body imprinted with A44 on cap in black ink contains white to off-white color granular powder. Capsules are supplied in bottles of 30 (NDC 0480-4490-56).
Storage and Handling
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Protect from exposure to light and moisture.
-
17 PATIENT COUNSELING INFORMATION
- Advise patients to limit their activities to preparing for going to bed after taking tasimelteon capsules because tasimelteon can potentially impair the performance of activities requiring complete mental alertness [see Warnings and Precautions (5.1)].
- Administration Information for tasimelteon capsules [see Dosage and Administration (2.2, 2.4)].
- Advise patients to take tasimelteon without food.
- Advise patients to take tasimelteon before bedtime at the same time every night.
- Advise patients to skip the dose that night if they cannot take tasimelteon at approximately the same time on a given night.
- Advise patients to swallow tasimelteon capsules whole.
- Advise patients that because of individual differences in circadian rhythms, daily use for several weeks or months may be necessary before benefit from tasimelteon capsules is observed [see Dosage and Administration (2.2)].
Manufactured in India By:
Watson Pharma Private Limited
Verna, Salcette Goa 403 722 INDIAManufactured For:
Teva Pharmaceuticals
Parsippany, NJ 07054Iss. 5/2022
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TASIMELTEON
tasimelteon capsule, gelatin coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0480-4490 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TASIMELTEON (UNII: SHS4PU80D9) (TASIMELTEON - UNII:SHS4PU80D9) TASIMELTEON 20 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C RED NO. 40 (UNII: WZB9127XOA) GELATIN (UNII: 2G86QN327L) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SHELLAC (UNII: 46N107B71O) Product Characteristics Color blue (light blue) Score no score Shape CAPSULE Size 19mm Flavor Imprint Code A44 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0480-4490-56 30 in 1 BOTTLE; Type 0: Not a Combination Product 12/29/2022 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA211601 12/29/2022 Labeler - Teva Pharmaceuticals, Inc. (022629579)
