Label: PARSABIV- etelcalcetide injection, solution



-
NDC Code(s):
55513-740-01,
55513-740-10,
55513-740-20,
55513-741-01, view more55513-741-10, 55513-741-20, 55513-742-01, 55513-742-10, 55513-742-20
- Packager: Amgen Inc
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated January 10, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PARSABIV safely and effectively. See full prescribing information for PARSABIV.
PARSABIV® (etelcalcetide) injection, for intravenous use
Initial U.S. Approval: 2017INDICATIONS AND USAGE
PARSABIV is a calcium-sensing receptor agonist indicated for:
- Secondary hyperparathyroidism (HPT) in adult patients with chronic kidney disease (CKD) on hemodialysis. (1)
Limitations of Use:
PARSABIV has not been studied in adult patients with parathyroid carcinoma, primary hyperparathyroidism, or with CKD who are not on hemodialysis and is not recommended for use in these populations.
DOSAGE AND ADMINISTRATION
- Ensure corrected serum calcium is at or above the lower limit of normal prior to initiation, dose increase, or re-initiation. (2.1)
- The recommended starting dose is 5 mg administered by intravenous bolus injection three times per week at the end of hemodialysis treatment. (2.1)
- The maintenance dose is individualized and determined by titration based on parathyroid hormone (PTH) and corrected serum calcium response. The dose range is 2.5 to 15 mg three times per week. (2.1)
- The dose may be increased in 2.5 mg or 5 mg increments no more frequently than every 4 weeks. (2.2)
- Measure serum calcium within 1 week after initiation or dose adjustment and every 4 weeks for maintenance. (2.2)
- Measure PTH after 4 weeks from initiation or dose adjustment. (2.2)
- Decrease or temporarily discontinue PARSABIV in individuals with PTH levels below the target range. (2.2)
- Consider decreasing or temporarily discontinuing PARSABIV or use concomitant therapies to increase corrected serum calcium in patients with a corrected serum calcium below the lower limit of normal but at or above 7.5 mg/dL without symptoms of hypocalcemia. (2.2)
- Stop PARSABIV and treat hypocalcemia if the corrected serum calcium falls below 7.5 mg/dL or patients report symptoms of hypocalcemia. (2.2)
- Do not mix or dilute prior to administration. (2.3)
- Administer by intravenous bolus injection into the venous line of the dialysis circuit after hemodialysis, during rinse back or intravenously after rinse back.
- Administer a sufficient volume of saline, e.g. 150 mL of rinse back, after injection into the dialysis tubing.
- If administered after rinse back, administer PARSABIV intravenously followed by at least 10 mL of saline flush. (2.3)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
PARSABIV is contraindicated in patients with known hypersensitivity to etelcalcetide or any of its excipients. (4)
WARNINGS AND PRECAUTIONS
- Hypocalcemia: Sometimes severe. Severe hypocalcemia can cause paresthesias, myalgias, muscle spasms, seizures, QT prolongation, and ventricular arrhythmias. Patients predisposed to QT interval prolongation, ventricular arrhythmias, and seizures may be at increased risk and require close monitoring. Educate patients on the symptoms of hypocalcemia and advise them to contact a healthcare provider if they occur. (5.1)
- Worsening Heart Failure: Reductions in corrected serum calcium may be associated with congestive heart failure, however, a causal relationship to PARSABIV could not be completely excluded. Closely monitor patients for worsening signs and symptoms of heart failure. (5.2)
- Upper Gastrointestinal (GI) Bleeding: Patients with risk factors for upper GI bleeding may be at increased risk. Monitor patients and promptly evaluate and treat any suspected GI bleeding. (5.3)
- Adynamic Bone: May develop if PTH levels are chronically suppressed. If PTH levels decrease below the recommended target range, the dose of PARSABIV should be reduced or discontinued. (5.4)
ADVERSE REACTIONS
The most common adverse reactions (≥ 5%) were blood calcium decreased, muscle spasms, diarrhea, nausea, vomiting, headache, hypocalcemia, and paresthesia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Monitoring and Dose Adjustment
2.3 Administration
2.4 Switching from Cinacalcet to PARSABIV
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypocalcemia
5.2 Worsening Heart Failure
5.3 Upper Gastrointestinal Bleeding
5.4 Adynamic Bone
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
- Ensure corrected serum calcium is at or above the lower limit of normal prior to PARSABIV initiation, a PARSABIV dose increase, or re-initiation of PARSABIV therapy after a dosing interruption [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
- The recommended starting dose of PARSABIV is 5 mg administered by intravenous (IV) bolus injection three times per week at the end of hemodialysis treatment [see Dosage and Administration (2.3)].
- The maintenance dose of PARSABIV is individualized and determined by titration based on parathyroid hormone (PTH) and corrected serum calcium response [see Dosage and Administration (2.2)]. The maintenance dose is the dose that maintains PTH levels within the recommended target range and corrected serum calcium within the normal range. The lowest maintenance dose of PARSABIV is 2.5 mg three times per week, and the highest maintenance dose of PARSABIV is 15 mg three times per week.
- Administer PARSABIV only at the end of hemodialysis treatment.
- If a regularly scheduled hemodialysis treatment is missed, DO NOT administer any missed doses. Resume PARSABIV at the end of the next hemodialysis treatment at the prescribed dose. If doses of PARSABIV are missed for more than 2 weeks, re-initiate PARSABIV at the recommended starting dose of 5 mg (or 2.5 mg if that was the patient's last dose).
2.2 Monitoring and Dose Adjustment
- Monitor corrected serum calcium and PTH levels during dose initiation, dose adjustment, and dose maintenance according to the schedule in Table 1.
Table 1: Recommended Schedule for Monitoring Corrected Serum Calcium and Parathyroid Hormone Levels during PARSABIV Treatment Dose Initiation or Dose Adjustment Maintenance Corrected Serum Calcium Levels 1 week after Every 4 weeks Parathyroid Hormone Levels 4 weeks after Per clinical practice - Titrate PARSABIV dose based on PTH and corrected serum calcium response. At the maintenance dose, PTH levels should be within the recommended target range and corrected serum calcium within the normal range.
- Increase the dose of PARSABIV in 2.5 mg or 5 mg increments in individuals with corrected serum calcium within the normal range and PTH levels above the recommended target range based on the patient's PTH levels no more frequently than every 4 weeks up to a maximum dose of 15 mg three times per week.
- Decrease or temporarily discontinue PARSABIV dosing in individuals with PTH levels below the target range. In individuals with a corrected serum calcium below the lower limit of normal but at or above 7.5 mg/dL without symptoms of hypocalcemia, consider decreasing or temporarily discontinuing PARSABIV or use concomitant therapies to increase corrected serum calcium [see Warnings and Precautions (5.1)]. If the dose is stopped, then re-initiate PARSABIV at a lower dose when the PTH is within the target range and hypocalcemia has been corrected.
- Stop PARSABIV and treat hypocalcemia if the corrected serum calcium falls below 7.5 mg/dL or patients report symptoms of hypocalcemia [see Warnings and Precautions (5.1)]. When the corrected serum calcium is within normal limits, symptoms of hypocalcemia have resolved, and predisposing factors for hypocalcemia have been addressed, re-initiate PARSABIV at a dose 5 mg lower than the last administered dose. If the last administered dose of PARSABIV was 2.5 mg or 5 mg, re-initiate at a dose of 2.5 mg.
2.3 Administration
- Do not mix or dilute PARSABIV prior to administration. The solution is clear and colorless. Inspect PARSABIV for particulate matter and discoloration prior to administration. Do not use PARSABIV vials if particulate matter or discoloration is observed.
- PARSABIV is removed by the dialyzer membrane and must be administered after blood is no longer circulating through the dialyzer.
- Administer PARSABIV by intravenous bolus injection into the venous line of the dialysis circuit after hemodialysis during rinse back or intravenously after rinse back.
- Administer a sufficient volume of saline, e.g. 150 mL of rinse back, after PARSABIV injection into the dialysis tubing.
- If PARSABIV is administered after rinse back, administer PARSABIV intravenously followed by at least 10 mL of saline flush.
2.4 Switching from Cinacalcet to PARSABIV
- Discontinue cinacalcet for at least 7 days prior to starting PARSABIV, and initiate PARSABIV treatment at a starting dose of 5 mg. Ensure corrected serum calcium is at or above the lower limit of normal prior to PARSABIV initiation [see Warnings and Precautions (5.1)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Hypersensitivity
PARSABIV is contraindicated in patients with known hypersensitivity to etelcalcetide or any of its excipients. Hypersensitivity reactions, including face edema and anaphylactic reaction, have occurred with PARSABIV [see Adverse Reactions (6)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypocalcemia
PARSABIV lowers serum calcium [see Adverse Reactions (6.1)] and can lead to hypocalcemia, sometimes severe. Significant lowering of serum calcium can cause paresthesias, myalgias, muscle spasms, seizures, QT interval prolongation, and ventricular arrhythmia.
QT Interval Prolongation and Ventricular Arrhythmia
In the combined placebo-controlled studies, more patients treated with PARSABIV experienced a maximum increase from baseline of greater than 60 msec in the QTcF interval (0% placebo versus 1.2% PARSABIV). In these studies, the incidence of a maximum post-baseline predialysis QTcF > 500 msec in the placebo and PARSABIV groups was 1.9% and 4.8%, respectively [see Adverse Reactions (6.1)]. Patients with congenital long QT syndrome, history of QT interval prolongation, family history of long QT syndrome or sudden cardiac death, and other conditions that predispose to QT interval prolongation and ventricular arrhythmia may be at increased risk for QT interval prolongation and ventricular arrhythmias if they develop hypocalcemia due to PARSABIV. Closely monitor corrected serum calcium and QT interval in patients at risk receiving PARSABIV.
Seizures
Significant reductions in corrected serum calcium may lower the threshold for seizures. Patients with a history of seizure disorder may be at increased risk for seizures if they develop hypocalcemia due to PARSABIV. Monitor corrected serum calcium in patients with seizure disorders receiving PARSABIV.
Risk of Hypocalcemia with Other Serum Calcium Lowering Products
Concurrent administration of PARSABIV with another oral calcium-sensing receptor agonist could result in severe, life-threatening hypocalcemia. Patients switching from cinacalcet to PARSABIV should discontinue cinacalcet for at least 7 days prior to initiating PARSABIV [see Dosage and Administration (2.4)]. Closely monitor corrected serum calcium in patients receiving PARSABIV and concomitant therapies known to lower serum calcium.
Monitoring Serum Calcium and Patient Education
Measure corrected serum calcium prior to initiation of PARSABIV. Do not initiate in patients if the corrected serum calcium is less than the lower limit of normal. Monitor corrected serum calcium within 1 week after initiation or dose adjustment and every 4 weeks during treatment with PARSABIV [see Dosage and Administration (2.2)]. Educate patients on the symptoms of hypocalcemia and advise them to contact a healthcare provider if they occur.
Management of Hypocalcemia
If corrected serum calcium falls below the lower limit of normal or symptoms of hypocalcemia develop, start or increase calcium supplementation (including calcium, calcium-containing phosphate binders, and/or vitamin D sterols or increases in dialysate calcium concentration). PARSABIV dose reduction or discontinuation of PARSABIV may be necessary [see Dosage and Administration (2.2)].
5.2 Worsening Heart Failure
In clinical studies with PARSABIV, cases of hypotension, congestive heart failure, and decreased myocardial performance have been reported. In clinical studies, heart failure requiring hospitalization occurred in 2% of PARSABIV-treated patients and 1% of placebo-treated patients. Reductions in corrected serum calcium may be associated with congestive heart failure, however, a causal relationship to PARSABIV could not be completely excluded. Closely monitor patients treated with PARSABIV for worsening signs and symptoms of heart failure.
5.3 Upper Gastrointestinal Bleeding
In clinical studies, two patients treated with PARSABIV in 1253 patient-years of exposure had upper gastrointestinal (GI) bleeding noted at the time of death while no patient in the control groups in 384 patient-years of exposure had upper GI bleeding noted at the time of death. The exact cause of GI bleeding in these patients is unknown, and there were too few cases to determine whether these cases were related to PARSABIV.
Patients with risk factors for upper GI bleeding (such as known gastritis, esophagitis, ulcers, or severe vomiting) may be at increased risk for GI bleeding while receiving PARSABIV treatment. Monitor patients for worsening of common GI adverse reactions of nausea and vomiting associated with PARSABIV [see Adverse Reactions (6.1)] and for signs and symptoms of GI bleeding and ulcerations during PARSABIV therapy. Promptly evaluate and treat any suspected GI bleeding.
5.4 Adynamic Bone
Adynamic bone may develop if PTH levels are chronically suppressed. If PTH levels decrease below the recommended target range, the dose of vitamin D sterols and/or PARSABIV should be reduced or therapy discontinued. After discontinuation, resume therapy at a lower dose to maintain PTH levels in the target range [see Dosage and Administration (2.1)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Hypocalcemia [see Warnings and Precautions (5.1)]
- Worsening Heart Failure [see Warnings and Precautions (5.2)]
- Upper Gastrointestinal Bleeding [see Warnings and Precautions (5.3)]
- Adynamic Bone [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The data in Table 2 are derived from two placebo-controlled clinical studies in patients with chronic kidney disease and secondary hyperparathyroidism on hemodialysis. The data reflect exposure of 503 patients to PARSABIV with a mean duration of exposure to PARSABIV of 23.6 weeks. The mean age of patients was approximately 58 years, and 60% of the patients were male. Of the total patients, 67% were Caucasian, 28% were Black or African American, 2.6% were Asian, 1.2% were Native Hawaiian or Other Pacific Islander, and 1.6% were categorized as Other.
Table 2 shows common adverse reactions associated with the use of PARSABIV in the pool of placebo-controlled studies. These adverse reactions occurred more commonly on PARSABIV than on placebo and were reported in at least 5% of patients treated with PARSABIV.
Table 2: Adverse Reactions Reported in ≥ 5% of PARSABIV-Treated Patients Adverse Reaction* Placebo
(N = 513)PARSABIV
(N = 503)- *
- Included adverse reactions reported with at least 1% greater incidence in the PARSABIV group compared to the placebo group
- †
- Asymptomatic reductions in calcium below 7.5 mg/dL or clinically significant asymptomatic reductions in corrected serum calcium between 7.5 and < 8.3 mg/dL (that required medical management)
- ‡
- Symptomatic reductions in corrected serum calcium < 8.3 mg/dL
- §
- Paresthesia includes preferred terms of paresthesia and hypoesthesia
Blood calcium decreased† 10% 64% Muscle spasms 7% 12% Diarrhea 9% 11% Nausea 6% 11% Vomiting 5% 9% Headache 6% 8% Hypocalcemia‡ 0.2% 7% Paresthesia§ 1% 6% Other adverse reactions associated with the use of PARSABIV but reported in < 5% of patients in the PARSABIV group in the two placebo-controlled clinical studies were:
- Hyperkalemia: 3% and 4% for placebo and PARSABIV, respectively.
- Hospitalization for Heart Failure: 1% and 2% for placebo and PARSABIV, respectively.
- Myalgia: 0.2% and 2% for placebo and PARSABIV, respectively.
- Hypophosphatemia: 0.2% and 1% for placebo and PARSABIV, respectively.
Description of Selected Adverse Reactions
Hypocalcemia
In the combined placebo-controlled studies, a higher proportion of patients on PARSABIV developed at least one corrected serum calcium value below 7.0 mg/dL (7.6% PARSABIV, 3.1% placebo), below 7.5 mg/dL (27% PARSABIV, 5.5% placebo), and below 8.3 mg/dL (79% PARSABIV, 19% placebo). In the combined placebo-controlled studies, 1% of patients in the PARSABIV group and 0% of patients in the placebo group discontinued treatment due to an adverse reaction attributed to a low corrected serum calcium.
Hypophosphatemia
In the combined placebo-controlled studies, 18% of patients treated with PARSABIV and 8.2% of patients treated with placebo had at least one measured phosphorus level below the lower normal limit (i.e., 2.2 mg/dL).
QTc Interval Prolongation Secondary to Hypocalcemia
In the combined placebo-controlled studies, more patients treated with PARSABIV experienced a maximum increase from baseline of greater than 60 msec in the QTcF interval (0% placebo versus 1.2% PARSABIV). The patient incidence of maximum post-baseline predialysis QTcF > 500 msec in the placebo and PARSABIV groups was 1.9% and 4.8%, respectively.
Hypersensitivity
In the combined placebo-controlled studies, the subject incidence of adverse reactions potentially related to hypersensitivity was 4.4% in the PARSABIV group and 3.7% in the placebo group. Hypersensitivity reactions in the PARSABIV group were pruritic rash, urticaria, and face edema.
6.2 Immunogenicity
As with all peptide therapeutics, there is potential for immunogenicity. The detection of anti-drug binding antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to etelcalcetide with the incidence of antibodies to other products may be misleading.
In clinical studies, 7.1% (71 out of 995) of patients with secondary hyperparathyroidism treated with PARSABIV for up to 6 months tested positive for binding anti-etelcalcetide antibodies. Fifty-seven out of 71 had pre-existing anti-etelcalcetide antibodies. No evidence of altered pharmacokinetic profile, clinical response, or safety profile was associated with pre-existing or developing anti-etelcalcetide antibodies.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postmarketing use of PARSABIV. Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Anaphylactic reaction
- Hypocalcemia in patients who were administered etelcalcetide concomitantly with other products known to lower serum calcium (e.g. cinacalcet, denosumab)
- Seizures secondary to hypocalcemia
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on the use of PARSABIV in pregnant women. In animal reproduction studies, effects were seen at doses associated with maternal toxicity that included hypocalcemia. In a pre- and post-natal study in rats administered etelcalcetide during organogenesis through delivery and weaning, there was a slight increase in perinatal pup mortality, delay in parturition, and transient effects on pup growth at exposures 1.8 times the human exposure for the clinical dose of 15 mg three times per week. There was no effect on sexual maturation, neurobehavioral, or reproductive function in the rat offspring. In embryo-fetal studies, when rats and rabbits were administered etelcalcetide during organogenesis, reduced fetal growth was observed at exposures 2.7 and 7 times exposures for the clinical dose, respectively.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
There were no effects on embryo-fetal development in Sprague-Dawley rats when etelcalcetide was dosed at 0.75, 1.5, and 3 mg/kg/day by the intravenous route during organogenesis (pre-mating to gestation day 17) at exposures up to 1.8 times human exposures at the clinical dose of 15 mg three times per week based on AUC. No effects on embryo-fetal development were observed in New Zealand White rabbits at doses of etelcalcetide of 0.375, 0.75, and 1.5 mg/kg by the intravenous route (gestation day 7 to 19), representing up to 4.3 times human exposures based on AUC. In separate studies at higher doses of 4.5 mg/kg in rats (gestation days 6 to 17) and 2.25 mg/kg in rabbits (gestation days 7 to 20), representing 2.7- and 7-fold clinical exposures, respectively, there was reduced fetal growth associated with maternal toxicities of hypocalcemia, tremoring, and reductions in body weight and food consumption.
In a pre- and post-natal development study in Sprague-Dawley rats administered etelcalcetide at 0.75, 1.5, and 3 mg/kg/day by the intravenous route (gestation day 7 to lactation day 20), there was a slight increase in perinatal pup mortality, delay in parturition, and transient reductions in post-natal growth at 3 mg/kg/day (representing 1.8-fold human exposures at the clinical dose of 15 mg three times per week based on AUC), associated with maternal toxicities of hypocalcemia, tremoring, and reductions in body weight and food consumption. There were no effects on sexual maturation, neurobehavioral, or reproductive function at up to 3 mg/kg/day, representing exposures up to 1.8-fold human exposure based on AUC.
8.2 Lactation
Risk Summary
There are no data regarding the presence of PARSABIV in human milk or effects on the breastfed infant or on milk production. Studies in rats showed [14C]-etelcalcetide was present in the milk at concentrations similar to plasma. Because of the potential for PARSABIV to cause adverse effects in breastfed infants including hypocalcemia, advise women that use of PARSABIV is not recommended while breastfeeding.
Data
Presence in milk was assessed following a single intravenous dose of [14C]-etelcalcetide in lactating rats at maternal exposures similar to the exposure at the human clinical dose of 15 mg three times per week. [14C]-etelcalcetide-derived radioactivity was present in milk at levels similar to plasma.
8.4 Pediatric Use
The safety and efficacy of PARSABIV have not been established in pediatric patients.
8.5 Geriatric Use
Of the 503 patients in placebo-controlled studies who received PARSABIV, 177 patients (35.2%) were ≥ 65 years old and 72 patients (14%) were ≥ 75 years old. No clinically significant differences in safety or efficacy were observed between patients ≥ 65 years and younger patients (≥ 18 and < 65 years old). No differences in plasma concentrations of etelcalcetide were observed between patients ≥ 65 years and younger patients (≥ 18 and < 65 years old).
-
10 OVERDOSAGE
There is no clinical experience with PARSABIV overdosage. Overdosage of PARSABIV may lead to hypocalcemia with or without clinical symptoms and may require treatment. Although PARSABIV is cleared by dialysis, hemodialysis has not been studied as a treatment for PARSABIV overdosage. In the event of overdosage, corrected serum calcium should be checked and patients should be monitored for symptoms of hypocalcemia, and appropriate measures should be taken [see Warnings and Precautions (5.1)].
-
11 DESCRIPTION
PARSABIV (etelcalcetide) is a synthetic peptide calcium-sensing receptor agonist. Etelcalcetide is a white to off-white powder with a molecular formula of C38H73N21O10S2∙xHCl (4 ≤ x ≤ 5) and a molecular weight of 1047.5 g/mol (monoisotopic; free base). It is soluble in water. The hydrochloride salt of etelcalcetide is described chemically as N-acetyl-D-cysteinyl-S-(L-cysteine disulfide)-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide hydrochloride.

PARSABIV (etelcalcetide) injection is supplied in a single-dose vial containing 5 mg/mL of etelcalcetide as a sterile, preservative-free, ready-to-use clear and colorless solution for intravenous injection.
Each PARSABIV single-dose vial contains 2.5 mg etelcalcetide (equivalent to 2.88 mg etelcalcetide as hydrochloride salt) or 5 mg etelcalcetide (equivalent to 5.77 mg etelcalcetide as hydrochloride salt) or 10 mg etelcalcetide (equivalent to 11.54 mg etelcalcetide as hydrochloride salt). PARSABIV single-dose vial is formulated with 0.85% (w/v) sodium chloride, 10 mM succinic acid, and adjusted to pH 3.3 with sodium hydroxide and/or hydrochloric acid.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Etelcalcetide is a calcimimetic agent that allosterically modulates the calcium-sensing receptor (CaSR). Etelcalcetide binds to the CaSR and enhances activation of the receptor by extracellular calcium. Activation of the CaSR on parathyroid chief cells decreases PTH secretion.
12.2 Pharmacodynamics
Following a single intravenous bolus administration of etelcalcetide, PTH levels decreased within 30 minutes post-dose. In the single-dose study, the extent and duration of the reduction in PTH increased with increasing dose. Reduction in PTH levels correlated with plasma etelcalcetide concentrations in hemodialysis patients. The reduction in PTH resulted in reductions in calcium and attenuation of post-dialytic phosphate elevation. The effect of reducing PTH levels was maintained throughout the 6-month dosing period when etelcalcetide was administered by intravenous bolus three times a week.
12.3 Pharmacokinetics
The pharmacokinetics of etelcalcetide is linear and does not change over time following single (5 to 60 mg) and multiple intravenous doses (2.5 to 20 mg) in chronic kidney disease patients with secondary hyperparathyroidism requiring hemodialysis. Etelcalcetide exhibited tri-exponential decay following intravenous administration. Based on population pharmacokinetic analysis, following three times a week intravenous dosing at the end of each 3- to 6-hour hemodialysis session in chronic kidney disease patients, etelcalcetide plasma levels reached steady state in 7-8 weeks after dosing with a predicted accumulation ratio of 3- to 4-fold, and the effective half-life was 3 to 4 days.
Distribution
In the population pharmacokinetics model, volume of distribution at steady state (Vss) was approximately 796 L. Etelcalcetide is predominately bound to plasma albumin by reversible covalent binding. Non-covalent binding of etelcalcetide to plasma proteins is low with a fraction unbound ratio of 0.53. The ratio of blood-to-plasma [14C]-etelcalcetide concentrations is approximately 0.6.
Elimination
Metabolism
Etelcalcetide is not metabolized by CYP450 enzymes. Etelcalcetide is biotransformed in blood by reversible disulfide exchange with endogenous thiols to predominantly form conjugates with serum albumin. Following a single radiolabeled dose of etelcalcetide in chronic kidney disease patients with secondary hyperparathyroidism requiring hemodialysis, the plasma exposure of biotransformation products is approximately 5-fold higher than that of etelcalcetide and their concentration-time course parallels that of etelcalcetide.
Excretion
Etelcalcetide is cleared by renal excretion in patients with normal renal function, while hemodialysis is the predominant elimination pathway in chronic kidney disease patients requiring hemodialysis. In chronic kidney disease patients on hemodialysis, etelcalcetide was removed with a hemodialysis clearance value of 7.66 L/hr. Following a single radiolabeled dose of etelcalcetide in chronic kidney disease patients with secondary hyperparathyroidism requiring hemodialysis, approximately 60% of [14C]-etelcalcetide was recovered in dialysate, and approximately 7% recovered in urine and feces combined over 175 days of collection period.
Specific Populations
Effects of Body Weight, Gender, Race, and Age
Results of population pharmacokinetic analyses indicate that body weight (29 to 163 kg), gender, race, and age (20 to 93 years of age) do not influence the pharmacokinetics of etelcalcetide. The pharmacokinetics of etelcalcetide in patients ≥ 65 years of age and in patients < 65 years of age is similar.
Drug Interactions
In Vitro Assessment of Drug Interactions
Etelcalcetide did not inhibit or induce CYP450 enzymes, and is not a substrate of CYP450 enzymes. Etelcalcetide was not a substrate of efflux and uptake transporter proteins (P-glycoprotein [Pgp], breast cancer resistance protein [BCRP], organic anion transporter [OAT] 1 and 3, organic anion polypeptide transporter [OATP] 1B1 and 1B3, organic cation transporter [OCT] 2, and peptide transporter [PEPT] 1 and 2). Etelcalcetide was also not an inhibitor of common transporter proteins (Pgp, BCRP, OAT1, OAT3, OATP1B1, OATP1B3, OCT2, or bile salt export pump [BSEP]).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No drug-related tumors were observed in Tg rasH2 transgenic mice when etelcalcetide was administered at doses of 0.3, 1, and 3 mg/kg in females and 0.375, 0.75, and 1.5 mg/kg in males once daily for 26 weeks by the subcutaneous route, representing up to 0.4-fold clinical exposure at the maximum human dose of 15 mg three times per week based on AUC. Etelcalcetide administration did not produce drug-related tumors when Sprague-Dawley rats were dosed at 0.2, 0.4, 0.8, and 1.6 mg/kg/day by the subcutaneous route for 89 weeks in females and 92 weeks in males, representing up to 0.4-fold clinical exposures achieved in patients receiving etelcalcetide at 15 mg three times per week, based on AUC.
Etelcalcetide was mutagenic in some strains of bacteria (Ames), but was not genotoxic in two in vitro and two in vivo mammalian genotoxicity assays.
There was no effect on male or female fertility when etelcalcetide was dosed at 0.75, 1.5, and 3 mg/kg/day by the intravenous route to rats at exposure levels up to 1.8-fold higher than exposures achieved in patients receiving etelcalcetide at 15 mg three times per week based on AUC.
-
14 CLINICAL STUDIES
The efficacy and safety of PARSABIV for the treatment of secondary hyperparathyroidism in patients with chronic kidney disease receiving hemodialysis three times per week were evaluated in two 26-week, randomized, double-blind, placebo-controlled studies (Study 1 and Study 2).
The starting dose of PARSABIV was 5 mg three times per week administered at the end of hemodialysis. PARSABIV dose was titrated every 4 weeks until week 17 to a maximum dose of 15 mg three times per week to target a PTH level of less than or equal to 300 pg/mL. PARSABIV was suspended temporarily if two consecutive PTH levels were less than 100 pg/mL. The dose of PARSABIV was not increased if PTH levels were less than or equal to 300 pg/mL, corrected serum calcium was less than 8.3 mg/dL, symptomatic hypocalcemia occurred, or the investigator judged that no dose increase was needed.
The average dose of PARSABIV at the time of the efficacy assessment (weeks 20 through 27, inclusive) was 7.2 mg three times per week. Patients with lower screening PTH levels were on lower doses with a mean average dose of 5.7 mg, 7.4 mg, and 8.7 mg three times per week for patients with screening PTH levels less than 600 pg/mL, 600 to less than or equal to 1000 pg/mL, and greater than 1000 pg/mL, respectively. Throughout the study, patients were maintained on a dialysate calcium concentration of greater than or equal to 2.25 meq/L.
In each study, the primary outcome measure was the proportion of patients with a greater than 30% reduction in PTH levels from baseline to the efficacy assessment phase (mean PTH levels for weeks 20 through 27, inclusive). The other outcome measures were the proportion of patients with a mean PTH of less than or equal to 300 pg/mL, percent change from baseline in PTH, corrected serum calcium, and phosphate levels.
Study 1 enrolled 508 patients (254 PARSABIV, 254 placebo). Baseline demographic and disease characteristics were balanced between groups. The mean age of the patients was 58 years, and 57% were male. Of enrolled patients, 69% were White, 28% were Black, 2% were Asian, and 13% were Hispanic/Latino in ethnicity. The mean baseline PTH level was 834.2 pg/mL, the mean baseline corrected serum calcium was 9.6 mg/dL, and the average duration of hemodialysis prior to study entry (minimum to maximum) was 5.5 (0.1 to 32.2) years. Sixty-six percent of patients had a mean screening PTH level greater than or equal to 600 pg/mL, 74% patients were receiving vitamin D sterols, and 84% patients were receiving phosphate binders.
Study 2 enrolled 515 patients (255 PARSABIV, 260 placebo). Baseline demographic and disease characteristics were balanced between groups. The mean age of the patients was 59 years, and 64% were male. Of enrolled patients, 65% were White, 28% were Black, 4% were Asian, and 13% were Hispanic/Latino in ethnicity. The mean baseline PTH level was 848.4 pg/mL, the mean baseline corrected serum calcium was 9.7 mg/dL, and the average duration of hemodialysis prior to study entry (minimum to maximum) was 5.4 (0.3 to 32.1) years. Sixty-seven percent of patients had a mean screening PTH level greater than or equal to 600 pg/mL, 62% patients were receiving vitamin D sterols, and 81% patients were receiving phosphate binders.
In both studies, a significantly higher proportion of patients treated with PARSABIV achieved a greater than 30% reduction in PTH levels from baseline to the efficacy assessment phase (mean PTH levels for weeks 20 through 27, inclusive) than the proportion of patients treated with placebo. In both studies, reduction in mean PTH, corrected serum calcium, and serum phosphate levels from baseline to the end of study were observed in the PARSABIV arm, and differences between PARSABIV and placebo were statistically significant. Results for each individual study are shown in Table 3.
Table 3: Effects of PARSABIV on Parathyroid Hormone, Corrected Serum Calcium, and Serum Phosphate Measured at Weeks 20 to 27 in Two 6-Month, Placebo-Controlled Studies in Chronic Kidney Disease Patients with Secondary Hyperparathyroidism on Hemodialysis Study 1 Study 2 PARSABIV
(N = 254)Placebo
(N = 254)PARSABIV
(N = 255)Placebo
(N = 260)EAP = Efficacy Assessment Phase; weeks 20 through 27, inclusive - *
- Results from a multiple imputation method for missing data using information from patients who discontinued treatment but had final assessment during the EAP. For Study 1, the percentages of patients with no PTH assessment during the EAP were 10% and 14% for PARSABIV and placebo, respectively; for Study 2, they were 11% and 9%, respectively. The corresponding percentages were the same for corrected serum calcium and one to two percentage points higher for serum phosphate.
- †
- Other outcome measures for Studies 1 and 2.
- ‡
- p < 0.001 vs. placebo
- §
- Main outcome measure for Studies 1 and 2.
- ¶
- p = 0.08 vs. placebo
- #
- p < 0.01 vs. placebo
Parathyroid Hormone Baseline (pg/mL): Median 706 706 740 726 Mean (SE) 849 (33) 820 (24) 845 (29) 852 (34) Mean EAP (pg/mL) (SE)* 424 (37) 898 (34) 416 (32) 971 (46) Mean Percent Change, % (SE)†,* -49.4 (3.4)‡ 14.9 (3.6) -47.8 (3.7)‡ 18.6 (3.5) Patients with > 30% Reduction in PTH during the EAP, multiple imputation, n (%)§,* 196 (77)‡ 28 (11) 201 (79)‡ 29 (11) Patients with ≤ 300 pg/mL in PTH during the EAP, n (%)†,* 131 (52)‡ 16 (6) 142 (56)‡ 14 (5) Corrected Serum Calcium Mean Baseline (mg/dL) (SE) 9.7 (0.04) 9.6 (0.04) 9.6 (0.04) 9.7 (0.04) Mean EAP (mg/dL) (SE)* 9.0 (0.1) 9.7 (0.04) 9.0 (0.1) 9.7 (0.05) Mean Percent Change, % (SE)†,* -7.0 (0.6)‡ 0.9 (0.6) -7.0 (0.7)‡ -0.8 (0.6) Serum Phosphate Mean Baseline (mg/dL) (SE) 6.0 (0.1) 5.8 (0.1) 5.8 (0.1) 5.8 (0.1) Mean EAP (mg/dL) (SE)* 5.4 (0.1) 5.5 (0.1) 5.2 (0.1) 5.6 (0.1) Mean Percent Change, % (SE)†,* -8.8 (2.5)¶ -3.6 (2.8) -7.2 (2.5)# -0.3 (2.2) PARSABIV decreased PTH levels regardless of baseline PTH, duration of dialysis, whether or not patients had been previously treated with cinacalcet, and whether or not patients were receiving vitamin D sterols. Reductions in PTH levels, corrected serum calcium, and serum phosphate were maintained for up to 78 weeks of treatment in those patients who elected to participate in the extension phase of both studies.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
PARSABIV (etelcalcetide) injection is supplied in a single-dose vial (type I glass) with stopper (fluoropolymer laminated elastomeric) and an aluminum seal with flip-off dust cover containing 5 mg/mL of etelcalcetide as a ready-to-use clear and colorless solution in the following strengths:
2.5 mg/0.5 mL Carton of 10 single-dose vials NDC 55513-740-10
NDC 55513-740-205 mg/mL Carton of 10 single-dose vials NDC 55513-741-10
NDC 55513-741-2010 mg/2 mL Carton of 10 single-dose vials NDC 55513-742-10
NDC 55513-742-20Storage
Store in the original carton in refrigerator at 2°C to 8°C (36°F to 46°F) to protect from light. Once removed from the refrigerator:
- Do not expose to temperatures above 25°C (77°F).
- Use within 7 days if stored in the original carton.
- Use within 4 hours and do not expose to direct sunlight if removed from the original carton.
-
17 PATIENT COUNSELING INFORMATION
Hypocalcemia
Advise patients to report symptoms of hypocalcemia, including paresthesias, myalgias, muscle spasms, and seizures, to their healthcare provider [see Warnings and Precautions (5.1)].
Heart Failure
Advise patients with heart failure that use of PARSABIV may worsen their heart failure and additional monitoring may be required [see Warnings and Precautions (5.2)].
Upper Gastrointestinal Bleeding
Advise patients to report any symptoms of upper gastrointestinal bleeding to their healthcare provider [see Warnings and Precautions (5.3)].
Laboratory Monitoring
Inform patients of the importance of regular blood tests, in order to monitor the safety and efficacy of PARSABIV therapy.
Lactation
Advise women that use of PARSABIV is not recommended while breastfeeding [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 2.5 mg/0.5 mL Vial Carton
10 x 0.5 mL Single-dose Vials
NDC 55513-740-10AMGEN®
Parsabiv®
(etelcalcetide)
Injection2.5 mg/
0.5 mLFor Intravenous Use after Hemodialysis
Single-dose vial. Discard unused portion
Store at 2°C to 8°C (36°F to 46°F) in the original carton in order to
protect from light.
Sterile, preservative-free solutionRx Only

-
PRINCIPAL DISPLAY PANEL - 5 mg/mL Vial Carton
10 x 1 mL Single-dose Vials
NDC 55513-741-10AMGEN®
Parsabiv®
(etelcalcetide)
Injection5
mg/mLFor Intravenous Use after Hemodialysis
Single-dose vial. Discard unused portion
Store at 2°C to 8°C (36°F to 46°F) in the original carton in order to
protect from light.
Sterile, preservative-free solutionRx Only

-
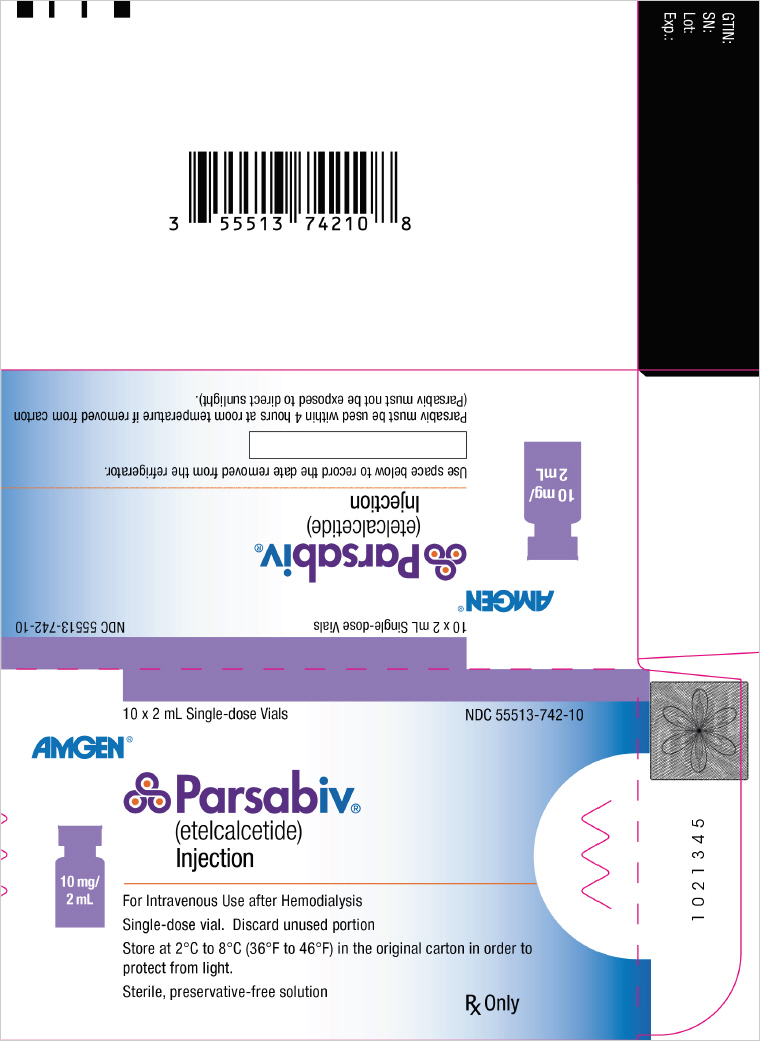
PRINCIPAL DISPLAY PANEL - 10 mg/2 mL Vial Carton
10 x 2 mL Single-dose Vials
NDC 55513-742-10AMGEN®
Parsabiv®
(etelcalcetide)
Injection10 mg/
2 mLFor Intravenous Use after Hemodialysis
Single-dose vial. Discard unused portion
Store at 2°C to 8°C (36°F to 46°F) in the original carton in order to
protect from light.
Sterile, preservative-free solutionRx Only
-
INGREDIENTS AND APPEARANCE
PARSABIV
etelcalcetide injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55513-740 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ETELCALCETIDE HYDROCHLORIDE (UNII: 72PT5993DU) (ETELCALCETIDE - UNII:60ME133FJB) ETELCALCETIDE 2.5 mg in 0.5 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 4.3 mg in 0.5 mL SUCCINIC ACID (UNII: AB6MNQ6J6L) 0.6 mg in 0.5 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55513-740-10 10 in 1 CARTON 04/03/2017 1 NDC:55513-740-01 0.5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:55513-740-20 10 in 1 CARTON 08/01/2025 2 NDC:55513-740-01 0.5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208325 04/03/2017 PARSABIV
etelcalcetide injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55513-741 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ETELCALCETIDE HYDROCHLORIDE (UNII: 72PT5993DU) (ETELCALCETIDE - UNII:60ME133FJB) ETELCALCETIDE 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 8.5 mg in 1 mL SUCCINIC ACID (UNII: AB6MNQ6J6L) 1.2 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55513-741-10 10 in 1 CARTON 04/03/2017 1 NDC:55513-741-01 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:55513-741-20 10 in 1 CARTON 08/01/2025 2 NDC:55513-741-01 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208325 04/03/2017 PARSABIV
etelcalcetide injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55513-742 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ETELCALCETIDE HYDROCHLORIDE (UNII: 72PT5993DU) (ETELCALCETIDE - UNII:60ME133FJB) ETELCALCETIDE 10 mg in 2 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 17 mg in 2 mL SUCCINIC ACID (UNII: AB6MNQ6J6L) 2.4 mg in 2 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55513-742-10 10 in 1 CARTON 04/03/2017 1 NDC:55513-742-01 2 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:55513-742-20 10 in 1 CARTON 08/01/2025 2 NDC:55513-742-01 2 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208325 04/03/2017 Labeler - Amgen Inc (039976196)