Label: BESPONSA- inotuzumab ozogamicin injection, powder, lyophilized, for solution


- NDC Code(s): 0008-0100-01
- Packager: Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated March 14, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BESPONSA™ safely and effectively. See full prescribing information for BESPONSA.
BESPONSA (inotuzumab ozogamicin) for injection, for intravenous use
Initial U.S. Approval: 2017WARNING: HEPATOTOXICITY, INCLUDING HEPATIC VENO-OCCLUSIVE DISEASE (VOD) (ALSO KNOWN AS SINUSOIDAL OBSTRUCTION SYNDROME) and INCREASED RISK OF POST- HEMATOPOIETIC STEM CELL TRANSPLANT (HSCT) NON-RELAPSE MORTALITY
See full prescribing information for complete boxed warning.
RECENT MAJOR CHANGES
Indications and Usage (1)
3/2024
Dosage and Administration, Instructions for Reconstitution, Dilution and Administration (2.4)
3/2024
Warnings and Precautions, Hepatotoxicity, Including Hepatic Veno-occlusive Disease (VOD) (also known as Sinusoidal Obstruction Syndrome) (5.1)
3/2024
Warnings and Precautions, Increased Risk of Post-Transplant Non-Relapse Mortality (5.2)
3/2024
Warnings and Precautions, Myelosuppression (5.3)
3/2024
Warnings and Precautions, Infusion Related Reactions (5.4)
3/2024
Warnings and Precautions, QT Interval Prolongation (5.5)
3/2024
INDICATIONS AND USAGE
BESPONSA is a CD22-directed antibody and cytotoxic drug conjugate indicated for the treatment of relapsed or refractory CD22-positive B-cell precursor acute lymphoblastic leukemia (ALL) in adult and pediatric patients 1 year and older. (1)
DOSAGE AND ADMINISTRATION
- •
- Administer by intravenous infusion only. (2.1)
- •
- Pre-medicate with a corticosteroid, antipyretic, and antihistamine prior to all infusions. (2.2)
- •
- Dosing regimens for Cycle 1 and subsequent cycles, depending on the response to treatment, are shown below. See full prescribing information for dosing details. (2)
- *
- For patients who achieve a CR or a CRi, and/or to allow for recovery from toxicity, the cycle length may be extended up to 28 days (i.e., 7-day treatment-free interval starting on Day 21).
Day 1
Day 8
Day 15
Dosing regimen for Cycle 1
All patients:
Dose
0.8 mg/m2
0.5 mg/m2
0.5 mg/m2
Cycle length
21 days*
Dosing regimen for subsequent cycles depending on response to treatment
Patients who have achieved a CR or CRi:
Dose
0.5 mg/m2
0.5 mg/m2
0.5 mg/m2
Cycle length
28 days
Patients who have not achieved a CR or CRi:
Dose
0.8 mg/m2
0.5 mg/m2
0.5 mg/m2
Cycle length
28 days
- •
- See full prescribing information for instructions on reconstitution of lyophilized powder, and preparation and administration of reconstituted drug. (2.4)
DOSAGE FORMS AND STRENGTHS
For injection: 0.9 mg lyophilized powder in a single-dose vial for reconstitution and further dilution. (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- •
- Myelosuppression: Monitor complete blood counts; for signs and symptoms of infection; bleeding/hemorrhage; or other effects of myelosuppression during treatment; manage appropriately. (5.3)
- •
- Infusion related reactions: Monitor for infusion related reactions during and for at least 1 hour after infusion ends. (5.4)
- •
- QT interval prolongation: Obtain electrocardiograms (ECGs) and electrolytes at baseline and monitor during treatment. Monitor more frequently when using concomitant mediations known to prolong QT interval. (5.5)
- •
- Embryo-fetal toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.6, 8.1, 8.3)
ADVERSE REACTIONS
The most common (≥ 20%) adverse reactions, including laboratory abnormalities, in adult and pediatric patients are thrombocytopenia, pyrexia, neutropenia, infection, anemia, vomiting, leukopenia, hemorrhage, fatigue, nausea, febrile neutropenia, headache, transaminases increased, abdominal pain, and gamma-glutamyltransferase increased, and hyperbilirubinemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HEPATOTOXICITY, INCLUDING HEPATIC VENO-OCCLUSIVE DISEASE (VOD) (ALSO KNOWN AS SINUSOIDAL OBSTRUCTION SYNDROME) and INCREASED RISK OF POST-HEMATOPOIETIC STEM CELL TRANSPLANT (HSCT) NON-RELAPSE MORTALITY
1. INDICATIONS AND USAGE
2. DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Recommended Pre-medications and Cytoreduction
2.3 Dosage Modifications for Adverse Reactions
2.4 Instructions for Reconstitution, Dilution, and Administration
3. DOSAGE FORMS AND STRENGTHS
4. CONTRAINDICATIONS
5. WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity, Including Hepatic Veno-occlusive Disease (VOD) (also known as Sinusoidal Obstruction Syndrome)
5.2 Increased Risk of Post-Transplant Non-Relapse Mortality
5.3 Myelosuppression
5.4 Infusion Related Reactions
5.5 QT Interval Prolongation
5.6 Embryo-Fetal Toxicity
6. ADVERSE REACTIONS
6.1 Clinical Trials Experience
7. DRUG INTERACTIONS
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
11. DESCRIPTION
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14. CLINICAL STUDIES
15. REFERENCES
16. HOW SUPPLIED/STORAGE AND HANDLING
17. PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HEPATOTOXICITY, INCLUDING HEPATIC VENO-OCCLUSIVE DISEASE (VOD) (ALSO KNOWN AS SINUSOIDAL OBSTRUCTION SYNDROME) and INCREASED RISK OF POST-HEMATOPOIETIC STEM CELL TRANSPLANT (HSCT) NON-RELAPSE MORTALITY
HEPATOTOXICITY, INCLUDING VOD
- •
- Hepatotoxicity, including fatal and life-threatening VOD occurred in patients with relapsed or refractory acute lymphoblastic leukemia (ALL) who received BESPONSA. The risk of VOD was greater in patients who underwent HSCT after BESPONSA treatment; use of HSCT conditioning regimens containing 2 alkylating agents and last total bilirubin level ≥ upper limit of normal (ULN) before HSCT were significantly associated with an increased risk of VOD.
- •
- Other risk factors for VOD in patients treated with BESPONSA included ongoing or prior liver disease, prior HSCT, increased age, later salvage lines, and a greater number of BESPONSA treatment cycles.
- •
- Elevation of liver tests may require dosing interruption, dose reduction, or permanent discontinuation of BESPONSA. Permanently discontinue treatment if VOD occurs. If severe VOD occurs, treat according to standard medical practice [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
INCREASED RISK OF POST-HSCT NON-RELAPSE MORTALITY
- •
- There was higher post-HSCT non-relapse mortality rate in patients receiving BESPONSA, resulting in a higher Day 100 post-HSCT mortality rate [see Warnings and Precautions (5.2)].
- 1. INDICATIONS AND USAGE
-
2. DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
- •
- Pre-medicate before each dose [see Dosage and Administration (2.2)].
- •
- Administer by intravenous infusion only.
- •
- For the first cycle, the recommended total dose of BESPONSA for all patients is 1.8 mg/m2 per cycle, administered as 3 divided doses on Day 1 (0.8 mg/m2), Day 8 (0.5 mg/m2), and Day 15 (0.5 mg/m2). Cycle 1 is 3 weeks in duration, but may be extended to 4 weeks if the patient achieves a complete remission (CR) or complete remission with incomplete hematologic recovery (CRi), and/or to allow recovery from toxicity.
- •
- For subsequent cycles:
- •
- In patients who achieve a CR or CRi, the recommended total dose of BESPONSA is 1.5 mg/m2 per cycle, administered as 3 divided doses on Day 1 (0.5 mg/m2), Day 8 (0.5 mg/m2), and Day 15 (0.5 mg/m2). Subsequent cycles are 4 weeks in duration.
OR - •
- In patients who do not achieve a CR or CRi, the recommended total dose of BESPONSA is 1.8 mg/m2 per cycle given as 3 divided doses on Day 1 (0.8 mg/m2), Day 8 (0.5 mg/m2), and Day 15 (0.5 mg/m2). Subsequent cycles are 4 weeks in duration. Patients who do not achieve a CR or CRi within 3 cycles should discontinue treatment.
- •
- For patients proceeding to hematopoietic stem cell transplant (HSCT), the recommended duration of treatment with BESPONSA is 2 cycles. A third cycle may be considered for those patients who do not achieve CR or CRi and minimal residual disease (MRD) negativity after 2 cycles [see Warnings and Precautions (5.1)].
- •
- For patients not proceeding to HSCT, additional cycles of treatment, up to a maximum of 6 cycles, may be administered.
Table 1 shows the recommended dosing regimens.
Table 1. Dosing Regimen for Cycle 1 and Subsequent Cycles Depending on Response to Treatment Abbreviations: CR=complete remission; CRi=complete remission with incomplete hematologic recovery. - *
- +/- 2 days (maintain minimum of 6 days between doses).
- †
- Dose is based on the patient's body surface area (m2).
- ‡
- For patients who achieve a CR or a CRi, and/or to allow for recovery from toxicity, the cycle length may be extended up to 28 days (i.e., 7-day treatment-free interval starting on Day 21).
- §
- CR is defined as < 5% blasts in the bone marrow and the absence of peripheral blood leukemic blasts, full recovery of peripheral blood counts (platelets ≥ 100 × 109/L and absolute neutrophil counts [ANC] ≥ 1 × 109/L) and resolution of any extramedullary disease.
- ¶
- CRi is defined as < 5% blasts in the bone marrow and the absence of peripheral blood leukemic blasts, incomplete recovery of peripheral blood counts (platelets < 100 × 109/L and/or ANC < 1 × 109/L) and resolution of any extramedullary disease.
- #
- 7-day treatment-free interval starting on Day 21.
Day 1
Day 8*
Day 15*
Dosing regimen for Cycle 1
All patients:
Dose†
0.8 mg/m2
0.5 mg/m2
0.5 mg/m2
Cycle length
21 days‡
Dosing regimen for subsequent cycles depending on response to treatment
Dose†
0.5 mg/m2
0.5 mg/m2
0.5 mg/m2
Cycle length
28 days#
Dose†
0.8 mg/m2
0.5 mg/m2
0.5 mg/m2
Cycle length
28 days#
2.2 Recommended Pre-medications and Cytoreduction
- •
- Premedication with a corticosteroid, antipyretic, and antihistamine is recommended prior to dosing. Patients should be observed during and for at least 1 hour after the end of infusion for symptoms of infusion related reactions [see Warnings and Precautions (5.4)].
- •
- For patients with circulating lymphoblasts, cytoreduction with a combination of hydroxyurea, steroids, and/or vincristine to a peripheral blast count of less than or equal to 10,000/mm3 is recommended prior to the first dose.
2.3 Dosage Modifications for Adverse Reactions
Modify the dose of BESPONSA for toxicities (see Tables 2–4). BESPONSA doses within a treatment cycle (i.e., Days 8 and/or 15) do not need to be interrupted due to neutropenia or thrombocytopenia, but dosing interruptions within a cycle are recommended for non-hematologic toxicities. If the dose is reduced due to BESPONSA-related toxicity, the dose must not be re-escalated.
Table 2. BESPONSA Dosage Modifications for Hematologic Toxicities [see Warnings and Precautions (5.3)]
Criteria BESPONSA Dosage Modification(s) Abbreviation: ANC=absolute neutrophil count. - *
- Platelet count used for dosing should be independent of blood transfusion.
If prior to BESPONSA treatment ANC was greater than or equal to 1 × 109/L
If ANC decreases, then interrupt the next cycle of treatment until recovery of ANC to greater than or equal to 1 × 109/L. Discontinue BESPONSA if low ANC persists for greater than 28 days and is suspected to be related to BESPONSA.
If prior to BESPONSA treatment platelet count was greater than or equal to 50 × 109/L*
If platelet count decreases, then interrupt the next cycle of treatment until platelet count recovers to greater than or equal to 50 × 109/L*. Discontinue BESPONSA if low platelet count persists for greater than 28 days and is suspected to be related to BESPONSA.
If prior to BESPONSA treatment ANC was less than 1 × 109/L and/or platelet count was less than 50 × 109/L*
If ANC or platelet count decreases, then interrupt the next cycle of treatment until at least one of the following occurs:
- -
- ANC and platelet counts recover to at least baseline levels for the prior cycle, or
- -
- ANC recovers to greater than or equal to 1 × 109/L and platelet count recovers to greater than or equal to 50 × 109/L*, or
- -
- Stable or improved disease (based on most recent bone marrow assessment) and the ANC and platelet count decrease is considered to be due to the underlying disease (not considered to be BESPONSA-related toxicity).
Table 3. BESPONSA Dosage Modifications for Non-hematologic Toxicities Non-hematologic Toxicity Dosage Modification(s) Abbreviations: ALT=alanine aminotransferase; AST=aspartate aminotransferase; ULN=upper limit of normal; VOD=veno‑occlusive disease. - *
- Severity grade according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 3.0.
VOD or other severe liver toxicity
Permanently discontinue treatment [see Warnings and Precautions (5.1)].
Total bilirubin greater than 1.5 × ULN and AST/ALT greater than 2.5 × ULN
Interrupt dosing until recovery of total bilirubin to less than or equal to 1.5 × ULN and AST/ALT to less than or equal to 2.5 × ULN prior to each dose unless due to Gilbert's syndrome or hemolysis. Permanently discontinue treatment if total bilirubin does not recover to less than or equal to 1.5 × ULN or AST/ALT does not recover to less than or equal to 2.5 × ULN [see Warnings and Precautions (5.1)].
Infusion related reaction
Interrupt the infusion and institute appropriate medical management. Depending on the severity of the infusion related reaction, consider discontinuation of the infusion or administration of steroids and antihistamines. For severe or life-threatening infusion reactions, permanently discontinue treatment [see Warnings and Precautions (5.4)].
Non-hematologic toxicity greater than or equal to Grade 2*
Interrupt treatment until recovery to Grade 1 or pre-treatment grade levels prior to each dose.
Table 4. BESPONSA Dosage Modifications Depending on Duration of Dosing Interruption Due to Non-Hematologic Toxicity Toxicities Duration of Dose Interruption Due to Toxicity Dosage Modification(s) Less than 7 days (within a cycle)
Interrupt the next dose (maintain a minimum of 6 days between doses).
Greater than or equal to 7 days
Omit the next dose within the cycle.
Greater than or equal to 14 days
Once adequate recovery is achieved, decrease the total dose by 25% for the subsequent cycle. If further dose modification is required, then reduce the number of doses to 2 per cycle for subsequent cycles. If a 25% decrease in the total dose followed by a decrease to 2 doses per cycle is not tolerated, then permanently discontinue treatment.
Greater than 28 days
Consider permanent discontinuation of treatment.
2.4 Instructions for Reconstitution, Dilution, and Administration
Protect the reconstituted and diluted BESPONSA solutions from light. Do not freeze the reconstituted or diluted solution.
The maximum time from reconstitution through the end of administration should be less than or equal to 8 hours, with less than or equal to 4 hours between reconstitution and dilution.
Reconstitution:
- •
- BESPONSA is a hazardous drug. Follow applicable special handling and disposal procedures.1
- •
- Calculate the dose (mg) and number of vial(s) of BESPONSA required.
- •
- Reconstitute each vial with 4 mL of Sterile Water for Injection, USP, to obtain a concentration of 0.25 mg/mL of BESPONSA that delivers 3.6 mL (0.9 mg).
- •
- Gently swirl the vial to aid dissolution. DO NOT SHAKE.
- •
- Inspect the reconstituted solution for particulates and discoloration. The reconstituted solution should be clear to opalescent, colorless to slightly yellow, and essentially free of visible foreign matter.
- •
- See Table 6 for storage times and conditions for the reconstituted solution.
Dilution:
- •
- Withdraw the required volume of the reconstituted solution from the vial(s) needed to obtain the appropriate dose according to the patient’s body surface area. Discard any unused reconstituted BESPONSA solution left in the vial.
- •
- Dilute the reconstituted BESPONSA solution in 0.9% Sodium Chloride Injection, USP, in the appropriate infusion container per Table 5:
Table 5. Infusion Container Information Infusion Bag Administration
Syringe Administration
- •
- For calculated doses greater than or equal to 0.5 mg
- •
- Ensure a final prepared concentration of 0.01 mg/mL to 0.1 mg/mL in a total volume of 50 mL
- •
- For calculated doses less than 0.5 mg
- •
- Ensure a final prepared concentration of 0.025 mg/mL to 0.1 mg/mL in a total volume between 2 mL to 50 mL
- •
- Gently invert the infusion container to mix the diluted solution. DO NOT SHAKE.
- •
- PROTECT FROM LIGHT.
- •
- See Table 6 for storage times and conditions for the diluted solution.
Administration:
- •
- See Table 6 for storage times and conditions for prior to and during administration of the diluted solution.
- •
- For syringe infusions, a syringe pump and micro-bore IV tubing must be used.
- •
- Filtration of the diluted solution is not required. However, if the diluted solution is filtered, polyethersulfone (PES)-, polyvinylidene fluoride (PVDF),- or hydrophilic polysulfone (HPS) -based filters are recommended. Do not use filters made of nylon or mixed cellulose ester (MCE).
- •
- Infuse the diluted solution as an intravenous infusion over one hour. Flush the intravenous infusion line with 0.9% Sodium Chloride Injection, USP, to ensure the complete dose is administered.
Do not mix BESPONSA or administer as an infusion with other medicinal products.
Table 6 shows the storage times and conditions for reconstitution, dilution, and administration of BESPONSA.
Table 6. Storage Times and Conditions for Reconstituted and Diluted BESPONSA Solution - *
- Maximum time from reconstitution through end of administration less than or equal to 8 hours with less than or equal to 4 hours between reconstitution and dilution.
Storage Time and Conditions*
Reconstituted Solution
- •
- BESPONSA contains no bacteriostatic preservatives. Use reconstituted solution immediately or store refrigerated at (2°C-8°C; 36°F-46°F) for up to 4 hours.
- •
- PROTECT FROM LIGHT. DO NOT FREEZE.
Diluted Solution
- •
- Use diluted solution immediately or store at room temperature (20°C-25°C; 68°F-77°F) or refrigerated (2°C-8°C; 36°F-46°F) for up to 6 hours.
- •
- If the diluted solution is refrigerated (2°C-8°C; 36°F-46°F), allow it to equilibrate at room temperature (20°C-25°C; 68°F-77°F) for approximately 1 hour prior to administration.
- •
- Administer diluted solution within 8 hours of reconstitution including the 1 hour equilibration and 1 hour infusion.
- •
- PROTECT FROM LIGHT. DO NOT FREEZE.
- 3. DOSAGE FORMS AND STRENGTHS
- 4. CONTRAINDICATIONS
-
5. WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity, Including Hepatic Veno-occlusive Disease (VOD) (also known as Sinusoidal Obstruction Syndrome)
BESPONSA can cause hepatotoxicity, including VOD. In adult patients in the INO-VATE ALL trial, hepatotoxicity, including severe, life-threatening, and sometimes fatal hepatic VOD occurred in 23/164 patients (14%) in the BESPONSA arm during or following treatment or following a HSCT after completion of treatment. VOD occurred up to 56 days after the final dose during treatment or during follow-up without an intervening HSCT. The median time from subsequent HSCT to onset of VOD was 15 days (range: 3-57 days).
In the BESPONSA arm, among the 79 patients who proceeded to a subsequent HSCT, VOD occurred in 18/79 patients (23%), and among all 164 patients treated, VOD occurred in 5/164 patients (3%) during study therapy or in follow-up without an intervening HSCT.
The risk of VOD was greater in patients who underwent HSCT after BESPONSA treatment; use of HSCT conditioning regimens containing 2 alkylating agents (e.g., busulfan in combination with other alkylating agents) and last total bilirubin level greater than or equal to the ULN before HSCT are significantly associated with an increased risk of VOD. Other risk factors for VOD in patients treated with BESPONSA included ongoing or prior liver disease, prior HSCT, increased age, later salvage lines, and a greater number of BESPONSA treatment cycles. Patients who have experienced prior VOD or have serious ongoing hepatic liver disease (e.g., cirrhosis, nodular regenerative hyperplasia, active hepatitis) are at an increased risk for worsening of liver disease, including developing VOD, following treatment with BESPONSA.
In Study WI203581 (ITCC-059) VOD occurred in 8/53 (15%) of pediatric patients treated with single agent BESPONSA. Among the 26 pediatric patients who underwent HSCT, VOD occurred in 5 (19%) patients [see Adverse Reactions (6.1)].
Monitor closely for signs and symptoms of VOD including elevations in total bilirubin, hepatomegaly (which may be painful), rapid weight gain, and ascites. Due to the risk of VOD, for patients proceeding to HSCT, the recommended duration of treatment with BESPONSA is 2 cycles; a third cycle may be considered for those patients who do not achieve a CR or CRi and MRD negativity after 2 cycles [see Dosage and Administration (2.1)]. For patients who proceed to HSCT, monitor liver tests at least weekly during the first month post-HSCT, then less frequently thereafter, according to standard medical practice.
In adult patients in the INO-VATE ALL trial, increases in liver test abnormalities occurred. Grade 3 or 4 AST, ALT, and total bilirubin abnormal liver tests occurred in 7/160 (4%), 7/161 (4%), and 8/161 patients (5%), respectively.
In pediatric patients in Study WI203581 (ITCC-059), liver test abnormalities occurred, with Grade 3 or 4 increases in AST, ALT, and blood bilirubin in 11/53 (21%), 11/53 (21%), and 5/53 (9%) of patients, respectively [see Adverse Reactions (6.1)].
In all patients, monitor liver tests, including ALT, AST, total bilirubin, and alkaline phosphatase, prior to and following each dose of BESPONSA. Based on elevations of liver tests withhold, reduce dose, or permanently discontinue BESPONSA [see Dosage and Administration (2.3)].
5.2 Increased Risk of Post-Transplant Non-Relapse Mortality
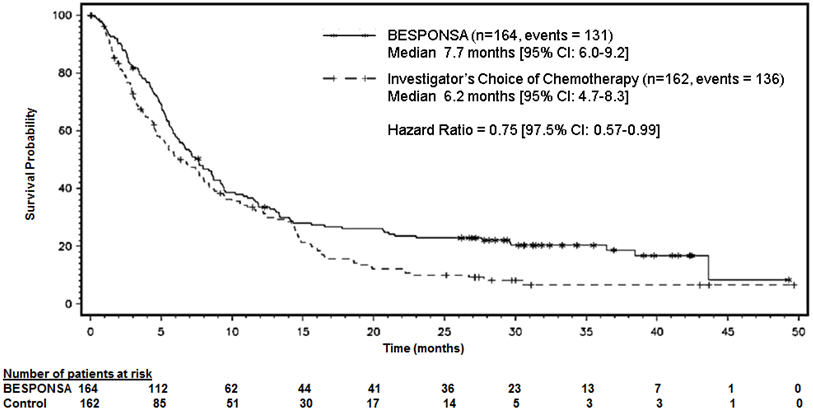
In adult patients in the INO-VATE ALL trial, a higher post-HSCT non-relapse mortality rate was observed in patients receiving BESPONSA compared to the Investigator's choice of chemotherapy arm, resulting in a higher Day 100 post-HSCT mortality rate.
Overall, 79/164 patients (48%) in the BESPONSA arm and 35/162 patients (22%) in the Investigator's choice of chemotherapy arm had a follow-up HSCT. The post-HSCT non-relapse mortality rate was 31/79 (39%) and 8/35 (23%) in the BESPONSA arm compared to the Investigator's choice of chemotherapy arm, respectively.
In the BESPONSA arm, the most common causes of post-HSCT non-relapse mortality included VOD and infections. Five of the 18 VOD events that occurred post-HSCT were fatal. In the BESPONSA arm, among patients with ongoing VOD at time of death, 6 patients died due to multiorgan failure (MOF) or infection (3 patients died due to MOF, 2 patients died due to infection, and 1 patient died due to MOF and infection).
In pediatric patients in Study WI203581 (ITCC-059), 26/53 patients (49%) had a follow-up HSCT. The post-HSCT non-relapse mortality rate was 7/26 (27%).
Monitor for toxicities post-HSCT, including signs and symptoms of infection and VOD [see Warnings and Precautions (5.1, 5.3)].
5.3 Myelosuppression
BESPONSA can cause myelosuppression, including thrombocytopenia and neutropenia [see Adverse Reactions (6.1)].
In adult patients in the INO-VATE ALL trial, thrombocytopenia and neutropenia occurred in 83/164 patients (51%) and 81/164 patients (49%), respectively. Grade 3 thrombocytopenia and neutropenia occurred in 23/164 patients (14%) and 33/164 patients (20%), respectively. Grade 4 thrombocytopenia and neutropenia occurred in 46/164 patients (28%) and 45/164 patients (27%), respectively. Febrile neutropenia, which may be life-threatening, occurred in 43/164 patients (26%). For patients who were in CR or CRi at the end of treatment, the recovery of platelet counts to > 50,000/mm3 was later than 45 days after the final dose in 15/164 patients (9%) who received BESPONSA and 3/162 patients (2%) who received Investigator's choice of chemotherapy.
Complications associated with myelosuppression (including infections and bleeding/hemorrhage) occurred in patients receiving BESPONSA [see Adverse Reactions (6.1)]. Infections, including serious infections, some of which were life-threatening or fatal, occurred in 79/164 patients (48%). Fatal infections, including pneumonia, neutropenic sepsis, sepsis, septic shock, and pseudomonal sepsis, occurred in 8/164 patients (5%). Bacterial, viral, and fungal infections occurred.
Hemorrhage occurred in 54/164 patients (33%). Grade 3 or 4 hemorrhage occurred in 8/164 patients (5%), including a fatality in 1/164 patients (1%) (intra-abdominal hemorrhage). The most common type of hemorrhage was epistaxis which occurred in 24/164 patients (15%).
In pediatric patients in Study WI203581 (ITCC-059), Grade 3 or 4 thrombocytopenia occurred in 24/53 (45%) patients. Grade 3 or 4 neutropenia occurred in 21/53 (40%) patients. Infections occurred in 23/53 (43%) patients, and hemorrhage occurred in 22/53 (42%) patients. The most common types of hemorrhage were hematoma in 8/53 (15%), mouth hemorrhage in 6/53 (11%), and epistaxis in 6/53 (11%) patients [see Adverse Reactions (6.1)].
Monitor complete blood counts prior to each dose of BESPONSA and monitor for signs and symptoms of infection, bleeding/hemorrhage, or other effects of myelosuppression during treatment with BESPONSA. As appropriate, administer prophylactic anti-infectives and employ surveillance testing during and after treatment with BESPONSA. Based on the severity of myelosuppression, reduce dose, temporarily withhold, or permanently discontinue BESPONSA [see Dosage and Administration (2.3)].
5.4 Infusion Related Reactions
BESPONSA can cause infusion related reactions. In adult patients in the INO-VATE ALL trial, infusion related reactions occurred in patients who received BESPONSA. Infusion related reactions (all Grade 2) occurred in 4/164 patients (2%) [see Adverse Reactions (6.1)]. Infusion related reactions generally occurred in Cycle 1 shortly after the end of the BESPONSA infusion and resolved spontaneously or with medical management.
In pediatric patients in Study WI203581 (ITCC-059), infusion related reactions occurred in 4/53 (8%) patients [see Adverse Reactions (6.1)].
Premedicate with a corticosteroid, antipyretic, and antihistamine prior to dosing [see Dosage and Administration (2.2)].
Monitor patients closely during and for at least 1 hour after the end of infusion for the potential onset of infusion related reactions, including symptoms such as fever, chills, rash, or breathing problems. Interrupt infusion and institute appropriate medical management if an infusion related reaction occurs. Depending on the severity of the infusion related reaction, consider discontinuation of the infusion or administration of steroids and antihistamines. For severe or life-threatening infusion reactions, permanently discontinue BESPONSA [see Dosage and Administration (2.3)].
5.5 QT Interval Prolongation
BESPONSA can cause QT interval prolongation. In adult patients in the INO-VATE ALL trial, increases in QT interval corrected for heart rate using Fridericia’s formula (QTcF) of ≥ to 60 msec from baseline occurred in 4/162 patients (3%). Grade 2 QT prolongation was reported in 2/164 patients (1%) [see Adverse Reactions (6.1) and Clinical Pharmacology (12.2)].
In pediatric patients in Study WI203581 (ITCC-059), increases in QTcF of > 60 msec from baseline occurred in 7/49 (14%) patients. 3/52 (6%) of patients had QTcF values > 500 msec [see Adverse Reactions (6.1)].
Administer BESPONSA with caution in patients who have a history of or predisposition for QTc prolongation, who are taking medicinal products that are known to prolong QT interval [see Drug Interactions (7)], and in patients with electrolyte disturbances [see Drug Interactions (7)]. Obtain electrocardiograms (ECGs) and electrolytes prior to the start of treatment, after initiation of any drug known to prolong QTc, and periodically monitor as clinically indicated during treatment [see Drug Interactions (7), Clinical Pharmacology (12.2)]).
5.6 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, BESPONSA can cause embryo-fetal harm when administered to a pregnant woman. In animal studies, inotuzumab ozogamicin caused embryo-fetal toxicities, starting at a dose that was approximately 0.4 times the exposure in patients at the maximum recommended dose, based on the area under the concentration-time curve (AUC). Advise females of reproductive potential to use effective contraception during treatment with BESPONSA and for 8 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with BESPONSA and for 5 months after the last dose. Advise pregnant women of the potential risk to the fetus. Advise women to contact their healthcare provider if they become pregnant or if pregnancy is suspected during treatment with BESPONSA [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)].
-
6. ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- •
- Hepatotoxicity, including hepatic VOD (also known as SOS) [see Warnings and Precautions (5.1)]
- •
- Increased risk of post-transplant non-relapse mortality [see Warnings and Precautions (5.2)]
- •
- Myelosuppression [see Warnings and Precautions (5.3)]
- •
- Infusion related reactions [see Warnings and Precautions (5.4)]
- •
- QT interval prolongation [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Relapsed or Refractory B-cell Precursor ALL
Adult Patients
The safety of BESPONSA was evaluated in adult patients with relapsed or refractory B-cell precursor ALL in the INO-VATE ALL trial. The study was a randomized clinical study of BESPONSA (n=164) versus Investigator’s choice of chemotherapy (fludarabine + cytarabine + granulocyte colony-stimulating factor [FLAG], mitoxantrone + cytarabine [MXN/Ara-C], or high dose cytarabine [HIDAC]) (n=143) [see Clinical Studies (14)].
Of the 164 patients who received BESPONSA, the median age was 47 years (range: 18–78 years), 56% were male, 68% had received 1 prior treatment regimen for ALL, 31% had received 2 prior treatment regimens for ALL, 68% were White, 19% were Asian, and 2% were Black.
In patients who received BESPONSA, the median duration of treatment was 8.9 weeks (range: 0.1–26.4 weeks), with a median of 3 treatment cycles started in each patient. In patients who received Investigator's choice of chemotherapy, the median duration of treatment was 0.9 weeks (range: 0.1–15.6 weeks), with a median of 1 treatment cycle started in each patient.
In patients who received BESPONSA, the most common (≥ 20%) adverse reactions were thrombocytopenia, neutropenia, infection, anemia, leukopenia, fatigue, hemorrhage, pyrexia, nausea, headache, febrile neutropenia, transaminases increased, abdominal pain, gamma-glutamyltransferase increased, and hyperbilirubinemia.
In patients who received BESPONSA, the most common (≥ 2%) serious adverse reactions were infection, febrile neutropenia, hemorrhage, abdominal pain, pyrexia, VOD, and fatigue.
In patients who received BESPONSA, the most common (≥ 2%) adverse reactions reported as the reason for permanent discontinuation were infection (6%), thrombocytopenia (2%), hyperbilirubinemia (2%), transaminases increased (2%), and hemorrhage (2%); the most common (≥ 5%) adverse reactions reported as the reason for dosing interruption were neutropenia (17%), infection (10%), thrombocytopenia (10%), transaminases increased (6%), and febrile neutropenia (5%); and the most common (≥ 1%) adverse reactions reported as the reason for dose reduction were neutropenia (1%), thrombocytopenia (1%), and transaminases increased (1%).
VOD was reported in 23/164 patients (14%) who received BESPONSA during or following treatment or following a HSCT after completion of treatment [see Warnings and Precautions (5.1)].
Table 7 shows the adverse reactions with ≥ 10% incidence reported in patients with relapsed or refractory ALL who received BESPONSA or Investigator's choice of chemotherapy.
Table 7. Adverse Reactions With ≥ 10% Incidence* in Adult Patients With Relapsed or Refractory B-Cell Precursor ALL Who Received BESPONSA or Investigator's Choice of Chemotherapy (FLAG, MXN/Ara-C, or HIDAC) Body System
Adverse ReactionBESPONSA
(N=164)FLAG, MXN/Ara-C, or HIDAC
(N=143†)All Grades ≥ Grade 3 All Grades ≥ Grade 3 % % % % Adverse reactions included treatment-emergent all-causality events that commenced on or after Cycle 1 Day 1 within 42 days after the final dose of BESPONSA, but prior to the start of a new anticancer treatment (including HSCT).
Preferred terms were retrieved by applying the Medical Dictionary for Regulatory Activities (MedDRA) version 18.1.
Severity grade of adverse reactions were according to NCI CTCAE version 3.0.
Abbreviations: ALL=acute lymphoblastic leukemia; FLAG=fludarabine + cytarabine + granulocyte colony-stimulating factor; HIDAC=high dose cytarabine; HSCT=hematopoietic stem cell transplant; MXN/Ara-C=mitoxantrone + cytarabine; N=number of patients; NCI CTCAE=National Cancer Institute Common Toxicity Criteria for Adverse Events.- *
- Only adverse reactions with ≥ 10% incidence in the BESPONSA arm are included.
- †
- 19 patients randomized to FLAG, MXN/Ara-C, or HIDAC did not receive treatment.
- ‡
- Infection includes any reported preferred terms for BESPONSA retrieved in the System Organ Class Infections and infestations.
- §
- Thrombocytopenia includes the following reported preferred terms: Platelet count decreased and Thrombocytopenia.
- ¶
- Neutropenia includes the following reported preferred terms: Neutropenia and Neutrophil count decreased.
- #
- Anemia includes the following reported preferred terms: Anemia and Hemoglobin decreased.
- Þ
- Leukopenia includes the following reported preferred terms: Leukopenia, Monocytopenia, and White blood cell count decreased.
- ß
- Lymphopenia includes the following reported preferred terms: B-lymphocyte count decreased, Lymphocyte count decreased, and Lymphopenia.
- à
- Headache includes the following reported preferred terms: Headache, Migraine, and Sinus headache.
- è
- Hemorrhage includes reported preferred terms for BESPONSA retrieved in the Standard MedDRA Query (narrow) for Hemorrhage terms (excluding laboratory terms), resulting in the following preferred terms: Conjunctival hemorrhage, Contusion, Ecchymosis, Epistaxis, Eyelid bleeding, Gastrointestinal hemorrhage, Gastritis hemorrhagic, Gingival bleeding, Hematemesis, Hematochezia, Hematotympanum, Hematuria, Hemorrhage intracranial, Hemorrhage subcutaneous, Hemorrhoidal hemorrhage, Intra-abdominal hemorrhage, Lip hemorrhage, Lower gastrointestinal hemorrhage, Mesenteric hemorrhage, Metrorrhagia, Mouth hemorrhage, Muscle hemorrhage, Oral mucosa hematoma, Petechiae, Post-procedural hematoma, Rectal hemorrhage, Shock hemorrhagic, Subcutaneous hematoma, Subdural hematoma, Upper gastrointestinal hemorrhage, and Vaginal hemorrhage.
- ð
- Abdominal pain includes the following reported preferred terms: Abdominal pain, Abdominal pain lower, Abdominal pain upper, Abdominal tenderness, Esophageal pain, and Hepatic pain.
- ø
- Stomatitis includes the following reported preferred terms: Aphthous ulcer, Mucosal inflammation, Mouth ulceration, Oral pain, Oropharyngeal pain, and Stomatitis.
- ý
- Fatigue includes the following reported preferred terms: Asthenia and Fatigue.
- £
- Transaminases increased includes the following reported preferred terms: Aspartate aminotransferase increased, Alanine aminotransferase increased, Hepatocellular injury, and Hypertransaminasemia.
Infections
Infection‡
48
28
76
54
Blood and lymphatic system disorders
Thrombocytopenia§
51
42
61
59
Neutropenia¶
49
48
45
43
Anemia#
36
24
59
47
LeukopeniaÞ
35
33
43
42
Febrile neutropenia
26
26
53
53
Lymphopeniaß
18
16
27
26
Metabolism and nutrition disorders
Decreased appetite
12
1
13
2
Nervous system disorders
Headacheà
28
2
27
1
Vascular disorders
Hemorrhageè
33
5
28
5
Gastrointestinal disorders
Nausea
31
2
46
0
Abdominal painð
23
3
23
1
Diarrhea
17
1
38
1
Constipation
16
0
24
0
Vomiting
15
1
24
0
Stomatitisø
13
2
26
3
Hepatobiliary disorders
Hyperbilirubinemia
21
5
17
6
General disorders and administration site conditions
Fatigueý
35
5
25
3
Pyrexia
32
3
42
6
Chills
11
0
11
0
Investigations
Transaminases increased£
26
7
13
5
Gamma-glutamyltransferase increased
21
10
8
4
Alkaline phosphatase increased
13
2
7
0
Additional adverse reactions (all grades) that were reported in less than 10% of patients treated with BESPONSA included: lipase increased (9%), abdominal distension (6%), amylase increased (5%), hyperuricemia (4%), ascites (4%), infusion related reaction (2%; includes the following: hypersensitivity and infusion related reaction), pancytopenia (2%; includes the following: bone marrow failure, febrile bone marrow aplasia, and pancytopenia), tumor lysis syndrome (2%), and electrocardiogram QT prolonged (1%).
Table 8 shows the clinically important laboratory abnormalities reported in patients with relapsed or refractory ALL who received BESPONSA or Investigator's choice of chemotherapy.
Table 8. Laboratory Abnormalities in Patients With Relapsed or Refractory B-Cell Precursor ALL Who Received BESPONSA or Investigator's Choice of Chemotherapy (FLAG, MXN/Ara-C, or HIDAC) BESPONSA FLAG, MXN/Ara-C, or HIDAC All Grades Grade 3/4 All Grades Grade 3/4 Laboratory Abnormality* N % % N % % Severity grade of laboratory abnormalities according to NCI CTCAE version 3.0.
Abbreviations: ALL=acute lymphoblastic leukemia; ALP=alkaline phosphatase; ALT=alanine aminotransferase; AST=aspartate aminotransferase; FLAG=fludarabine + cytarabine + granulocyte colony-stimulating factor; GGT=gamma-glutamyltransferase; HIDAC=high dose cytarabine; MXN/Ara-C=mitoxantrone + cytarabine; N=number of patients; NCI CTCAE=National Cancer Institute Common Toxicity Criteria for Adverse Events.- *
- Laboratory abnormalities were summarized up to the end of treatment + 42 days but prior to the start of a new anti-cancer therapy.
Hematology
Platelet count decreased
161
98
76
142
100
99
Hemoglobin decreased
161
94
40
142
100
70
Leukocytes decreased
161
95
82
142
99
98
Neutrophil count decreased
160
94
86
130
93
88
Lymphocytes (absolute) decreased
160
93
71
127
97
91
Chemistry
GGT increased
148
67
18
111
68
17
AST increased
160
71
4
134
38
4
ALP increased
158
57
1
133
52
3
ALT increased
161
49
4
137
46
4
Blood bilirubin increased
161
36
5
138
35
6
Lipase increased
139
32
13
90
20
2
Hyperuricemia
158
16
3
122
11
0
Amylase increased
143
15
2
102
9
1
Pediatric Patients
The safety of BESPONSA in pediatric patients 1 year and older with relapsed or refractory CD22-positive B-cell precursor ALL was evaluated in a multicenter, single-arm, open-label study (ITCC-059) [see Clinical Studies (14)]. Patients (n=53) received the recommended dosage of BESPONSA [see Dosage and Administration (2.1)] or BESPONSA at an initial dose of 1.4 mg/m2/cycle (approximately 0.78 times the recommended initial dosage). Patients received BESPONSA for a median of 2 (range: 1-4) cycles. The median age of patients who received BESPONSA was 9 years (range: 1-17), with 68% male.
Serious adverse reactions occurred in 62% of patients who received BESPONSA. Serious adverse reactions in > 2% of patients included infection (21%), febrile neutropenia (17%), VOD (15%), hemorrhage (4%), pyrexia (6%) and multiorgan failure (2%). Fatal adverse reactions occurred in 8% of patients who received BESPONSA, including multiorgan failure, lung infection, sepsis, and encephalopathy.
Permanent discontinuation of BESPONSA due to an adverse reaction occurred in 21% of patients. Adverse reactions which resulted in permanent discontinuation of BESPONSA in 2 or more patients included ALT increased and platelet count decreased.
Dosage interruptions of BESPONSA due to an adverse reaction occurred in 11% of patients. Adverse reactions which required dosage interruption of BESPONSA in 6 patients included increased transaminases, febrile neutropenia, and headache.
The most common adverse reactions (≥ 20%), including laboratory abnormalities, were thrombocytopenia, pyrexia, anemia, vomiting, infection, hemorrhage, neutropenia, nausea, leukopenia, febrile neutropenia, increased transaminases, abdominal pain, and headache.
Table 9 summarizes the adverse reactions in ITCC-059.
Table 9. Adverse Reactions (≥ 5%) in Pediatric Patients (N=53) With CD22-Positive Relapsed or Refractory ALL in Study WI203581 (ITCC-059) Severity grade of adverse reactions were according to NCI CTCAE version 4.03. - *
- Includes other related terms.
- †
- Infection includes any reported preferred terms for system organ class infections and infestations resulting in the following preferred terms: Acinetobacter bacteremia, bacteremia, candida infection, Cytomegalovirus infection, device related infection, device related sepsis, encephalitis, infectious enterocolitis, fungal infection, herpes virus infection, herpes zoster, influenza, kidney infection, mucosal infection, otitis media, paronychia, pneumonia, pneumonia fungal, respiratory tract infection, rhinitis, sepsis, sinusitis, skin infection, stoma site infection, upper respiratory tract infection, urinary tract inflammation, urinary tract infection, vaginal infection.
- ‡
- Hemorrhage includes any reported PT terms within the hemorrhage terms (excl laboratory terms) (SMQ) narrow, resulting in the following preferred terms: catheter site hemorrhage, diarrhea hemorrhagic, epistaxis, gingival bleeding, hematemesis, hematoma, hematuria, hemoptysis, hemorrhage intracranial, hemorrhoidal hemorrhage, lip hemorrhage, mouth hemorrhage, oral blood blister, petechiae, purpura, thrombotic thrombocytopenic purpura, and upper gastrointestinal hemorrhage.
- §
- Infusion related reaction includes the following preferred terms: infusion-related reaction and hypersensitivity.
Body System
Adverse Reaction
BESPONSA Monotherapy
(N=53)
All Grades
≥ Grade 3
%
%
General disorders and administration site conditions
Pyrexia
49
4
Edema*
19
0
Fatigue*
17
0
Pain
15
2
Chills
8
0
Blood and lymphatic system disorders
Anemia*
45
38
Febrile neutropenia
28
28
Gastrointestinal disorders
Vomiting
45
2
Nausea
32
0
Abdominal pain*
25
2
Constipation
19
2
Stomatitis*
17
6
Diarrhea
11
0
Infections and infestations
Infection†
43
23
Vascular disorders
Hemorrhage‡
42
6
Hypotension
6
4
Nervous system disorders
Headache*
21
0
Skin and subcutaneous tissue disorders
Rash*
19
4
Pruritis
9
0
Hyperhidrosis
6
0
Musculoskeletal and connective tissue disorders
Pain in extremity
19
2
Back pain
6
0
Neck pain
6
0
Muscular weakness
6
0
Respiratory, thoracic and mediastinal disorders
Cough
17
0
Dyspnea
8
2
Hypoxia
8
4
Hepatobiliary disorders
Veno-occlusive disease*
15
13
Hyperbilirubinemia*
9
8
Metabolism and nutrition disorders
Decreased appetite
11
4
Tumor lysis syndrome
11
11
Investigations
Weight increased
8
2
Injury, poisoning and procedural complications
Infusion related reaction§
8
0
Cardiac disorders
Sinus tachycardia
6
2
Psychiatric disorders
Anxiety
6
0
Table 10 summarizes select laboratory abnormalities in pediatric patients with CD22-positive relapsed/refractory ALL after receiving BESPONSA monotherapy in Study WI203581 (ITCC-059).
Table 10. Select Laboratory Abnormalities in Pediatric Patients with CD22-positive Relapsed/Refractory ALL after receiving BESPONSA Monotherapy in Study WI203581 (ITCC-059) Severity grade of laboratory abnormalities according to NCI CTCAE version 4.03. Abbreviations: N= number of subjects with valid post-baseline assessment; NCI CTCAE=National Cancer Institute Common Toxicity Criteria for Adverse Events. Laboratory Abnormality
BESPONSA Monotherapy
All Grades
Grade 3/4
N
%
%
Hematology
Platelet count decreased
53
100
85
Neutrophil count decreased
53
98
96
White blood cell decreased
53
98
89
Hemoglobin decreased
53
96
42
Lymphocyte count decreased
52
87
73
Chemistry
AST increased
53
87
21
ALT increased
53
83
21
GGT increased
33
79
27
Blood bilirubin increased
53
30
9
ALP increased
53
28
0
Lipase increased
48
23
4
Serum amylase increased
49
14
0
-
7. DRUG INTERACTIONS
Drugs That Prolong the QT Interval
Concomitant use of BESPONSA with drugs known to prolong the QT interval or induce Torsades de Pointes may increase the risk of a clinically significant QTc interval prolongation [see Clinical Pharmacology (12.2)]. Discontinue or use alternative concomitant drugs that do not prolong QT/QTc interval while the patient is using BESPONSA. When it is not feasible to avoid concomitant use of drugs known to prolong QT/QTc, obtain ECGs and electrolytes prior to the start of treatment, after initiation of any drug known to prolong QTc, and periodically monitor as clinically indicated during treatment [see Warnings and Precautions (5.5)].
-
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings from animal studies [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)], BESPONSA can cause embryo-fetal harm when administered to a pregnant woman. There are no available data on BESPONSA use in pregnant women to inform a drug-associated risk. In rat embryo-fetal development studies, inotuzumab ozogamicin caused embryo-fetal toxicity at maternal systemic exposures that were ≥ 0.4 times the exposure in patients at the maximum recommended dose, based on AUC [see Data]. Advise patients of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2–4% and 15–20%, respectively.
Animal Data
In embryo-fetal development studies in rats, pregnant animals received daily intravenous doses of inotuzumab ozogamicin up to 0.36 mg/m2 during the period of organogenesis. Embryo-fetal toxicities including increased resorptions and fetal growth retardation as evidenced by decreased live fetal weights and delayed skeletal ossification were observed at ≥ 0.11 mg/m2 (approximately 2 times the exposure in patients at the maximum recommended dose, based on AUC). Fetal growth retardation also occurred at 0.04 mg/m2 (approximately 0.4 times the exposure in patients at the maximum recommended dose, based on AUC).
In an embryo-fetal development study in rabbits, pregnant animals received daily intravenous doses up to 0.15 mg/m2 (approximately 3 times the exposure in patients at the maximum recommended dose, based on AUC) during the period of organogenesis. At a dose of 0.15 mg/m2, slight maternal toxicity was observed in the absence of any effects on embryo‑fetal development.
8.2 Lactation
Risk Summary
There are no data on the presence of inotuzumab ozogamicin or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with BESPONSA and for 2 months after the last dose.
8.3 Females and Males of Reproductive Potential
Based on its mechanism of action and findings from animal studies, BESPONSA can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating BESPONSA.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with BESPONSA and for 8 months after the last dose [see Nonclinical Toxicology (13.1)].
Males
Advise males with female partners of reproductive potential to use effective contraception during treatment with BESPONSA and for 5 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Females
Based on findings in animals, BESPONSA may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)].
Males
Based on findings in animals, BESPONSA may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of BESPONSA in pediatric patients 1 year and older with relapsed or refractory CD22-positive B-cell precursor ALL have been established. The use of BESPONSA for this indication is supported by evidence of safety and effectiveness in Study WI203581 (ITCC-059) [see Adverse Reactions (6.1), Clinical Studies (14.1)]. The study included patients in the following age groups: 2 patients 1 year to < 2 years old, 10 patients 2 years to < 6 years old, 20 patients 6 years to < 12 years old, and 20 patients 12 years to < 17 years old. Compared to adults, pediatric patients had a higher incidence of liver test abnormalities; with grade 3-4 increases in AST, ALT, and total bilirubin in 21%, 21%, and 9%, respectively, in pediatric patients treated with BESPONSA compared to 4%, 4%, and 5% in adults.
The safety and effectiveness of BESPONSA in patients < 1 year of age with relapsed or refractory CD22-positive B-cell precursor ALL have not been established.
8.5 Geriatric Use
In the INO-VATE ALL trial, 30/164 patients (18%) treated with BESPONSA were ≥ 65 years of age. No differences in responses were identified between older and younger patients.
Based on a population pharmacokinetic analysis in 765 patients, no adjustment to the starting dose is required based on age [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
Based on a population pharmacokinetic analysis, the clearance of inotuzumab ozogamicin in patients with mild hepatic impairment (total bilirubin less than or equal to ULN and AST greater than ULN, or total bilirubin greater than 1.0–1.5 × ULN and AST any level; n=150) was similar to patients with normal hepatic function (total bilirubin/AST less than or equal to ULN; n=611). In patients with moderate (total bilirubin greater than 1.5–3 × ULN and AST any level; n=3) and severe hepatic impairment (total bilirubin greater than 3 × ULN and AST any level; n=1), inotuzumab ozogamicin clearance did not appear to be reduced [see Clinical Pharmacology (12.3)].
No adjustment to the starting dose is required when administering BESPONSA to patients with total bilirubin less than or equal to 1.5 × ULN and AST/ALT less than or equal to 2.5 × ULN [see Dosage and Administration (2.3)]. There is limited safety information available in patients with total bilirubin greater than 1.5 × ULN and/or AST/ALT greater than 2.5 × ULN prior to dosing. Interrupt dosing until recovery of total bilirubin to less than or equal to 1.5 × ULN and AST/ALT to less than or equal to 2.5 × ULN prior to each dose unless due to Gilbert's syndrome or hemolysis. Permanently discontinue treatment if total bilirubin does not recover to less than or equal to 1.5 × ULN or AST/ALT does not recover to less than or equal to 2.5 × ULN [see Dosage and Administration (2.3), Warnings and Precautions (5.1)].
-
11. DESCRIPTION
Inotuzumab ozogamicin is a CD22-directed antibody and cytotoxic-drug conjugate (ADC) consisting of 3 components: 1) the recombinant humanized immunoglobulin class G subtype 4 (IgG4) kappa antibody inotuzumab, specific for human CD22, 2) N-acetyl-gamma-calicheamicin that causes double-stranded DNA breaks, and 3) an acid-cleavable linker composed of the condensation product of 4-(4'-acetylphenoxy)-butanoic acid (AcBut) and 3-methyl-3-mercaptobutane hydrazide (known as dimethylhydrazide) that covalently attaches N-acetyl-gamma-calicheamicin to inotuzumab.
Inotuzumab ozogamicin has an approximate molecular weight of 160 kDa. The average number of calicheamicin derivative molecules conjugated to each inotuzumab molecule is approximately 6 with a distribution from 2–8. Inotuzumab ozogamicin is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the semisynthetic calicheamicin derivative is produced by microbial fermentation followed by synthetic modification.
BESPONSA (inotuzumab ozogamicin) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized powder for intravenous administration. Each single-dose vial delivers 0.9 mg inotuzumab ozogamicin. Inactive ingredients are polysorbate 80 (0.36 mg), sodium chloride (2.16 mg), sucrose (180 mg), and tromethamine (8.64 mg). After reconstitution with 4 mL of Sterile Water for Injection, USP, the final concentration is 0.25 mg/mL of inotuzumab ozogamicin with a deliverable volume of 3.6 mL (0.9 mg) and a pH of approximately 8.0.
-
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Inotuzumab ozogamicin is a CD22-directed antibody drug conjugate (ADC). Inotuzumab recognizes human CD22. The small molecule, N-acetyl-gamma-calicheamicin, is a cytotoxic agent that is covalently attached to the antibody via a linker. Nonclinical data suggest that the anticancer activity of inotuzumab ozogamicin is due to the binding of the ADC to CD22-expressing tumor cells, followed by internalization of the ADC-CD22 complex, and the intracellular release of N-acetyl-gamma-calicheamicin dimethylhydrazide via hydrolytic cleavage of the linker. Activation of N-acetyl-gamma-calicheamicin dimethylhydrazide induces double-strand DNA breaks, subsequently inducing cell cycle arrest and apoptotic cell death.
12.2 Pharmacodynamics
During the treatment period, the pharmacodynamic response to BESPONSA was characterized by the depletion of CD22-positive leukemic blasts.
Cardiac Electrophysiology
In a randomized clinical study in patients with relapsed or refractory ALL, increases in QTcF of ≥ 60 msec from baseline were measured in 4/162 patients (3%) in the BESPONSA arm and 3/124 patients (2%) in the Investigator's choice of chemotherapy arm. Increases in QTcF of > 500 msec were observed in none of the patients in the BESPONSA arm and 1/124 patients (1%) in the Investigator's choice of chemotherapy arm. Central tendency analysis of the QTcF interval changes from baseline showed that the highest mean (upper bound of the 2-sided 90% CI) for QTcF was 15.3 (21.1) msec, which was observed at Cycle 4/Day 1/1 hour in the BESPONSA arm [see Warnings and Precautions (5.5)].
12.3 Pharmacokinetics
The mean Cmax of inotuzumab ozogamicin was 308 ng/mL. The mean simulated total AUC per cycle was 100,000 ng∙h/mL. In patients with relapsed or refractory ALL, steady-state drug concentration was achieved by Cycle 4. Following administration of multiple doses, a 5.3 times accumulation of inotuzumab ozogamicin was predicted by Cycle 4.
Distribution
N-acetyl-gamma-calicheamicin dimethylhydrazide is approximately 97% bound to human plasma proteins in vitro. In humans, the total volume of distribution of inotuzumab ozogamicin was approximately 12 L.
Elimination
The pharmacokinetics of inotuzumab ozogamicin was well characterized by a 2-compartment model with linear and time-dependent clearance components. In 234 patients with relapsed or refractory ALL, the clearance of inotuzumab ozogamicin at steady state was 0.0333 L/h and the terminal half-life (t½) was 12.3 days. Following administration of multiple doses, a 5.3 times accumulation of inotuzumab ozogamicin was predicted by Cycle 4.
Metabolism
In vitro, N-acetyl-gamma-calicheamicin dimethylhydrazide was primarily metabolized via nonenzymatic reduction. In humans, N-acetyl-gamma-calicheamicin dimethylhydrazide serum levels were typically below the limit of quantitation.
Specific Populations
The effect of intrinsic factors on inotuzumab ozogamicin pharmacokinetics was assessed using a population pharmacokinetic analysis unless otherwise specified. Age (18 to 92 years of age), sex, and race (Asian versus non-Asian [Caucasian, Black, and Unspecified]) had no clinically significant effect on the pharmacokinetics of inotuzumab ozogamicin. Body surface area was found to significantly affect inotuzumab ozogamicin disposition. BESPONSA is dosed based on body surface area [see Dosage and Administration (2.1)].
Patients with Renal Impairment
The clearance of inotuzumab ozogamicin in patients with mild renal impairment (creatinine clearance [CLcr; based on the Cockcroft-Gault formula] 60–89 mL/min; n=237), moderate renal impairment (CLcr 30–59 mL/min; n=122), or severe renal impairment (CLcr 15–29 mL/min; n=4) was similar to patients with normal renal function (CLcr ≥ 90 mL/min; n=402). The safety and efficacy of inotuzumab ozogamicin in patients with end stage renal disease with or without hemodialysis is unknown.
Patients with Hepatic Impairment
The clearance of inotuzumab ozogamicin in patients with mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin > 1.0–1.5 × ULN and AST any level; n=150) was similar to patients with normal hepatic function (total bilirubin/AST ≤ ULN; n=611). There is insufficient data in patients with moderate and severe hepatic impairment (total bilirubin > 1.5 ULN).
Pediatric Patients
The exposure of inotuzumab ozogamicin increases with decreasing body size at the recommended dosage. There was an approximately 30% increase in AUC of inotuzumab ozogamicin in pediatric patients 1 year and older compared with adults.
Drug Interactions
In vitro
Effect of Metabolic Pathways and Transporter Systems on BESPONSA
N-acetyl-gamma-calicheamicin dimethylhydrazide is a substrate of P-glycoprotein (P-gp).
Effect of BESPONSA on Metabolic Pathways and Transporter Systems
At clinically relevant concentrations, N-acetyl-gamma-calicheamicin dimethylhydrazide had a low potential to:
- •
- Inhibit cytochrome P450 (CYP 450) Enzymes: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5.
- •
- Induce CYP450 Enzymes: CYP1A2, CYP2B6, and CYP3A4.
- •
- Inhibit UGT Enzymes: UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7.
- •
- Inhibit Drug Transporters: P-gp, breast cancer resistance protein (BCRP), organic anion transporter (OAT)1 and OAT3, organic cation transporter (OCT)2, and organic anion transporting polypeptide (OATP)1B1 and OATP1B3.
At clinically relevant concentrations, inotuzumab ozogamicin had a low potential to:
- •
- Inhibit CYP450 Enzymes: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5.
- •
- Induce CYP450 Enzymes: CYP1A2, CYP2B6, and CYP3A4.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of inotuzumab ozogamicin.
In clinical studies of BESPONSA in patients with relapsed or refractory ALL, the immunogenicity of inotuzumab ozogamicin was evaluated using an electrochemiluminescence (ECL)‑based immunoassay to test for ADA. For patients whose sera tested positive for ADA, a cell-based assay was performed to detect neutralizing antibodies (NAb).
During a maximum 6 cycles of treatment period in clinical studies of BESPONSA in adult patients with relapsed or refractory ALL, 7/236 (3%) patients tested positive for ADA. No patients tested positive for NAb. In patients who tested positive for ADA, the presence of positive ADA did not affect clearance following inotuzumab ozogamicin treatment. Because of the low occurrence of ADA, the effect of these antibodies on safety and efficacy of inotuzumab ozogamicin is unknown.
During a maximum 4 cycles of treatment period in clinical Study WI203581 (ITCC-059) of BESPONSA in pediatric patients with relapsed or refractory ALL (n=51), no patients tested positive for ADA against inotuzumab ozogamcin.
-
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Formal carcinogenicity studies have not been conducted with inotuzumab ozogamicin. In toxicity studies, rats were dosed weekly for 4 or 26 weeks with inotuzumab ozogamicin at doses up to 4.1 mg/m2 and 0.73 mg/m2, respectively. After 26 weeks of dosing, rats developed hepatocellular adenomas in the liver at 0.73 mg/m2 (approximately 2 times the exposure in patients at the maximum recommended dose, based on AUC).
Inotuzumab ozogamicin was clastogenic in vivo in the bone marrow of male mice that received single doses ≥ 1.1 mg/m2. This is consistent with the known induction of DNA breaks by calicheamicin. N-acetyl-gamma-calicheamicin dimethylhydrazide (the cytotoxic agent released from inotuzumab ozogamicin) was mutagenic in an in vitro bacterial reverse mutation (Ames) assay.
In a female fertility and early embryonic development study, female rats were administered daily intravenous doses of inotuzumab ozogamicin up to 0.11 mg/m2 for 2 weeks before mating through Day 7 of pregnancy. An increase in the proportion of resorptions and decrease in the number of viable embryos and gravid uterine weights were observed at the 0.11 mg/m2 dose level (approximately 2 times the exposure in patients at the maximum recommended dose, based on AUC). Additional findings in female reproductive organs occurred in repeat-dose toxicology studies and included decreased ovarian and uterine weights, and ovarian and uterine atrophy. Findings in male reproductive organs occurred in repeat-dose toxicology studies and included decreased testicular weights, testicular degeneration, hypospermia, and prostatic and seminal vesicle atrophy. Testicular degeneration and hypospermia were nonreversible following a 4-week nondosing period. In the chronic studies of 26-weeks duration, adverse effects on reproductive organs occurred at ≥ 0.07 mg/m2 in male rats and at 0.73 mg/m2 in female monkeys [see Use in Specific Populations (8.3)].
-
14. CLINICAL STUDIES
Relapsed or Refractory ALL
INO-VATE ALL Study – Adult Patients
The safety and efficacy of BESPONSA were evaluated in INO-VATE ALL (NCT01564784) a randomized (1:1), open‑label, international, multicenter study in patients with relapsed or refractory ALL. Patients were stratified at randomization based on duration of first remission (< 12 months or ≥ 12 months, salvage treatment (Salvage 1 or 2) and patient age at randomization (< 55 or ≥ 55 years). Eligible patients were ≥ 18 years of age with Philadelphia chromosome-negative or Philadelphia chromosome-positive relapsed or refractory B-cell precursor ALL. All patients were required to have ≥ 5% bone marrow blasts and to have received 1 or 2 previous induction chemotherapy regimens for ALL. Patients with Philadelphia chromosome-positive B-cell precursor ALL were required to have disease that failed treatment with at least 1 tyrosine kinase inhibitor and standard chemotherapy. Table 1 shows the dosing regimen used to treat patients.
Among all 326 patients who were randomized to receive BESPONSA (N=164) or Investigator's choice of chemotherapy (N=162), 215 patients (66%) had received 1 prior treatment regimen for ALL and 108 patients (33%) had received 2 prior treatment regimens for ALL. The median age was 47 years (range: 18–79 years), 276 patients (85%) had Philadelphia chromosome-negative ALL, 206 patients (63%) had a duration of first remission < 12 months, and 55 patients (17%) had undergone a HSCT prior to receiving BESPONSA or Investigator's choice of chemotherapy. The two treatment groups were generally balanced with respect to the baseline demographics and disease characteristics.
All evaluable patients had B-cell precursor ALL that expressed CD22, with ≥ 90% of evaluable patients exhibiting ≥ 70% leukemic blast CD22 positivity prior to treatment, as assessed by flow cytometry performed at a central laboratory.
The efficacy of BESPONSA was established on the basis of CR, the duration of CR, and proportion of MRD-negative CR (< 1 × 10-4 of bone marrow nucleated cells by flow cytometry) in the first 218 patients randomized. CR, duration of remission (DoR), and MRD results in the initial 218 randomized patients were consistent with those seen in all 326 randomized patients.
Among the initial 218 randomized patients, 64/88 (73%) and 21/88 (24%) of responding patients per EAC achieved CR/CRi in Cycles 1 and 2, respectively, in the BESPONSA arm, and 29/32 (91%) and 1/32 (3%) of responding patients per EAC achieved a CR/CRi in Cycles 1 and 2, respectively, in the Investigator's choice of chemotherapy arm.
Table 11 shows the efficacy results from this study.
Table 11. Efficacy Results in Patients With Relapsed or Refractory B-Cell Precursor ALL Who Received BESPONSA or Investigator's Choice of Chemotherapy (FLAG, MXN/Ara-C, or HIDAC) CR* CRi† CR/CRi*,† BESPONSA
(N=109)HIDAC, FLAG, or MXN/Ara-C
(N=109)BESPONSA
(N=109)HIDAC, FLAG or MXN/Ara-C
(N=109)BESPONSA
(N=109)HIDAC, FLAG, or MXN/Ara-C
(N=109)Abbreviations: CI=confidence interval; CR=complete remission; CRi=complete remission with incomplete hematologic recovery; DoR=duration of remission; EAC=Endpoint Adjudication Committee; FLAG=fludarabine + cytarabine + granulocyte colony-stimulating factor; HIDAC=high-dose cytarabine; HR=hazard ratio; MRD=minimal residual disease; MXN/AraC=mitoxantrone + cytarabine; N/n=number of patients; OS=overall survival; PFS=progression-free survival. - *
- CR, per EAC, was defined as < 5% blasts in the bone marrow and the absence of peripheral blood leukemic blasts, full recovery of peripheral blood counts (platelets ≥ 100 × 109/L and absolute neutrophil counts [ANC] ≥ 1 × 109/L) and resolution of any extramedullary disease.
- †
- CRi, per EAC, was defined as < 5% blasts in the bone marrow and the absence of peripheral blood leukemic blasts, incomplete recovery of peripheral blood counts (platelets < 100 × 109/L and/or ANC < 1 × 109/L) and resolution of any extramedullary disease.
- ‡
- 1-sided p-value using Chi-squared test.
- §
- DoR, based on a later cutoff date than the CR/CRi, was defined for patients who achieved CR/CRi per Investigator's assessment as time since first response of CR* or CRi† per Investigator's assessment to the date of a PFS event or censoring date if no PFS event was documented.
- ¶
- MRD-negativity was defined by flow cytometry as leukemic cells comprising < 1 × 10-4 (< 0.01%) of bone marrow nucleated cells.
- #
- Rate was defined as the number of patients who achieved MRD negativity divided by the total number of patients who achieved CR/CRi per EAC.
Responding (CR/CRi) patients
n (%)
[95% CI]39 (35.8)
[26.8–45.5]19 (17.4)
[10.8–25.9]49 (45.0)
[35.4–54.8]13 (11.9)
[6.5–19.5]88 (80.7)
[72.1–87.7]32 (29.4)
[21.0–38.8]p-value‡
< 0.0001
DoR§
n
39
18
45
14
84
32
Median, months
[95% CI]8.0
[4.9–10.4]4.9
[2.9–7.2]4.6
[3.7–5.7]2.9
[0.6–5.7]5.4
[4.2–8.0]3.5
[2.9–6.6]MRD-negativity¶
n
35
6
34
3
69
9
Rate# (%)
[95% CI]35/39 (89.7)
[75.8–97.1]6/19 (31.6)
[12.6–56.6]34/49 (69.4)
[54.6–81.7]3/13 (23.1)
[5.0–53.8]69/88 (78.4)
[68.4–86.5]9/32 (28.1)
[13.7–46.7]Among the initial 218 patients, as per EAC assessment, 32/109 patients (29%) in the BESPONSA arm achieved complete remission with partial hematologic recovery (CRh; defined as < 5% blasts in the bone marrow, ANC > 0.5 × 109/L, and platelet counts > 50 × 109/L but not meeting full recovery of peripheral blood counts) versus 6/109 patients (6%) in the Investigator's choice of chemotherapy arm, and 71/109 patients (65%) in the BESPONSA arm achieved CR/CRh versus 25/109 patients (23%) in the Investigator's choice of chemotherapy arm.
Overall, 79/164 patients (48%) in the BESPONSA arm and 35/162 patients (22%) in the Investigator's choice of chemotherapy arm had a follow-up HSCT.
Figure 1 shows the analysis of overall survival (OS). The analysis of OS did not meet the pre-specified boundary for statistical significance.
Figure 1. Kaplan-Meier Curve for Overall Survival (Intent-to-Treat Population)
WI203581 (ITCC-059) – Pediatric Patients
BESPONSA was evaluated in a multicenter, single-arm, open-label study in 53 pediatric patients ≥ 1 and < 18 years of age with relapsed or refractory CD22-positive B-cell precursor ALL.
In 53 patients, there were two dose levels: an initial dose of 1.4 mg/m2/cycle (approximately 0.78 times the recommended initial dosage) in 12 patients and 1.8 mg/m2/cycle in 41 patients (premedications included methylprednisolone 1 mg/kg with a maximum of 50 mg, an antipyretic, and an antihistamine). Table 1 shows the dosing regimen used to treat patients. Patients received a median of 2 cycles of therapy (range: 1 to 4 cycles). The median age was 9 years (range: 1 to 17 years), and 55% of patients had second or greater relapsed B-cell precursor ALL.
Efficacy was established on the basis of the Complete Remission (CR) Rate [CR was defined as < 5% blasts in the bone marrow and the absence of peripheral blood leukemic blasts, full recovery of peripheral blood counts (platelets ≥ 100 × 109/L and ANC ≥ 1 × 109/L) and resolution of any extramedullary disease], duration of CR, and proportion of patients with MRD negative CR [MRD was defined by leukemic cells comprising < 1 × 10-4 (< 0.01%) of bone marrow nucleated cells by flow cytometry or by PCR]. In all patients, 22/53 (42%, 95% CI 28.1-55.9%) patients achieved CR, and the median duration of CR (DOCR) was 8.2 months (95% CI: 2.6-NE). The minimal residual disease (MRD) negativity rate in patients with CR was 21/22 [95.5% (95% CI: 77.2-99.9)] based on flow cytometry, and 19/22 [86.4% (95% CI: 65.1-97.1)] based on RQ-PCR.
- 15. REFERENCES
-
16. HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
BESPONSA (inotuzumab ozogamicin) for injection is supplied as a white to off-white lyophilized powder in a single-dose vial for reconstitution and further dilution. Each vial delivers 0.9 mg inotuzumab ozogamicin. Each carton (NDC 0008-0100-01) contains one single-dose vial.
Storage and Handling
Refrigerate (2-8°C; 36-46°F) BESPONSA vials and store in the original carton to protect from light. Do not freeze.
BESPONSA is a hazardous drug. Follow applicable special handling and disposal procedures.1
-
17. PATIENT COUNSELING INFORMATION
Hepatotoxicity, Including Hepatic Veno-occlusive Disease (VOD) (also known as Sinusoidal Obstruction Syndrome)
Inform patients that liver problems, including severe, life-threatening, or fatal VOD, and increases in liver tests may develop during BESPONSA treatment. Inform patients that they should seek immediate medical advice if they experience symptoms of VOD, which may include elevated bilirubin, rapid weight gain, and abdominal swelling that may be painful. Inform patients that they should carefully consider the benefit/risk of BESPONSA treatment if they have a prior history of VOD or serious ongoing liver disease [see Warnings and Precautions (5.1)].
Increased Risk of Post-HSCT Non-Relapse Mortality
Inform patients that there is an increased risk of post-HSCT non-relapse mortality after receiving BESPONSA, that the most common causes of post-HSCT non-relapse mortality included infection and VOD. Advise patients to report signs and symptoms of infection [see Warnings and Precautions (5.2)].
Myelosuppression
Inform patients that decreased blood counts, which may be life-threatening, may develop during BESPONSA treatment and that complications associated with decreased blood counts may include infections, which may be life-threatening or fatal, and bleeding/hemorrhage events. Inform patients that signs and symptoms of infection, bleeding/hemorrhage, or other effects of decreased blood counts should be reported during treatment with BESPONSA [see Warnings and Precautions (5.3)].
Infusion Related Reactions
Advise patients to contact their health care provider if they experience symptoms such as fever, chills, rash, or breathing problems during the infusion of BESPONSA [see Warnings and Precautions (5.4)].
QT Interval Prolongation
Inform patients of symptoms that may be indicative of significant QTc prolongation including dizziness, lightheadedness, and syncope. Advise patients to report these symptoms and the use of all medications to their healthcare provider [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
Advise males and females of reproductive potential to use effective contraception during BESPONSA treatment and for 5 and 8 months after the last dose, respectively [see Use in Specific Populations (8.3)]. Advise women to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with BESPONSA. Inform the patient of the potential risk to the fetus [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
Lactation
Advise women against breastfeeding while receiving BESPONSA and for 2 months after the last dose [see Use in Specific Populations (8.2)].
-
SPL UNCLASSIFIED SECTION
This product's label may have been updated. For current full prescribing information, please visit www.BESPONSA.com.
US License No. 003
LAB-0763-3.0
- PRINCIPAL DISPLAY PANEL - 0.9 mg Vial Label
- PRINCIPAL DISPLAY PANEL - 0.9 mg Vial Carton
-
INGREDIENTS AND APPEARANCE
BESPONSA
inotuzumab ozogamicin injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0008-0100 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength INOTUZUMAB OZOGAMICIN (UNII: P93RUU11P7) (INOTUZUMAB OZOGAMICIN - UNII:P93RUU11P7) INOTUZUMAB OZOGAMICIN 0.25 mg in 1 mL Inactive Ingredients Ingredient Name Strength TROMETHAMINE (UNII: 023C2WHX2V) SUCROSE (UNII: C151H8M554) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM CHLORIDE (UNII: 451W47IQ8X) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0008-0100-01 1 in 1 CARTON 08/18/2017 1 4 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761040 08/18/2017 Labeler - Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc. (113008515) Establishment Name Address ID/FEI Business Operations Pharmacia & Upjohn Company LLC 618054084 LABEL(0008-0100) , PACK(0008-0100) Establishment Name Address ID/FEI Business Operations Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC 174350868 ANALYSIS(0008-0100) , API MANUFACTURE(0008-0100) Establishment Name Address ID/FEI Business Operations Wyeth Pharmaceutical Division of Wyeth Holdings LLC 054065909 ANALYSIS(0008-0100) , API MANUFACTURE(0008-0100) , MANUFACTURE(0008-0100)