Label: LOVASTATIN tablet

-
NDC Code(s):
63629-1018-0,
63629-1018-1,
63629-1018-2,
63629-1018-3, view more63629-1018-4, 63629-1018-5, 63629-1018-6, 63629-1018-8, 63629-1018-9
- Packager: Bryant Ranch Prepack
- This is a repackaged label.
- Source NDC Code(s): 61442-141
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated May 31, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Lovastatin is a cholesterol lowering agent isolated from a strain of Aspergillus terreus. After oral ingestion, lovastatin, which is an inactive lactone, is hydrolyzed to the corresponding β-hydroxyacid form. This is a principal metabolite and an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. This enzyme catalyzes the conversion of HMG-CoA to mevalonate, which is an early and rate limiting step in the biosynthesis of cholesterol.
Lovastatin is [1 S -[1α( R *), 3α, 7β, 8β(2 S *,4 S *),8aβ]]-1,2,3, 7,8,8a-hexahydro-3,7-dimethyl-8-[2-(tetrahydro-4-hydroxy-6-oxo-2 H -pyran-2-yl)ethyl]-1-naphthalenyl 2-methylbutanoate. The empirical formula of lovastatin is C 24 H 36 O 5 and its molecular weight is 404.55. Its structural formula is:
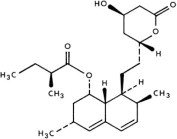
Lovastatin is a white, nonhygroscopic crystalline powder that is insoluble in water and sparingly soluble in ethanol, methanol, and acetonitrile.
Lovastatin tablets are supplied as 10 mg, 20 mg and 40 mg tablets for oral administration. In addition to the active ingredient lovastatin, each tablet contains the following inactive ingredients: lactose monohydrate, magnesium stearate, microcrystalline cellulose, poloxamer, pregelatinized starch, sodium starch glycolate, butylated hydroxyanisole and talc. Butylated hydroxyanisole (BHA) is added as a preservative.
-
CLINICAL PHARMACOLOGY
The involvement of low-density lipoprotein cholesterol (LDL-C) in atherogenesis has been well-documented in clinical and pathological studies, as well as in many animal experiments. Epidemiological and clinical studies have established that high LDL-C and low high-density lipoprotein cholesterol (HDL-C) are both associated with coronary heart disease. However, the risk of developing coronary heart disease is continuous and graded over the range of cholesterol levels and many coronary events do occur in patients with total cholesterol (total-C) and LDL-C in the lower end of this range.
Lovastatin has been shown to reduce both normal and elevated LDL-C concentrations. LDL is formed from very low-density lipoprotein (VLDL) and is catabolized predominantly by the high affinity LDL receptor. The mechanism of the LDL-lowering effect of lovastatin may involve both reduction of VLDL-C concentration, and induction of the LDL receptor, leading to reduced production and/or increased catabolism of LDL-C. Apolipoprotein B also falls during treatment with lovastatin.
Lovastatin is a specific inhibitor of HMG-CoA reductase, the enzyme which catalyzes the conversion of HMG-CoA to mevalonate. The conversion of HMG-CoA to mevalonate is an early step in the biosynthetic pathway for cholesterol.
Pharmacokinetics
Lovastatin is a lactone, which is readily hydrolyzed in vivo to the corresponding β-hydroxyacid, a strong. inhibitor of HMG-CoA reductase. Inhibition of HMG-CoA reductase is the basis for an assay in pharmacokinetic studies of the β-hydroxyacid metabolites (active inhibitors) and, following base hydrolysis, active plus latent inhibitors (total inhibitors) in plasma following administration of lovastatin.
Following an oral dose of 14 C-labeled lovastatin in man, 10% of the dose was excreted in urine and 83% in feces. The latter represents absorbed drug equivalents excreted in bile, as well as any unabsorbed drug. Plasma concentrations of total radioactivity (lovastatin plus 14 C-metabolites) peaked at 2 hours and declined rapidly to about 10% of peak by 24 hours postdose. Absorption of lovastatin, estimated relative to an intravenous reference dose, in each of four animal species tested, averaged about 30% of an oral dose. In animal studies, after oral dosing, lovastatin had high selectivity for the liver, where it achieved substantially higher concentrations than in non-target tissues. Lovastatin undergoes extensive first-pass extraction in the liver, its primary site of action, with subsequent excretion of drug equivalents in the bile. As a consequence of extensive hepatic extraction of lovastatin, the availability of drug to the general circulation is low and variable. In a single dose study in four hypercholesterolemic patients, it was estimated that less than 5% of an oral dose of lovastatin reaches the general circulation as active inhibitors. Following administration of lovastatin tablets the coefficient of variation, based on between-subject variability, was approximately 40% for the area under the curve (AUC) of total inhibitory activity in the general circulation.
Both lovastatin and its β-hydroxyacid metabolite are highly bound (>95%) to human plasma proteins. Animal studies demonstrated that lovastatin crosses the blood-brain and placental barriers.
The major active metabolites present in human plasma are the β-hydroxyacid of lovastatin, its 6'-hydroxy derivative, and two additional metabolites. Peak plasma concentrations of both active and total inhibitors were attained within 2 to 4 hours of dose administration. While the recommended therapeutic dose range is 10 to 80 mg/day, linearity of inhibitory activity in the general circulation was established by a single dose study employing lovastatin tablet dosages from 60 to as high as 120 mg. With a once-a-day dosing regimen, plasma concentrations of total inhibitors over a dosing interval achieved a steady state between the second and third days of therapy and were about 1.5 times those following a single dose. When lovastatin was given under fasting conditions, plasma concentrations of total inhibitors were on average about two-thirds those found when lovastatin was administered immediately after a standard test meal.
In a study of patients with severe renal insufficiency (creatinine clearance 10 to 30 mL/min), the plasma concentrations of total inhibitors after a single dose of lovastatin were approximately two-fold higher than those in healthy volunteers.
In a study including 16 elderly patients between 70 to 78 years of age who received lovastatin 80 mg/day, the mean plasma level of HMG-CoA reductase inhibitory activity was increased approximately 45% compared with 18 patients between 18 to 30 years of age (see PRECAUTIONS, Geriatric Use ).
Although the mechanism is not fully understood, cyclosporine has been shown to increase the AUC of HMG-CoA reductase inhibitors. The increase in AUC for lovastatin and lovastatin acid is presumably due, in part, to inhibition of CYP3A4.
The risk of myopathy is increased by high levels of HMG-CoA reductase inhibitory activity in plasma. Strong inhibitors of CYP3A4 can raise the plasma levels of HMG-CoA reductase inhibitory activity and increase the risk of myopathy (see WARNINGS, Myopathy/Rhabdomyolysis and PRECAUTIONS, Drug Interactions ).
Lovastatin is a substrate for cytochrome P450 isoform 3A4 (CYP3A4) (see PRECAUTIONS, Drug Interactions .) Grapefruit juice contains one or more components that inhibit CYP3A4 and can increase the plasma concentrations of drugs metabolized by CYP3A4. In one study 1, 10 subjects consumed 200 mL of double-strength grapefruit juice (one can of frozen concentrate diluted with one rather than 3 cans of water) three times daily for 2 days and an additional 200 mL double-strength grapefruit juice together with and 30 and 90 minutes following a single dose of 80 mg lovastatin on the third day. This regimen of grapefruit juice resulted in a mean increase in the serum concentration of lovastatin and its β-hydroxyacid metabolite (as measured by the area under the concentration-time curve) of 15-fold and 5-fold, respectively [as measured using a chemical assay-high performance liquid chromatography.] In a second study, 15 subjects consumed one 8 oz glass of single-strength grapefruit juice (one can of frozen concentrate diluted with 3 cans of water) with breakfast for 3 consecutive days and a single dose of 40 mg lovastatin in the evening of the third day. This regimen of grapefruit juice resulted in a mean increase in the plasma concentration (as measured by the area under the concentration-time curve) of active and total HMG-CoA reductase inhibitory activity [using an enzyme inhibition assay both before (for active inhibitors) and after (for total inhibitors) base hydrolysis] of 1.34-fold and 1.36-fold, respectively, and of lovastatin and its β-hydroxyacid metabolite [measured using a chemical assay-liquid chromatography/tandem mass spectrometry-different from that used in the first 1 study] of 1.94-fold and 1.57-fold, respectively. The effect of amounts of grapefruit juice between those used in these two studies on lovastatin pharmacokinetics has not been studied.
____________________________________
1 Kantola, T, et al., Clin Pharmacol Ther 1998; 63(4):397-402.
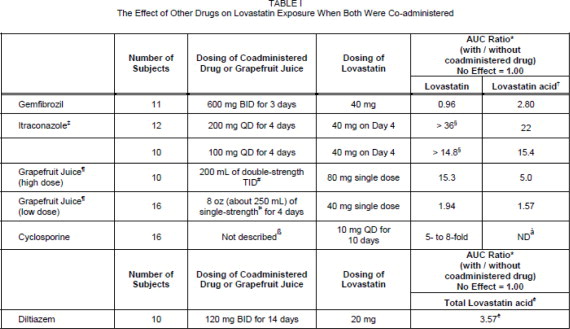
* Results based on a chemical assay.
† Lovastatin acid refers to the β-hydroxyacid of lovastatin.
‡ The mean total AUC of lovastatin without itraconazole phase could not be determined accurately. Results could be representative of strong CYP3A4 inhibitors such as ketoconazole, posaconazole, clarithromycin, telithromycin, HIV protease inhibitors, and nefazodone.
§ Estimated minimum change.
¶ The effect of amounts of grapefruit juice between those used in these two studies on lovastatin pharmacokinetics has not been studied.
# Double-strength: one can of frozen concentrate diluted with one can of water.
Grapefruit juice was administered TID for 2 days, and 200 mL together with single dose lovastatin and 30 and 90 minutes following single dose lovastatin on Day 3.
Þ Single-strength: one can of frozen concentrate diluted with 3 cans of water. Grapefruit juice was administered with breakfast for 3 days, and lovastatin was administered in the evening on Day 3.
ß Cyclosporine-treated patients with psoriasis or post kidney or heart transplant patients with stable graft function, transplanted at least 9 months prior to study.
à ND = Analyte not determined.
è Lactone converted to acid by hydrolysis prior to analysis. Figure represents total unmetabolized acid and lactone.
Clinical Studies in Adults
Lovastatin has been shown to reduce total-C and LDL-C in heterozygous familial and non-familial forms of primary hypercholesterolemia and in mixed hyperlipidemia. A marked response was seen within 2 weeks, and the maximum therapeutic response occurred within 4 to 6 weeks. The response was maintained during continuation of therapy. Single daily doses given in the evening were more effective than the same dose given in the morning, perhaps because cholesterol is synthesized mainly at night.
In multicenter, double-blind studies in patients with familial or non-familial hypercholesterolemia, lovastatin, administered in doses ranging from 10 mg q.p.m. to 40 mg b.i.d., was compared to placebo. Lovastatin significantly decreased plasma total-C, LDL-C, total-C/HDL-C ratio and LDL-C/HDL-C ratio. In addition, lovastatin produced increases of variable magnitude in HDL-C, and modestly decreased VLDL-C and plasma TG (see Tables II through IV for dose response results).
The results of a study in patients with primary hypercholesterolemia are presented in Table II.
TABLE II Lovastatin vs Placebo (Mean Percent Change from Baseline After 6 Weeks) DOSAGE N TOTAL-C LDL-C HDL-C LDL-C/
HDL-CTOTAL-C/
HDL/CTRIG. Placebo 33 -2 -1 -1 0 +1 +9 Lovastatin 10 mg q.p.m. 33 -16 -21 +5 -24 -19 -10 20 mg q.p.m. 33 -19 -27 +6 -30 -23 +9 10 mg b.i.d. 32 -19 -28 +8 -33 -25 -7 40 mg q.p.m. 33 -22 -31 +5 -33 -25 -8 20 mg b.i.d. 36 -24 -32 +2 -32 -24 -6 Lovastatin was compared to cholestyramine in a randomized open parallel study. The study was performed with patients with hypercholesterolemia who were at high risk of myocardial infarction. Summary results are presented in Table III.
TABLE III Lovastatin vs. Cholestyramine (Percent Change from Baseline After 12 Weeks) TREATMENT N TOTAL-C
(mean)LDL-C
(mean)HDL-C
(mean)LDL-C/
HDL-C
(mean)TOTAL-C/
HDL-C
(mean)VLDL-C
(median)TRIG.
(median)Lovastatin 20 mg b.i.d. 85 -27 -32 +9 -36 -31 -34 -21 40 mg b.i.d. 88 -34 -42 +8 -44 -37 -31 -27 Cholestyramine 12 g b.i.d. 88 -17 -23 +8 -27 -21 +2 +11 Lovastatin was studied in controlled trials in hypercholesterolemic patients with well-controlled non-insulin dependent diabetes mellitus with normal renal function. The effect of lovastatin on lipids and lipoproteins and the safety profile of lovastatin were similar to that demonstrated in studies in nondiabetics. Lovastatin had no clinically important effect on glycemic control or on the dose requirement of oral hypoglycemic agents.
Expanded Clinical Evaluation of Lovastatin (EXCEL) Study
Lovastatin was compared to placebo in 8,245 patients with hypercholesterolemia (total-C 240-300 mg/dL [6.2 mmol/L-7.6 mmol/L], LDL-C >160 mg/dL [4.1 mmol/L]) in the randomized, double-blind, parallel, 48-week EXCEL study. All changes in the lipid measurements (Table IV) in lovastatin treated patients were dose-related and significantly different from placebo (p≤0.001). These results were sustained throughout the study.
TABLE IV Lovastatin vs. Placebo (Percent Change from Baseline-- Average Values Between Weeks 12 and 48) **Patients enrolled
DOSAGE N ** TOTAL-C
(mean)LDL-C
(mean)HDL-C
(mean)LDL-C/
HDL-C
(mean)TOTAL-C/
HDL-C
(mean)TRIG.
(median)Placebo 1663 +0.7 +0.4 +2.0 +0.2 +0.6 +4 Lovastatin 20 mg q.p.m. 1642 -17 -24 +6.6 -27 -21 -10 40 mg q.p.m. 1645 -22 -30 +7.2 -34 -26 -14 20 mg b.i.d. 1646 -24 -34 +8.6 -38 -29 -16 40 mg b.i.d. 1649 -29 -40 +9.5 -44 -34 -19 Efficacy of Lovastatin in Adolescent Boys with Heterozygous Familial Hypercholesterolemia
In a double-blind, placebo-controlled study, 132 boys 10 to 17 years of age (mean age 12.7 yrs) with heterozygous familial hypercholesterolemia (heFH) were randomized to lovastatin (n=67) or placebo (n=65) for 48 weeks. Inclusion in the study required a baseline LDL-C level between 189 and 500 mg/dL and at least one parent with an LDL-C level >189 mg/dL. The mean baseline LDL-C value was 253.1 mg/dL (range: 171 to 379 mg/dL) in the lovastatin group compared to 248.2 mg/dL (range 158.5 to 413.5 mg/dL) in the placebo group. The dosage of lovastatin (once daily in the evening) was 10 mg for the first 8 weeks, 20 mg for the second 8 weeks, and 40 mg thereafter.
Lovastatin significantly decreased plasma levels of total-C, LDL-C and apolipoprotein B (see Table V).
TABLE V Lipid-lowering Effects of Lovastatin in Adolescent Boys with Heterozygous Familial Hypercholesterolemia (Mean Percent Change from Baseline at week 48 in Intention-to-Treat Population) *data presented as median percent changes
DOSAGE N TOTAL-C LDL-C HDL-C TG. * Apolipoprotein B Placebo 61 -1.1 -1.4 -2.2 -1.4 -4.4 Lovastatin 64 -19.3 -24.2 +1.1 -1.9 -21 The mean achieved LDL-C value was 190.9 mg/dL (range: 108 to 336 mg/dL) in the lovastatin group compared to 244.8 mg/dL (range: 135 to 404 mg/dL) in the placebo group.
Efficacy of Lovastatin in Post-menarchal Girls with Heterozygous Familial Hypercholesterolemia
In a double-blind, placebo-controlled study, 54 girls 10 to 17 years of age who were at least 1 year post-menarche with heFH were randomized to lovastatin (n=35) or placebo (n=19) for 24 weeks. Inclusion in the study required a baseline LDL-C level of 160 to 400 mg/dL and a parental history of familial hypercholesterolemia. The mean baseline LDL-C value was 218.3 mg/dL (range: 136.3 to 363.7 mg/dL) in the lovastatin group compared to 198.8 mg/dL (range: 151.5 to 283.1 mg/dL) in the placebo group. The dosage of lovastatin (once daily in the evening) was 20 mg for the first 4 weeks, and 40 mg thereafter.
Lovastatin significantly decreased plasma levels of total-C, LDL-C, and apolipoprotein B (see Table VI).
TABLE VI Lipid-lowering Effects of Lovastatin in Post-menarchal Girls with Heterozygous Familial Hypercholesterolemia (Mean Percent Change from Baseline at Week 24 in Intention-to-Treat Population) *data presented as median percent changes
DOSAGE N TOTAL-C LDL-C HDL-C TG. * Apolipoprotein B Placebo 18 +3.6 +2.5 +4.8 -3.0 +6.4 Lovastatin 35 -22.4 -29.2 +2.4 -22.7 -24.4 The mean achieved LDL-C value was 154.5 mg/dL (range: 82 to 286 mg/dL) in the lovastatin group compared to 203.5 mg/dL (range: 135 to 304 mg/dL) in the placebo group.
The safety and efficacy of doses above 40 mg daily have not been studied in children. The long-term efficacy of lovastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS)
The Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS), a double-blind, randomized, placebo-controlled, primary prevention study, demonstrated that treatment with lovastatin decreased the rate of acute major coronary events (composite endpoint of myocardial infarction, unstable angina, and sudden cardiac death) compared with placebo during a median of 5.1 years of follow-up. Participants were middle-aged and elderly men (ages 45 to 73) and women (ages 55 to 73) without symptomatic cardiovascular disease with average to moderately elevated total-C and LDL-C, below average HDL-C, and who were at high risk based on elevated total-C/HDL-C. In addition to age, 63% of the participants had at least one other risk factor (baseline HDL-C <35 mg/dL, hypertension, family history, smoking and diabetes).
AFCAPS/TexCaps enrolled 6,605 participants (5,608 men, 997 women) based on the following lipid entry criteria: total-C range of 180 to 264 mg/dL, LDL-C range of 130 to 190 mg/dL, HDL-C of ≤45 mg/dL for men and ≤47 mg/dL for women, and TG of ≤400 mg/dL. Participants were treated with standard care, including diet, and either lovastatin 20 to 40 mg daily (n= 3,304) or placebo (n= 3,301). Approximately 50% of the participants treated with lovastatin were titrated to 40 mg daily when their LDL-C remained >110 mg/dL at the 20-mg starting dose.
Lovastatin reduced the risk of a first acute major coronary event, the primary efficacy endpoint, by 37% (lovastatin 3.5%, placebo 5.5%; p<0.001; Figure 1). A first acute major coronary event was defined as myocardial infarction (54 participants on lovastatin, 94 on placebo) or unstable angina (54 vs. 80) or sudden cardiac death (8 vs. 9). Furthermore, among the secondary endpoints, lovastatin reduced the risk of unstable angina by 32% (1.8 vs. 2.6%; p=0.023), of myocardial infarction by 40% (1.7 vs. 2.9%; p=0.002), and of undergoing coronary revascularization procedures (e.g., coronary artery bypass grafting or percutaneous transluminal coronary angioplasty) by 33% (3.2 vs. 4.8%; p=0.001). Trends in risk reduction associated with treatment with lovastatin were consistent across men and women, smokers and non-smokers, hypertensives and non-hypertensives, and older and younger participants. Participants with ≥2 risk factors had risk reductions (RR) in both acute major coronary events (RR 43%) and coronary revascularization procedures (RR 37%). Because there were too few events among those participants with age as their only risk factor in this study, the effect of lovastatin on outcomes could not be adequately assessed in this subgroup.
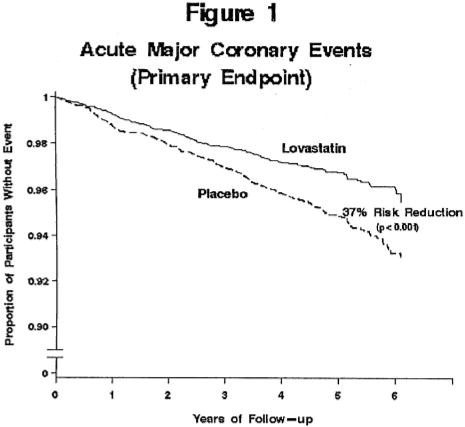
Atherosclerosis
In the Canadian Coronary Atherosclerosis Intervention Trial (CCAIT), the effect of therapy with lovastatin on coronary atherosclerosis was assessed by coronary angiography in hyperlipidemic patients. In the randomized, double-blind, controlled clinical trial, patients were treated with conventional measures (usually diet and 325 mg of aspirin every other day) and either lovastatin 20 to 80 mg daily or placebo. Angiograms were evaluated at baseline and at two years by computerized quantitative coronary angiography (QCA). Lovastatin significantly slowed the progression of lesions as measured by the mean change per-patient in minimum lumen diameter (the primary endpoint) and percent diameter stenosis, and decreased the proportions of patients categorized with disease progression (33% vs. 50%) and with new lesions (16% vs. 32%).
In a similarly designed trial, the Monitored Atherosclerosis Regression Study (MARS), patients were treated with diet and either lovastatin 80 mg daily or placebo. No statistically significant difference between lovastatin and placebo was seen for the primary endpoint (mean change per patient in percent diameter stenosis of all lesions), or for most secondary QCA endpoints. Visual assessment by angiographers who formed a consensus opinion of overall angiographic change (Global Change Score) was also a secondary endpoint. By this endpoint, significant slowing of disease was seen, with regression in 23% of patients treated with lovastatin compared to 11% of placebo patients.
In the Familial Atherosclerosis Treatment Study (FATS), either lovastatin or niacin in combination with a bile acid sequestrant for 2.5 years in hyperlipidemic subjects significantly reduced the frequency of progression and increased the frequency of regression of coronary atherosclerotic lesions by QCA compared to diet and, in some cases, low-dose resin.
The effect of lovastatin on the progression of atherosclerosis in the coronary arteries has been corroborated by similar findings in another vasculature. In the Asymptomatic Carotid Artery Progression Study (ACAPS), the effect of therapy with lovastatin on carotid atherosclerosis was assessed by B-mode ultrasonography in hyperlipidemic patients with early carotid lesions and without known coronary heart disease at baseline. In this double-blind, controlled clinical trial, 919 patients were randomized in a 2 x 2 factorial design to placebo, lovastatin 10 to 40 mg daily and/or warfarin. Ultrasonograms of the carotid walls were used to determine the change per patient from baseline to three years in mean maximum intimal-
medial thickness (IMT) of 12 measured segments. There was a significant regression of carotid lesions in patients receiving lovastatin alone compared to those receiving placebo alone (p=0.001). The predictive value of changes in IMT for stroke has not yet been established. In the lovastatin group there was a significant reduction in the number of patients with major cardiovascular events relative to the placebo group (5 vs. 14) and a significant reduction in all-cause mortality (1 vs. 8).
Eye
There was a high prevalence of baseline lenticular opacities in the patient population included in the early clinical trials with lovastatin. During these trials the appearance of new opacities was noted in both the lovastatin and placebo groups. There was no clinically significant change in visual acuity in the patients who had new opacities reported nor was any patient, including those with opacities noted at baseline, discontinued from therapy because of a decrease in visual acuity.
A three-year, double-blind, placebo-controlled study in hypercholesterolemic patients to assess the effect of lovastatin on the human lens demonstrated that there were no clinically or statistically significant differences between the lovastatin and placebo groups in the incidence, type or progression of lenticular opacities. There are no controlled clinical data assessing the lens available for treatment beyond three years.
-
INDICATIONS AND USAGE
Therapy with lovastatin should be a component of multiple risk factor intervention in those individuals with dyslipidemia at risk for atherosclerotic vascular disease. Lovastatin should be used in addition to a diet restricted in saturated fat and cholesterol as part of a treatment strategy to lower total-C and LDL-C to target levels when the response to diet and other nonpharmacological measures alone has been inadequate to reduce risk.
Primary Prevention of Coronary Heart Disease
In individuals without symptomatic cardiovascular disease, average to moderately elevated total-C and LDL-C, and below average HDL-C, lovastatin is indicated to reduce the risk of:
- Myocardial infarction
- Unstable angina
- Coronary revascularization procedures
Coronary Heart Disease
Lovastatin is indicated to slow the progression of coronary atherosclerosis in patients with coronary heart disease as part of a treatment strategy to lower total-C and LDL-C to target levels.
Hypercholesterolemia
Therapy with lipid-altering agents should be a component of multiple risk factor intervention in those individuals at significantly increased risk for artherosclerotic vascular disease due to hypercholesterolemia. Lovastatin is indicated as an adjunct to diet for the reduction of elevated total-C and LDL-C levels in patients with primary hypercholesterolemia (Types IIa and IIb 2), when the response to diet restricted in saturated fat and cholesterol and to other nonpharmacological measures alone has been inadequate.
Adolescent Patients with Heterozygous Familial Hypercholesterolemia
Lovastatin is indicated as an adjunct to diet to reduce total-C, LDL-C and apolipoprotein B levels in adolescent boys and girls who are at least one year post-menarche, 10 to 17 years of age, with heFH if after an adequate trial of diet therapy the following findings are present:
- LDL-C remains >189 mg/dL or
- LDL-C remains >160 mg/dL
and:
- there is a positive family history of premature cardiovascular disease or
- two or more other CVD risk factors are present in the adolescent patient
General Recommendations
Prior to initiating therapy with lovastatin, secondary causes for hypercholesterolemia (e.g., poorly controlled diabetes mellitus, hypothyroidism, nephrotic syndrome, dysproteinemias, obstructive liver disease, other drug therapy, alcoholism) should be excluded, and a lipid profile performed to measure total-C, HDL-C, and TG. For patients with TG less than 400 mg/dL (<4.5 mmol/L), LDL-C can be estimated using the following equation:
LDL-C = total-C - [0.2 × (TG) + HDL-C]
For TG levels >400 mg/dL (>4.5 mmol/L), this equation is less accurate and LDL-C concentrations should be determined by ultracentrifugation. In hypertriglyceridemic patients, LDL-C may be low or normal despite elevated total-C. In such cases, lovastatin is not indicated.
The National Cholesterol Education Program (NCEP) Treatment Guidelines are summarized below:
† CHD, coronary heart disease
†† Some authorities recommend use of LDL-lowering drugs in this category if an LDL-C level of <100 mg/dL cannot be achieved by therapeutic lifestyle changes. Others prefer use of drugs that primarily modify triglycerides and HDL-C, e.g., nicotinic acid or fibrate. Clinical judgment also may call for deferring drug therapy in this subcategory.
††† Almost all people with 0-1 risk factor have a 10-year risk <10%; thus, 10-year risk assessment in people with 0-1 risk factor is not necessary.
NCEP Treatment Guidelines:
LDL-C Goals and Cutpoints for Therapeutic Lifestyle Changes
and Drug Therapy in Different Risk CategoriesRisk Category LDL Goal
(mg/dL)LDL Level at Which to
Initiate Therapeutic
Lifestyle Changes
(mg/dL)LDL Level at Which to
Consider Drug Therapy
(mg/dL)CHD † or CHD risk equivalents
(10-year risk >20%)<100 ≥100 ≥130
(100-129: drug optional) ††2+ Risk factors
(10 year risk ≤20%)<130 ≥130 10-year risk 10-20%: ≥130
10-year risk <10%: ≥1600-1 Risk factor ††† <160 ≥160 ≥190
(160-189: LDL-lowering drug optional)2 Classification of Hyperlipoproteinemias Lipid Elevations Type Lipoproteins
elevatedmajor minor I chylomicrons TG ↑→C IIa LDL C -- IIb LDL, VLDL C TG III (rare) IDL C/TG -- IV VLDL TG ↑→C V (rare) chylomicrons, VLDL TG ↑→C IDL = intermediate-density lipoprotein. After the LDL-C goal has been achieved, if the TG is still ≥200 mg/dL, non-HDL-C (total-C minus HDL-C) becomes a secondary target of therapy. Non-HDL-C goals are set 30 mg/dL higher than LDL-C goals for each risk category.
At the time of hospitalization for an acute coronary event, consideration can be given to initiating drug therapy at discharge if the LDL-C is ≥130 mg/dL (see NCEP Guidelines above).
Since the goal of treatment is to lower LDL-C, the NCEP recommends that LDL-C levels be used to initiate and assess treatment response. Only if LDL-C levels are not available, should the total-C be used to monitor therapy.
Although lovastatin may be useful to reduce elevated LDL-C levels in patients with combined hypercholesterolemia and hypertriglyceridemia where hypercholesterolemia is the major abnormality (Type IIb hyperlipoproteinemia), it has not been studied in conditions where the major abnormality is elevation of chylomicrons, VLDL or IDL (i.e., hyperlipoproteinemia types I, III, IV, or V). ***
The NCEP classification of cholesterol levels in pediatric patients with a familial history of hypercholesterolemia or premature cardiovascular disease is summarized below:
Category Total-C (mg/dL) LDL-C (mg/dL) Acceptable <170 <110 Borderline 170-199 110-129 High ≥200 ≥130 Children treated with lovastatin in adolescence should be re-evaluated in adulthood and appropriate changes made to their cholesterol-lowering regimen to achieve adult goals for LDL-C.
-
CONTRAINDICATIONS
Hypersensitivity to any component of this medication.
Active liver disease or unexplained persistent elevations of serum transaminases (see WARNINGS ).
Concomitant administration with strong CYP3A4 inhibitors (e.g., itraconazole, ketoconazole, posaconazole, voriconazole, HIV protease inhibitors, boceprevir, telaprevir, erythromycin, clarithromycin, telithromycin ,nefazodone, and cobicistat-containing products) (see WARNINGS, Myopathy/Rhabdomyolysis) .
Pregnancy and lactation. (see PRECAUTIONS, Pregnancy and Nursing Mothers). Atherosclerosis is a chronic process and the discontinuation of lipid-lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hypercholesterolemia. Moreover, cholesterol and other products of the cholesterol biosynthesis pathway are essential components for fetal development, including synthesis of steroids and cell membranes. Because of the ability of inhibitors of HMG-CoA reductase such as lovastatin to decrease the synthesis of cholesterol and possibly other products of the cholesterol biosynthesis pathway, lovastatin is contraindicated during pregnancy and in nursing mothers. Lovastatin should be administered to women of childbearing age only when such patients are highly unlikely to conceive. If the patient becomes pregnant while taking this drug, lovastatin should be discontinued immediately and the patient should be apprised of the potential hazard to the fetus (see PRECAUTIONS, Pregnancy).
-
WARNINGS
Myopathy/Rhabdomyolysis
Lovastatin, like other inhibitors of HMG-CoA reductase, occasionally causes myopathy manifested as muscle pain, tenderness or weakness with creatine kinase (CK) above 10 times the upper limit of normal (ULN). Myopathy sometimes takes the form of rhabdomyolysis with or without acute renal failure secondary to myoglobinuria, and rare fatalities have occurred. The risk of myopathy is increased by high levels of HMG-CoA reductase inhibitory activity in plasma.
The risk of myopathy/rhabdomyolysis is dose related. In a clinical study (EXCEL) in which patients were carefully monitored and some interacting drugs were excluded, there was one case of myopathy among 4933 patients randomized to lovastatin 20 to 40 mg daily for 48 weeks, and 4 among 1649 patients randomized to 80 mg daily.
All patients starting therapy with lovastatin, or whose dose of lovastatin is being increased, should be advised of the risk of myopathy and told to report promptly any unexplained muscle pain, tenderness or weakness particularly if accompanied by malaise or fever or if muscle signs and symptoms persist after discontinuing Lovastatin. Lovastatin therapy should be discontinued immediately if myopathy is diagnosed or suspected. In most cases, muscle symptoms and CK increases resolved when treatment was promptly discontinued. Periodic CK determinations may be considered in patients starting therapy with lovastatin or whose dose is being increased, but there is no assurance that such monitoring will prevent myopathy.
Many of the patients who have developed rhabdomyolysis on therapy with lovastatin have had complicated medical histories, including renal insufficiency usually as a consequence of long-standing diabetes mellitus. Such patients merit closer monitoring. Lovastatin therapy should be discontinued if markedly elevated CPK levels occur or myopathy is diagnosed or suspected. Lovatsatin therapy should also be temporarily withheld in any patient experiencing an acute or serious condition predisposing to the development of renal failure secondary to rhabdomyolysis, e.g., sepsis; hypotension; major surgery; trauma; severe metabolic, endocrine, or electrolyte disorders; or uncontrolled epilepsy.
The risk of myopathy/rhabdomyolysis is increased by concomitant use of lovastatin with the following:
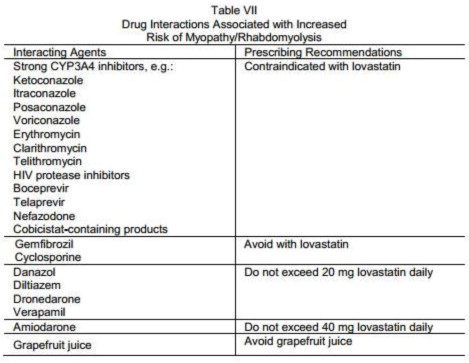
Strong inhibitors of CYP3A4: Lovastatin, like several other inhibitors of HMG-CoA reductase, is a substrate of cytochrome P450 3A4 (CYP3A4). Certain drugs which inhibit this metabolic pathway can raise the plasma levels of lovastatin and may increase the risk of myopathy. These include itraconazole, ketoconazole, and posaconazole, voriconazole, the macrolide antibiotics erythromycin and clarithromycin, the ketolide antibiotic telithromycin, HIV protease inhibitors, boceprevir, telaprevir, the antidepressant nefazodone, or cobicistat-containing products. Combination of these drugs with lovastatin is contraindicated. If short-term treatment with strong CYP3A4 inhibitors is unavoidable, therapy with lovastatin should be suspended during the course of treatment (see CONTRAINDICATIONS; PRECAUTIONS, Drug Interactions).
Other lipid-lowering drugs (other fibrates or ≥1 g/day of niacin): Caution should be used when prescribing other fibrates or lipid-lowering doses (≥1 g/day) of niacin with lovastatin, as these agents can cause myopathy when given alone. The benefit of further alterations in lipid levels by the combined use of lovastatin with other fibrates or niacin should be carefully weighed against the potential risks of these combinations.
Danazol, diltiazem, dronedarone or verapamil with higher doses of lovastatin: The dose of lovastatin should not exceed 20 mg daily in patients receiving concomitant medication with danazol, diltiazem, dronedarone, or verapamil. The benefits of the use of lovastatin in patients receiving danazol, diltiazem, dronedarone, or verapamil should be carefully weighed against the risks of these combinations.
Amiodarone: The dose of lovastatin should not exceed 40 mg daily in patients receiving concomitant medication with amiodarone. The combined use of lovastatin at doses higher than 40 mg daily with amiodarone should be avoided unless the clinical benefit is likely to outweigh the increased risk of myopathy. The risk of myopathy/rhabdomyolysis is increased when amiodarone is used concomitantly with higher doses of a closely related member of the HMG-CoA reductase inhibitor class.
Colchicine: Cases of myopathy, including rhabdomyolysis, have been reported with lovastatin coadministered with colchicine, and caution should be exercised when prescribing lovastatin with colchicine (see PRECAUTIONS, Drug Interactions).
Ranolazine: The risk of myopathy, including rhabdomyolysis, may be increased by concomitant administration of ranolazine. Dose adjustment of lovastatin may be considered during coadministration with ranolazine.
Prescribing recommendations for interacting agents are summarized in Table VII (see also CLINICAL PHARMACOLOGY, Pharmacokinetics; PRECAUTIONS, Drug Interactions; DOSAGE AND ADMINISTRATION).
Immune-Mediated Necrotizing Myopathy
There have been rare reports of immune-mediated necrotizing myopathy (IMNM), an autoimmune myopathy, associated with statin use. IMNM is characterized by: proximal muscle weakness and elevated serum creatine kinase, which persist despite discontinuation of statin treatment; positive anti-HMG CoA reductase antibody; muscle biopsy showing necrotizing myopathy; and improvement with immunosuppressive agents. Additional neuomuscular and serologic testing may be necessary. Treatment with immunosuppressive agents may be required. Consider risk of IMNM carefully prior to initiation of a different statin. If therapy is initiated with a different statin, monitor for signs and symptoms of IMNM.
Liver Dysfunction
Persistent increases (to more than 3 times the upper limit of normal) in serum transaminases occurred in 1.9% of adult patients who received lovastatin for at least one year in early clinical trials (see ADVERSE REACTIONS). When the drug was interrupted or discontinued in these patients, the transaminase levels usually fell slowly to pretreatment levels. The increases usually appeared 3 to 12 months after the start of therapy with lovastatin, and were not associated with jaundice or other clinical signs or symptoms. There was no evidence of hypersensitivity. In the EXCEL study (see CLINICAL PHARMACOLOGY , Clinical Studies ), the incidence of persistent increases in serum transaminases over 48 weeks was 0.1% for placebo, 0.1% at 20 mg/day, 0.9% at 40 mg/day, and 1.5% at 80 mg/day in patients on lovastatin. However, in post-marketing experience with lovastatin, symptomatic liver disease has been reported rarely at all dosages (see ADVERSE REACTIONS ).
In AFCAPS/TexCAPS, the number of participants with consecutive elevations of either alanine aminotransferase (ALT) or aspartate aminotransferase (AST) (> 3 times the upper limit of normal), over a median of 5.1 years of follow-up, was not significantly different between the lovastatin and placebo groups (18 [0.6%] vs. 11 [0.3%]). The starting dose of lovastatin was 20 mg/day; 50% of the lovastatin treated participants were titrated to 40 mg/day at Week 18. Of the 18 participants on lovastatin with consecutive elevations of either ALT or AST, 11 (0.7%) elevations occurred in participants taking 20 mg/day, while 7 (0.4%) elevations occurred in participants titrated to 40 mg/day. Elevated transaminases resulted in discontinuation of 6 (0.2%) participants from therapy in the lovastatin group (n=3,304) and 4 (0.1%) in the placebo group (n=3,301).
It is recommended that liver enzyme tests be obtained prior to initiating therapy with Lovastatin and repeated as clinically indicated.
There have been rare postmarketing reports of fatal and non-fatal hepatic failure in patients taking statins, including lovastatin. If serious liver injury with clinical symptoms and/or hyperbilirubinemia or jaundice occurs during treatment with Lovastatin, promptly interrupt therapy. If an alternate etiology is not found do not restart Lovastatin.
The drug should be used with caution in patients who consume substantial quantities of alcohol and/or have a past history of liver disease. Active liver disease or unexplained transaminase elevations are contraindications to the use of lovastatin.
Moderate (less than three times the upper limit of normal) elevations of serum transaminases have been reported following therapy with lovastatin (see ADVERSE REACTIONS ). These changes appeared soon after initiation of therapy with lovastatin, were often transient, were not accompanied by any symptoms and interruption of treatment was not required.
-
PRECAUTIONS
General
Lovastatin may elevate creatine phosphokinase and transaminase levels (see WARNINGS and ADVERSE REACTIONS ). This should be considered in the differential diagnosis of chest pain in a patient on therapy with lovastatin.
Homozygous Familial Hypercholesterolemia
Lovastatin is less effective in patients with the rare homozygous familial hypercholesterolemia, possibly because these patients have no functional LDL receptors. Lovastatin appears to be more likely to raise serum transaminases (see ADVERSE REACTIONS) in these homozygous patients.
Information for Patients
Patients should be advised about substances they should not take concomitantly with lovastatin and be advised to report promptly unexplained muscle pain, tenderness, or weakness particularly if accompanied by malaise or fever or if muscle signs and symptoms persist after discountinuing Lovastatin (see list below and WARNINGS , Myopathy/Rhabdomyolysis ) . Patients should also be advised to inform other physicians prescribing a new medication that they are taking lovastatin.
It is recommended that liver enzymes be checked before starting therapy, and if signs or symptoms of liver injury occur. All patients treated with Lovastatin should be advised to report promptly any symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice.
Drug Interactions
CYP3A4 Interactions
Lovastatin is metabolized by CYP3A4 but has no CYP3A4 inhibitory activity; therefore it is not expected to affect the plasma concentrations of other drugs metabolized by CYP3A4. Strong inhibitors of CYP3A4 (e.g., itraconazole, ketoconazole, posaconazole, voriconazole, clarithromycin, telithromycin, HIV protease inhibitors, boceprevir, telaprevir, nefazodone, erythromycin, and cobicistat-containing products), and grapefruit juice increase the risk of myopathy by reducing the elimination of lovastatin. (See CONTRAINDICATIONS, WARNINGS, Myopathy/Rhabdomyolysis, and CLINICAL PHARMACOLOGY, Pharmacokinetics.)
Interactions With Lipid-Lowering Drugs That Can Cause Myopathy When Given Alone
The risk of myopathy is also increased by the following lipid-lowering drugs that are not strong CYP3A4 inhibitors, but which can cause myopathy when given alone.
-
Gemfibrozil
-
Other fibrates
-
Niacin (nicotinic acid) (≥1 g/day)
Other drug interactions
Cyclosporine: The risk of myopathy/rhabdomyolysis is increased by concomitant administration of cyclosporine (see WARNINGS, Myopathy/Rhabdomyolysis).
Danazol, Diltiazem, Dronedarone, or Verapamil: The risk of myopathy/rhabdomyolysis is increased by concomitant administration of danazol, diltiazem, dronedarone or verapamil particularly with higher doses of lovastatin (see WARNINGS, Myopathy/Rhabdomyolysis; CLINICAL PHARMACOLOGY, Pharmacokinetics).
Amiodarone: The risk of myopathy/rhabdomyolysis is increased when amiodarone is used concomitantly with a closely related member of the HMG-CoA reductase inhibitor class (see WARNINGS, Myopathy/Rhabdomyolysis).
Coumarin Anticoagulants: In a small clinical trial in which lovastatin was administered to warfarin treated patients; no effect on prothrombin time was detected. However, another HMG-CoA reductase inhibitor has been found to produce a less than two seconds increase in prothrombin time in healthy volunteers receiving low doses of warfarin. Also, bleeding and/or increased prothrombin time have been reported in a few patients taking coumarin anticoagulants concomitantly with lovastatin. It is recommended that in patients taking anticoagulants, prothrombin time be determined before starting lovastatin and frequently enough during early therapy to insure that no significant alteration of prothrombin time occurs. Once a stable prothrombin time has been documented, prothrombin times can be monitored at the intervals usually recommended for patients on coumarin anticoagulants. If the dose of lovastatin is changed, the same procedure should be repeated. Lovastatin therapy has not been associated with bleeding or with changes in prothrombin time in patients not taking anticoagulants.
Colchicine: Cases of myopathy, including rhabdomyolysis, have been reported with lovastatin coadministered with colchicine. See WARNINGS, Myopathy/Rhabdomyolysis.
Ranolazine: The risk of myopathy, including rhabdomyolysis, may be increased by concomitant administration of ranolazine. See WARNINGS, Myopathy/Rhabdomyolysis.
Propranolol: In normal volunteers, there was no clinically significant pharmacokinetic or pharmacodynamic interaction with concomitant administration of single doses of lovastatin and propranolol.
Digoxin: In patients with hypercholesterolemia, concomitant administration of lovastatin and digoxin resulted in no effect on digoxin plasma concentrations.
Oral Hypoglycemic Agents: In pharmacokinetic studies of lovastatin in hypercholesterolemic non-insulin dependent diabetic patients, there was no drug interaction with glipizide or with chlorpropamide (see CLINICAL PHARMACOLOGY, Clinical Studies ).
Endocrine Function
Increases in HbA1c and fasting serum glucose levels have been reported with HMG-CoA reductase inhibitors, including Lovastatin.
HMG-CoA reductase inhibitors interfere with cholesterol synthesis and as such might theoretically blunt adrenal and/or gonadal steroid production. Results of clinical trials with drugs in this class have been inconsistent with regard to drug effects on basal and reserve steroid levels. However, clinical studies have shown that lovastatin does not reduce basal plasma cortisol concentration or impair adrenal reserve, and does not reduce basal plasma testosterone concentration. Another HMG-CoA reductase inhibitor has been shown to reduce the plasma testosterone response to HCG. In the same study, the mean testosterone response to HCG was slightly but not significantly reduced after treatment with lovastatin 40 mg daily for 16 weeks in 21 men. The effects of HMG-CoA reductase inhibitors on male fertility have not been studied in adequate numbers of male patients. The effects, if any, on the pituitary-gonadal axis in premenopausal women are unknown. Patients treated with lovastatin who develop clinical evidence of endocrine dysfunction should be evaluated appropriately. Caution should also be exercised if an HMG-CoA reductase inhibitor or other agent used to lower cholesterol levels is administered to patients also receiving other drugs (e.g., spironolactone, cimetidine) that may decrease the levels or activity of endogenous steroid hormones.
CNS Toxicity
Lovastatin produced optic nerve degeneration (Wallerian degeneration of retinogeniculate fibers) in clinically normal dogs in a dose-dependent fashion starting at 60 mg/kg/day, a dose that produced mean plasma drug levels about 30 times higher than the mean drug level in humans taking the highest recommended dose (as measured by total enzyme inhibitory activity). Vestibulocochlear Wallerian-like degeneration and retinal ganglion cell chromatolysis were also seen in dogs treated for 14 weeks at 180 mg/kg/day, a dose which resulted in a mean plasma drug level (C max) similar to that seen with the 60 mg/kg/day dose.
CNS vascular lesions, characterized by perivascular hemorrhage and edema, mononuclear cell infiltration of perivascular spaces, perivascular fibrin deposits and necrosis of small vessels, were seen in dogs treated with lovastatin at a dose of 180 mg/kg/day, a dose which produced plasma drug levels (C max) which were about 30 times higher than the mean values in humans taking 80 mg/day.
Similar optic nerve and CNS vascular lesions have been observed with other drugs of this class.
Cataracts were seen in dogs treated for 11 and 28 weeks at 180 mg/kg/day and 1 year at 60 mg/kg/day.
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 21-month carcinogenic study in mice, there was a statistically significant increase in the incidence of hepatocellular carcinomas and adenomas in both males and females at 500 mg/kg/day. This dose produced a total plasma drug exposure 3 to 4 times that of humans given the highest recommended dose of lovastatin (drug exposure was measured as total HMG-CoA reductase inhibitory activity in extracted plasma). Tumor increases were not seen at 20 and 100 mg/kg/day, doses that produced drug exposures of 0.3 to 2 times that of humans at the 80 mg/day dose. A statistically significant increase in pulmonary adenomas was seen in female mice at approximately 4 times the human drug exposure. (Although mice were given 300 times the human dose [HD] on a mg/kg body weight basis, plasma levels of total inhibitory activity were only 4 times higher in mice than in humans given 80 mg of lovastatin.)
There was an increase in incidence of papilloma in the non-glandular mucosa of the stomach of mice beginning at exposures of 1 to 2 times that of humans. The glandular mucosa was not affected. The human stomach contains only glandular mucosa.
In a 24-month carcinogenicity study in rats, there was a positive dose response relationship for hepatocellular carcinogenicity in males at drug exposures between 2 to 7 times that of human exposure at 80 mg/day (doses in rats were 5, 30 and 180 mg/kg/day).
An increased incidence of thyroid neoplasms in rats appears to be a response that has been seen with other HMG-CoA reductase inhibitors.
A chemically similar drug in this class was administered to mice for 72 weeks at 25, 100, and 400 mg/kg body weight, which resulted in mean serum drug levels approximately 3, 15, and 33 times higher than the mean human serum drug concentration (as total inhibitory activity) after a 40 mg oral dose. Liver carcinomas were significantly increased in high dose females and mid- and high dose males, with a maximum incidence of 90 percent in males. The incidence of adenomas of the liver was significantly increased in mid- and high dose females. Drug treatment also significantly increased the incidence of lung adenomas in mid- and high dose males and females. Adenomas of the Harderian gland (a gland of the eye of rodents) were significantly higher in high dose mice than in controls.
No evidence of mutagenicity was observed in a microbial mutagen test using mutant strains of Salmonella typhimurium with or without rat or mouse liver metabolic activation. In addition, no evidence of damage to genetic material was noted in an in vitro alkaline elution assay using rat or mouse hepatocytes, a V-79 mammalian cell forward mutation study, an in vitro chromosome aberration study in CHO cells, or an in vivo chromosomal aberration assay in mouse bone marrow.
Drug-related testicular atrophy, decreased spermatogenesis, spermatocytic degeneration and giant cell formation were seen in dogs starting at 20 mg/kg/day. Similar findings were seen with another drug in this class. No drug-related effects on fertility were found in studies with lovastatin in rats. However, in studies with a similar drug in this class, there was decreased fertility in male rats treated for 34 weeks at 25 mg/kg body weight, although this effect was not observed in a subsequent fertility study when this same dose was administered for 11 weeks (the entire cycle of spermatogenesis, including epididymal maturation). In rats treated with this same reductase inhibitor at 180 mg/kg/day, seminiferous tubule degeneration (necrosis and loss of spermatogenic epithelium) was observed. No microscopic changes were observed in the testes from rats of either study. The clinical significance of these findings is unclear.
Pregnancy
Pregnancy Category X
See CONTRAINDICATIONS.
Safety in pregnant women has not been established.
Lovastatin has been shown to produce skeletal malformations in offspring of pregnant mice and rats dosed during gestation at 80 mg/kg/day (affected mouse fetuses/total: 8/307 compared to 4/289 in the control group; affected rat fetuses/total: 6/324 compared to 2/308 in the control group). Female rats dosed before mating through gestation at 80 mg/kg/day also had fetuses with skeletal malformations (affected fetuses/total: 1/152 compared to 0/171 in the control group). The 80 mg/kg/day dose in mice is 7 times the human dose based on body surface area and in rats results in 5 times the human exposure based on AUC. In pregnant rats given doses of 2, 20, or 200 mg/kg/day and treated through lactation, the following effects were observed: neonatal mortality (4.1%, 3.5%, and 46%, respectively, compared to 0.6% in the control group), decreased pup body weights throughout lactation (up to 5%, 8%, and 38%, respectively, below control), supernumerary ribs in dead pups (affected fetuses/total: 0/7, 1/17, and11/79, respectively, compared to 0/5 in the control group), delays in ossification in dead pups (affected fetuses/total: 0/7, 0/17, and 1/79, respectively, compared to 0/5 in the control group) and delays in pup development (delays in the appearance of an auditory startle response at 200 mg/kg/day and free-fall righting reflexes at 20 and 200 mg/kg/day).
Direct dosing of neonatal rats by subcutaneous injection with 10 mg/kg/day of the open hydroxyacid form of lovastatin resulted in delayed passive avoidance learning in female rats (mean of 8.3 trials to criterion, compared to 7.3 and 6.4 in untreated and vehicle-treated controls; no effects on retention 1 week later) at exposures 4 times the human systemic exposure at 80 mg/day based on AUC. No effect was seen in male rats. No evidence of malformations was observed when pregnant rabbits were given 5 mg/kg/day (doses equivalent to a human dose of 80 mg/day based on body surface area) or a maternally toxic dose of 15 mg/kg/day (3 times the human dose of 80 mg/day based on body surface area).
Rare clinical reports of congenital anomalies following intrauterine exposure to HMG-CoA reductase inhibitors have been received. However, in an analysis† of greater than 200 prospectively followed pregnancies exposed during the first trimester to lovastatin or another closely related HMG-CoA reductase inhibitor, the incidence of congenital anomalies was comparable to that seen in the general population. This number of pregnancies was sufficient to exclude a 3-fold or greater increase in congenital anomalies over the background incidence.
Maternal treatment with lovastatin may reduce the fetal levels of mevalonate, which is a precursor of cholesterol biosynthesis. Atherosclerosis is a chronic process, and ordinarily discontinuation of lipid-lowering drugs during pregnancy should have little impact on the long-term risk associated with primary hypercholesterolemia. For these reasons, lovastatin should not be used in women who are pregnant, or can become pregnant (see CONTRAINDICATIONS). Lovastatin should be administered to women of childbearing potential only when such patients are highly unlikely to conceive and have been informed of the potential hazards. Treatment should be immediately discontinued as soon as pregnancy is recognized.
Nursing Mothers
It is not known whether lovastatin is excreted in human milk. Because a small amount of another drug in this class is excreted in human breast milk and because of the potential for serious adverse reactions in nursing infants, women taking lovastatin should not nurse their infants (see CONTRAINDICATIONS).
Pediatric Use
Safety and effectiveness in patients 10 to 17 years of age with heFH have been evaluated in controlled clinical trials of 48 weeks duration in adolescent boys and controlled clinical trials of 24 weeks duration in girls who were at least 1 year post-menarche. Patients treated with lovastatin had an adverse experience profile generally similar to that of patients treated with placebo. Doses greater than 40 mg have not been studied in this population. In these limited controlled studies, there was no detectable effect on growth or sexual maturation in the adolescent boys or on menstrual cycle length in girls. See CLINICAL PHARMACOLOGY, Clinical Studies in Adolescent Patients ; ADVERSE REACTIONS , Adolescent Patients ; and DOSAGE AND ADMINISTRATION , Adolescent Patients (10 to 17 years of age) with Heterozygous Familial Hypercholesterolemia. Adolescent females should be counseled on appropriate contraceptive methods while on lovastatin therapy (see CONTRAINDICATIONS and PRECAUTIONS , Pregnancy ). Lovastatin has not been studied in pre-pubertal patients or patients younger than 10 years of age.
Geriatric Use
A pharmacokinetic study with lovastatin showed the mean plasma level of HMG-CoA reductase inhibitory activity to be approximately 45% higher in elderly patients between 70 to 78 years of age compared with patients between 18 to 30 years of age; however, clinical study experience in the elderly indicates that dosage adjustment based on this age-related pharmacokinetic difference is not needed. In the two large clinical studies conducted with lovastatin (EXCEL and AFCAPS/TexCAPS), 21% (3094/14850) of patients were ≥65 years of age. Lipid-lowering efficacy with lovastatin was at least as great in elderly patients compared with younger patients, and there were no overall differences in safety over the 20 to 80 mg/day dosage range (see CLINICAL PHARMACOLOGY ). Because advanced age (≥65 years) is a predisposing factor for myopathy, including rhabdomyolysis, Lovastatin should be prescribed with caution in the elderly.
-
ADVERSE REACTIONS
____________________________________
3 Manson, J.M., Freyssinges, C., Ducrocq, M.B., Stephenson, W.P., Postmarketing Surveillance of Lovastatin and Simvastatin Exposure During Pregnancy. Reproductive Toxicology. 10(6):439-446. 1996.
Phase III Clinical Studies
In Phase III controlled clinical studies involving 613 patients treated with lovastatin, the adverse experience profile was similar to that shown below for the 8,245-patient EXCEL study (see Expanded Clinical Evaluation of Lovastatin [EXCEL] Study ).
Persistent increases of serum transaminases have been noted (see WARNINGS, Liver Dysfunction ). About 11% of patients had elevations of CK levels of at least twice the normal value on one or more occasions. The corresponding values for the control agent cholestyramine was 9 percent. This was attributable to the noncardiac fraction of CK. Large increases in CK have sometimes been reported (see WARNINGS, Myopathy/Rhabdomyolysis ).
Expanded Clinical Evaluation of Lovastatin (EXCEL) Study
Lovastatin was compared to placebo in 8,245 patients with hypercholesterolemia (total-C 240-300 mg/dL [6.2-7.8 mmol/L]) in the randomized, double-blind, parallel, 48-week EXCEL study. Clinical adverse experiences reported as possibly, probably or definitely drug-related in ≥1% in any treatment group are shown in the table below. For no event was the incidence on drug and placebo statistically different.
Placebo
(N=1663)
%Lovastatin
20 mg q.p.m.
(N=1642)
%Lovastatin
40 mg q.p.m.
(N=1645)
%Lovastatin
20 mg b.i.d.
(N=1646)
%Lovastatin
40 mg b.i.d.
(N=1649)
%Body As a Whole Asthenia 1.4 1.7 1.4 1.5 1.2 Gastrointestinal Abdominal pain 1.6 2.0 2.0 2.2 2.5 Constipation 1.9 2.0 3.2 3.2 3.5 Diarrhea 2.3 2.6 2.4 2.2 2.6 Dyspepsia 1.9 1.3 1.3 1.0 1.6 Flatulence 4.2 3.7 4.3 3.9 4.5 Nausea 2.5 1.9 2.5 2.2 2.2 Musculoskeletal Muscle cramps 0.5 0.6 0.8 1.1 1.0 Myalgia 1.7 2.6 1.8 2.2 3.0 Nervous System/Psychiatric Dizziness 0.7 0.7 1.2 0.5 0.5 Headache 2.7 2.6 2.8 2.1 3.2 Skin Rash 0.7 0.8 1.0 1.2 1.3 Special Senses Blurred vision 0.8 1.1 0.9 0.9 1.2 Other clinical adverse experiences reported as possibly, probably or definitely drug-related in 0.5 to 1.0 percent of patients in any drug-treated group are listed below. In all these cases the incidence on drug and placebo was not statistically different. Body as a Whole: chest pain; Gastrointestinal: acid regurgitation, dry mouth, vomiting; Musculoskeletal: leg pain, shoulder pain, arthralgia; Nervous System/Psychiatric: insomnia, paresthesia; Skin: alopecia, pruritus; Special Senses: eye irritation.
In the EXCEL study (see CLINICAL PHARMACOLOGY, Clinical Studies ), 4.6% of the patients treated up to 48 weeks were discontinued due to clinical or laboratory adverse experiences which were rated by the investigator as possibly, probably or definitely related to therapy with lovastatin. The value for the placebo group was 2.5%.
Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS)
In AFCAPS/TexCAPS (see CLINICAL PHARMACOLOGY , Clinical Studies ) involving 6,605 participants treated with 20 to 40 mg/day of lovastatin (n=3,304) or placebo (n=3,301), the safety and tolerability profile of the group treated with lovastatin was comparable to that of the group treated with placebo during a median of 5.1 years of follow-up. The adverse experiences reported in AFCAPS/TexCAPS were similar to those reported in EXCEL (see ADVERSE REACTIONS , Expanded Clinical Evaluation of Lovastatin (EXCEL) Study ).
Concomitant Therapy
In controlled clinical studies in which lovastatin was administered concomitantly with cholestyramine, no adverse reactions peculiar to this concomitant treatment were observed. The adverse reactions that occurred were limited to those reported previously with lovastatin or cholestyramine. Other lipid-lowering agents were not administered concomitantly with lovastatin during controlled clinical studies. Preliminary data suggests that the addition of gemfibrozil to therapy with lovastatin is not associated with greater reduction in LDL-C than that achieved with lovastatin alone. In uncontrolled clinical studies, most of the patients who have developed myopathy were receiving concomitant therapy with cyclosporine, gemfibrozil or niacin (nicotinic acid). The combined use of lovastatin with cyclosporine or gemfibrozil should be avoided. Caution should be used when prescribing other fibrates or lipid-lowering doses (≥1 g/day) of niacin with lovastatin (see WARNINGS, Myopathy/Rhabdomyolysis).
The following effects have been reported with drugs in this class. Not all the effects listed below have necessarily been associated with lovastatin therapy.
Skeletal: muscle cramps, myalgia, myopathy, rhabdomyolysis, arthralgias.
There have been rare reports of immune-mediated necrotizing myopathy associated with statin use [see WARNINGS, Myopathy/Rhabdomyolysis].
Neurological: dysfunction of certain cranial nerves (including alteration of taste, impairment of extra-ocular movement, facial paresis), tremor, dizziness, vertigo, paresthesia, peripheral neuropathy, peripheral nerve palsy, psychic disturbances, anxiety, insomnia, depression.
There have been rare postmarketing reports of cognitive impairment (e.g., memory loss, forgetfulness, amnesia, memory impairment, confusion) associated with statin use. These cognitive issues have been reported for all statins. The reports are generally nonserious, and reversible upon statin discontinuation, with variable times to symptom onset (1 day to years) and symptom resolution (median of 3 weeks).
Hypersensitivity Reactions: An apparent hypersensitivity syndrome has been reported rarely which has included one or more of the following features: anaphylaxis, angioedema, lupus erythematous-like syndrome, polymyalgia rheumatica, dermatomyositis, vasculitis, purpura, thrombocytopenia, leukopenia, hemolytic anemia, positive ANA, ESR increase, eosinophilia, arthritis, arthralgia, urticaria, asthenia, photosensitivity, fever, chills, flushing, malaise, dyspnea, toxic epidermal necrolysis, erythema multiforme, including Stevens-Johnson syndrome.
Gastrointestinal: pancreatitis, hepatitis, including chronic active hepatitis, cholestatic jaundice, fatty change in liver; and rarely, cirrhosis, fulminant hepatic necrosis, and hepatoma; anorexia, vomiting, fatal and non-fatal hepatic failure.
Skin: alopecia, pruritus. A variety of skin changes (e.g., nodules, discoloration, dryness of skin/mucous membranes, changes to hair/nails) have been reported.
Reproductive: gynecomastia, loss of libido, erectile dysfunction.
Eye: progression of cataracts (lens opacities), ophthalmoplegia.
Laboratory Abnormalities: elevated transaminases, alkaline phosphatase, γ-glutamyl transpeptidase, and bilirubin; thyroid function abnormalities.
Respiratory: interstitial lung disease
Adolescent Patients (ages 10 to 17 years)
In a 48-week controlled study in adolescent boys with heFH (n=132) and a 24-week controlled study in girls who were at least 1 year post-menarche with heFH (n=54), the safety and tolerability profile of the groups treated with lovastatin (10 to 40 mg daily) was generally similar to that of the groups treated with placebo (see CLINICAL PHARMACOLOGY, Clinical Studies in Adolescent Patients and PRECAUTIONS, Pediatric Use ).
-
OVERDOSAGE
After oral administration of lovastatin to mice, the median lethal dose observed was >15 g/m 2.
Five healthy human volunteers have received up to 200 mg of lovastatin as a single dose without clinically significant adverse experiences. A few cases of accidental overdosage have been reported; no patients had any specific symptoms, and all patients recovered without sequelae. The maximum dose taken was 5 to 6 g.
Until further experience is obtained, no specific treatment of overdosage with lovastatin can be recommended.
The dialyzability of lovastatin and its metabolites in man is not known at present.
-
DOSAGE AND ADMINISTRATION
The patient should be placed on a standard cholesterol-lowering diet before receiving lovastatin and should continue on this diet during treatment with lovastatin (see NCEP Treatment Guidelines for details on dietary therapy). Lovastatin should be given with meals.
Adult Patients
The usual recommended starting dose is 20 mg once a day given with the evening meal. The recommended dosing range is 10 to 80 mg/day in single or two divided doses; the maximum recommended dose is 80 mg/day. Doses should be individualized according to the recommended goal of therapy (see NCEP Guidelines and CLINICAL PHARMACOLOGY ). Patients requiring reductions in LDL-C of 20% or more to achieve their goal (see INDICATIONS AND USAGE) should be started on 20 mg/day of lovastatin. A starting dose of 10 mg may be considered for patients requiring smaller reductions. Adjustments should be made at intervals of 4 weeks or more.
Cholesterol levels should be monitored periodically and consideration should be given to reducing the dosage of lovastatin if cholesterol levels fall significantly below the targeted range.
-
Dosage in Patients taking Danazol, Diltiazem, Dronedarone, or Verapamil
In patients taking danazol, diltiazem, dronedarone or verapamil concomitantly with lovastatin, therapy should begin with 10 mg of lovastatin and should not exceed 20 mg /day (see CLINICAL PHARMACOLOGY, Pharmacokinetics, WARNINGS, Myopathy/Rhabdomyolysis, PRECAUTIONS, Drug Interactions, Other Drug Interactions).
Dosage in Patients taking Amiodarone
In patients taking amiodarone concomitantly with lovastatin, the dose should not exceed 40 mg/day (see WARNINGS , Myopathy/Rhabdomyolysis and PRECAUTIONS , Drug Interactions , Other drug interactions ).
Adolescent Patients (10 to 17 years of age) with Heterozygous Familial Hypercholesterolemia
The recommended dosing range is 10 to 40 mg/day; the maximum recommended dose is 40 mg/day. Doses should be individualized according to the recommended goal of therapy (see NCEP Pediatric Panel Guidelines ††, CLINICAL PHARMACOLOGY , and INDICATIONS AND USAGE ). Patients requiring reductions in LDL-C of 20% or more to achieve their goal should be started on 20 mg/day of lovastatin. A starting dose of 10 mg may be considered for patients requiring smaller reductions. Adjustments should be made at intervals of 4 weeks or more.
Concomitant Lipid-Lowering Therapy
Lovastatin is effective alone or when used concomitantly with bile-acid sequestrants (see WARNINGS, Myopathy/Rhabdomyolysis and PRECAUTIONS, Drug Interactions).
Dosage in Patients with Renal Insufficiency
In patients with severe renal insufficiency (creatinine clearance <30 mL/min), dosage increases above 20 mg/day should be carefully considered and, if deemed necessary, implemented cautiously (see CLINICAL PHARMACOLOGY and WARNINGS, Myopathy/Rhabdomyolysis ).
-
HOW SUPPLIED
Lovastatin Tablets USP (white to off white round, unscored tablets) containing 10mg of lovastatin and engraved with “CTI 141"
Bottle of 60..................................................................................................................................(NDC 63629-1018-6)
Bottle of 90..................................................................................................................................(NDC 63629-1018-9)
Bottle of 100................................................................................................................................(NDC 63629-1018-1)
Bottle of 500................................................................................................................................(NDC 63629-1018-5)
Bottle of 1,000.............................................................................................................................(NDC 63629-1018-0)
Bottle of 30..................................................................................................................................(NDC 63629-1018-2)
Bottle of 300................................................................................................................................(NDC 63629-1018-3)
Bottle of 120................................................................................................................................(NDC 63629-1018-4)
Bottle of 800................................................................................................................................(NDC 63629-1018-8)
4 National Cholesterol Education Program (NCEP): Highlights of the Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Pediatrics . 89(3):495-501, 1992.
Storage
Store at 20º to 25º C (68º to 77º F). [See USP Controlled Room Temperature.] Lovastatin Tablets must be protected from light and stored in a well-closed, light-resistant container. - PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LOVASTATIN
lovastatin tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63629-1018(NDC:61442-141) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LOVASTATIN (UNII: 9LHU78OQFD) (LOVASTATIN - UNII:9LHU78OQFD) LOVASTATIN 10 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLOXAMER 188 (UNII: LQA7B6G8JG) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) STARCH, CORN (UNII: O8232NY3SJ) TALC (UNII: 7SEV7J4R1U) BUTYLATED HYDROXYANISOLE (UNII: REK4960K2U) Product Characteristics Color white (WHITE) Score no score Shape ROUND (ROUND) Size 6mm Flavor Imprint Code CTI;141 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63629-1018-1 100 in 1 BOTTLE; Type 0: Not a Combination Product 10/21/2021 2 NDC:63629-1018-2 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/21/2021 3 NDC:63629-1018-3 300 in 1 BOTTLE; Type 0: Not a Combination Product 10/21/2021 4 NDC:63629-1018-4 120 in 1 BOTTLE; Type 0: Not a Combination Product 12/16/2019 5 NDC:63629-1018-5 500 in 1 BOTTLE; Type 0: Not a Combination Product 12/10/2019 6 NDC:63629-1018-6 60 in 1 BOTTLE; Type 0: Not a Combination Product 10/21/2021 7 NDC:63629-1018-8 800 in 1 BOTTLE; Type 0: Not a Combination Product 10/21/2021 8 NDC:63629-1018-9 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/21/2021 9 NDC:63629-1018-0 1000 in 1 BOTTLE; Type 0: Not a Combination Product 10/21/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA075991 11/25/2002 Labeler - Bryant Ranch Prepack (171714327) Registrant - Bryant Ranch Prepack (171714327) Establishment Name Address ID/FEI Business Operations Bryant Ranch Prepack 171714327 repack(63629-1018) , relabel(63629-1018)
